Characterization of Iron Core–Gold Shell Nanoparticles for Anti-Cancer Treatments: Chemical and Structural Transformations During Storage and Use

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
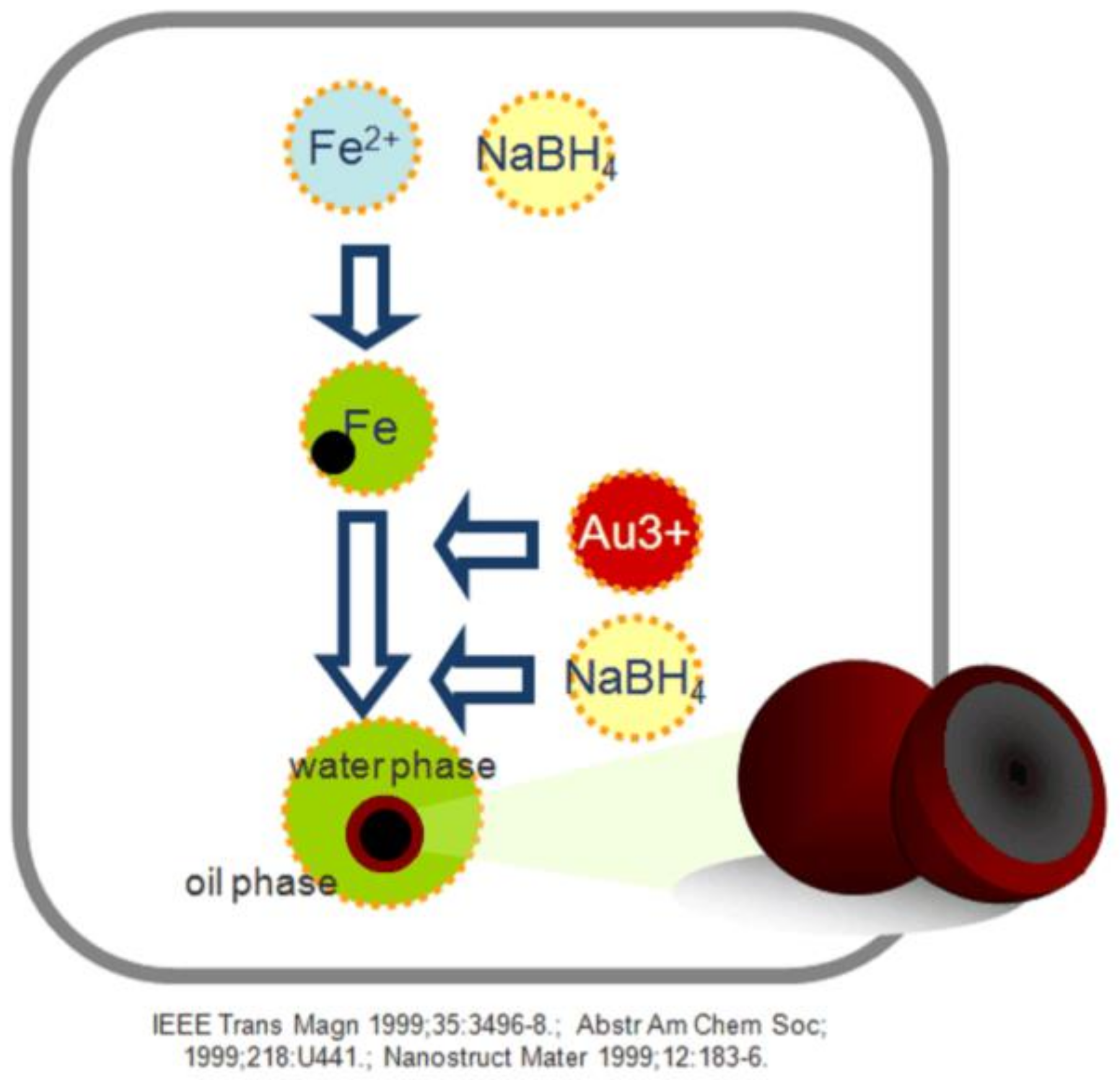
2.2. Synthesis of Fe@Au Nanoparticles
2.3. Characterization of Fe@Au
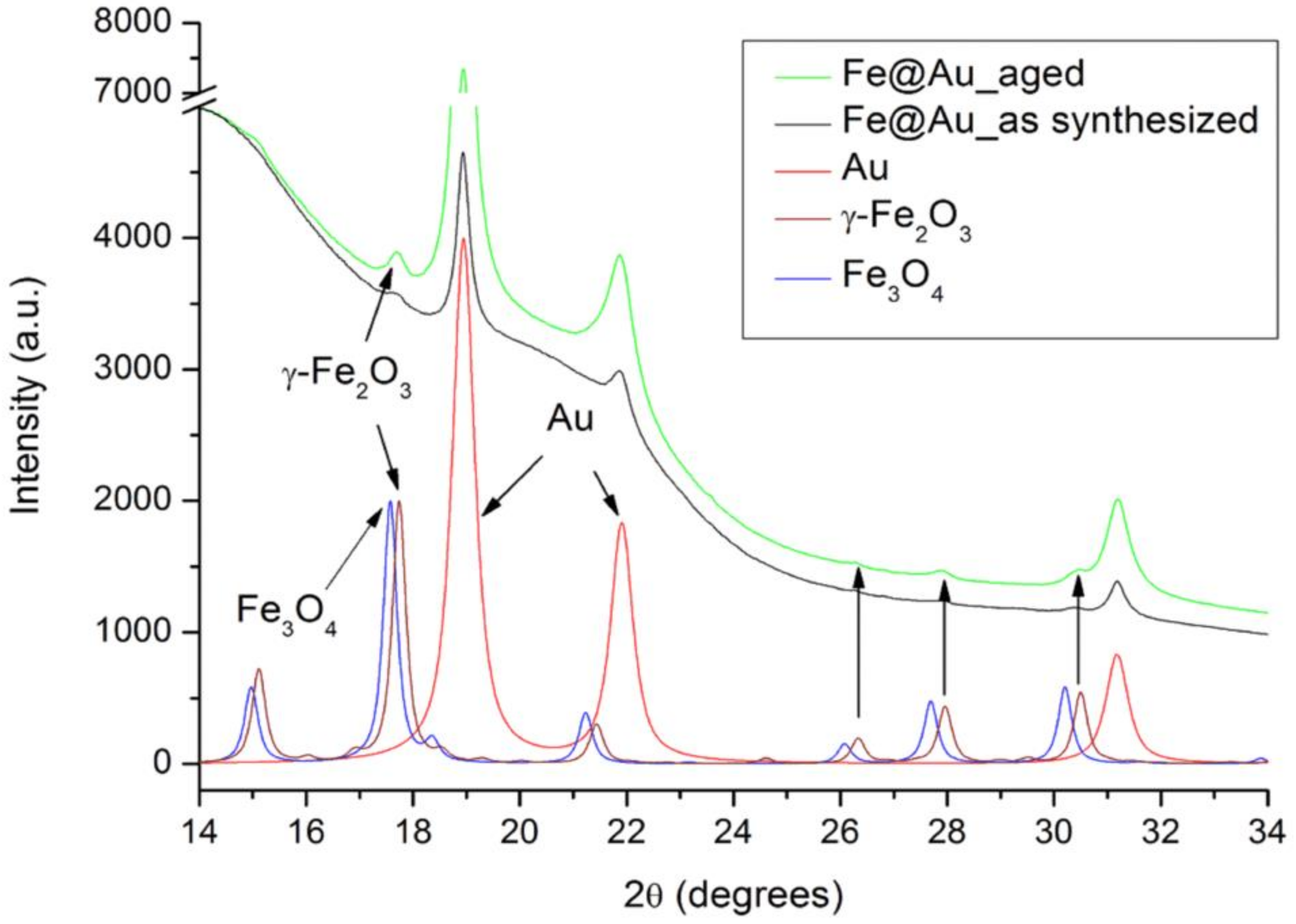
2.4. X-ray Diffraction Analysis of Fe@Au
2.5. Cell Culture
2.6. WST-1 Cell Viability Assay
2.7. Transmission Electron Microscopy of Treated Cells
2.8. Statistical Analysis
3. Results
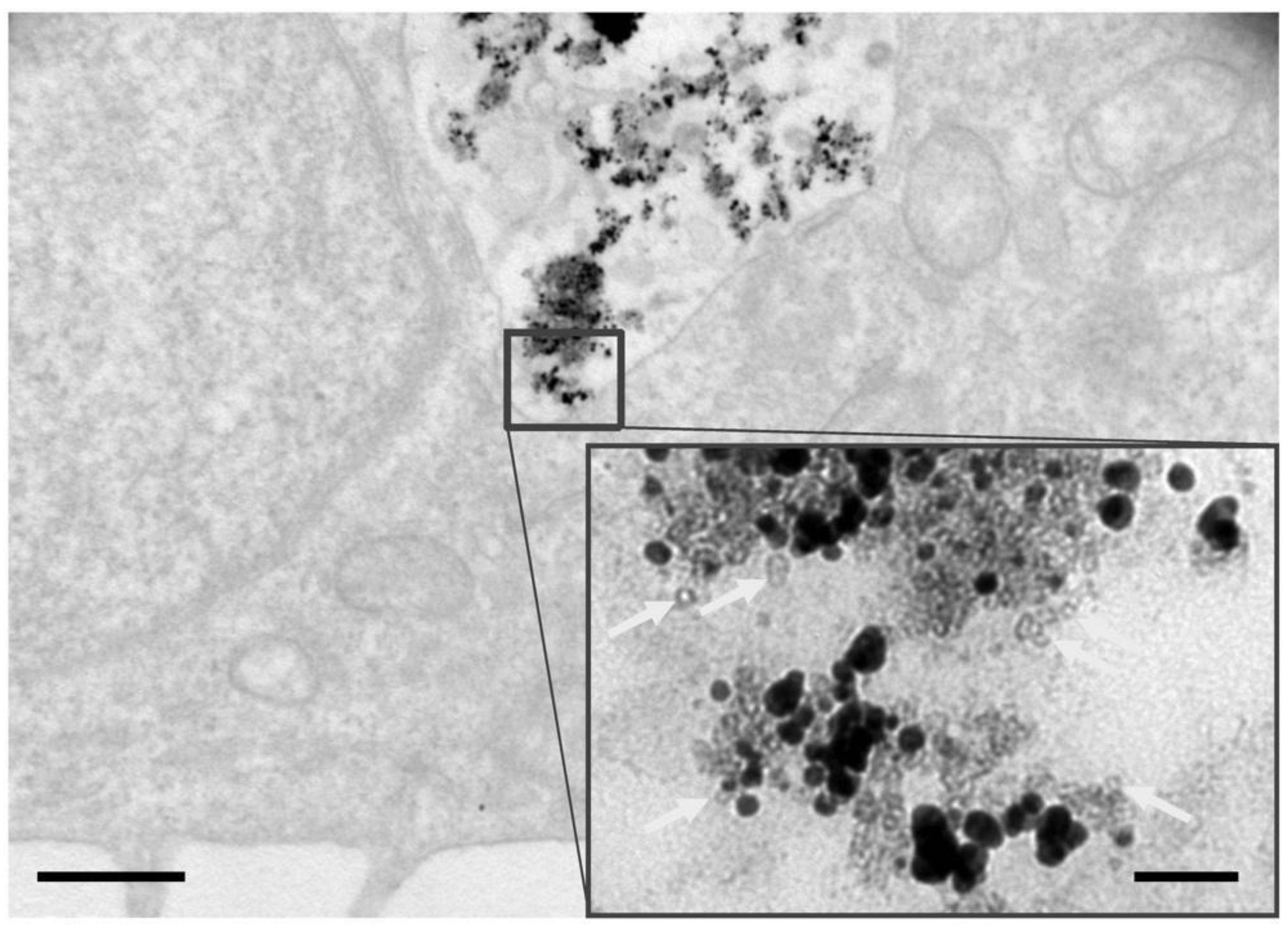
3.1. Structural and Chemical Transformations of Fe@Au In Vitro and In Vivo
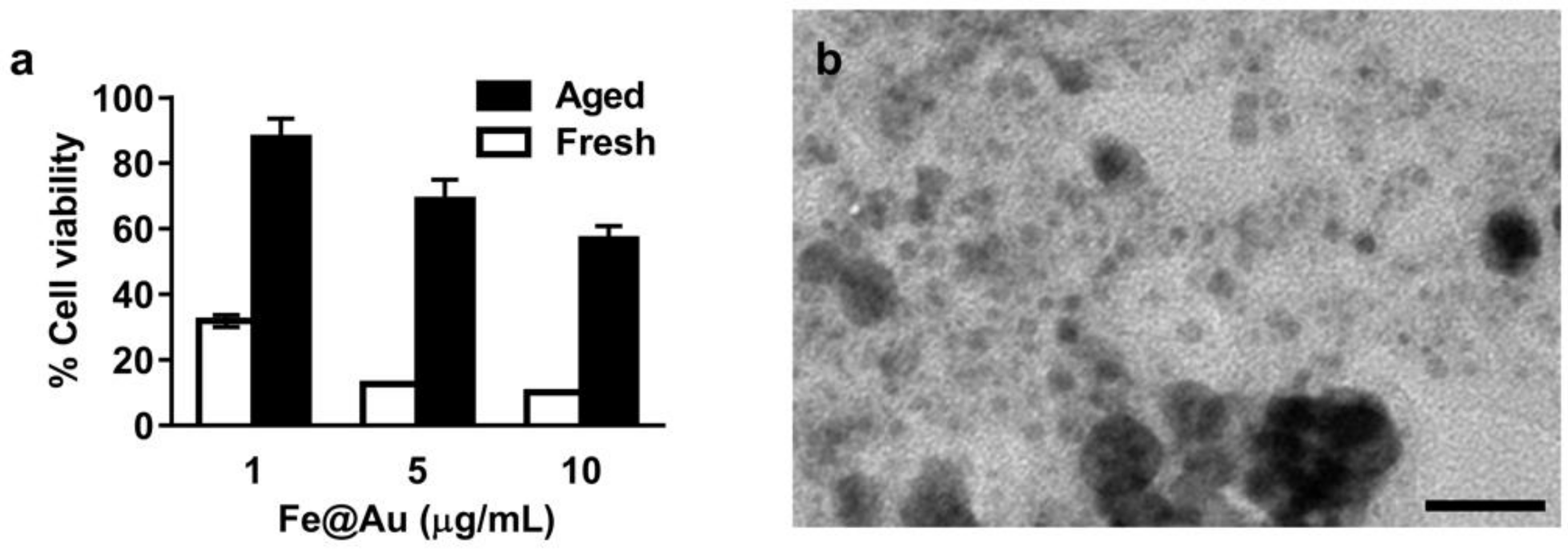
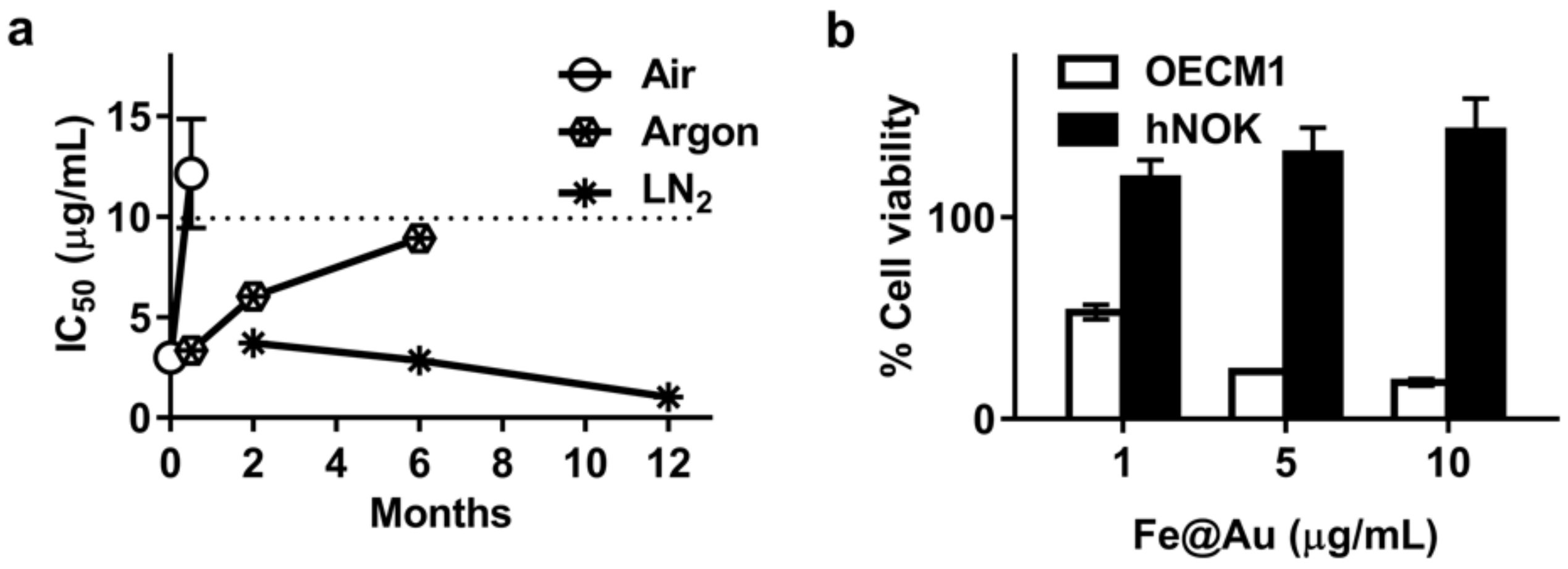
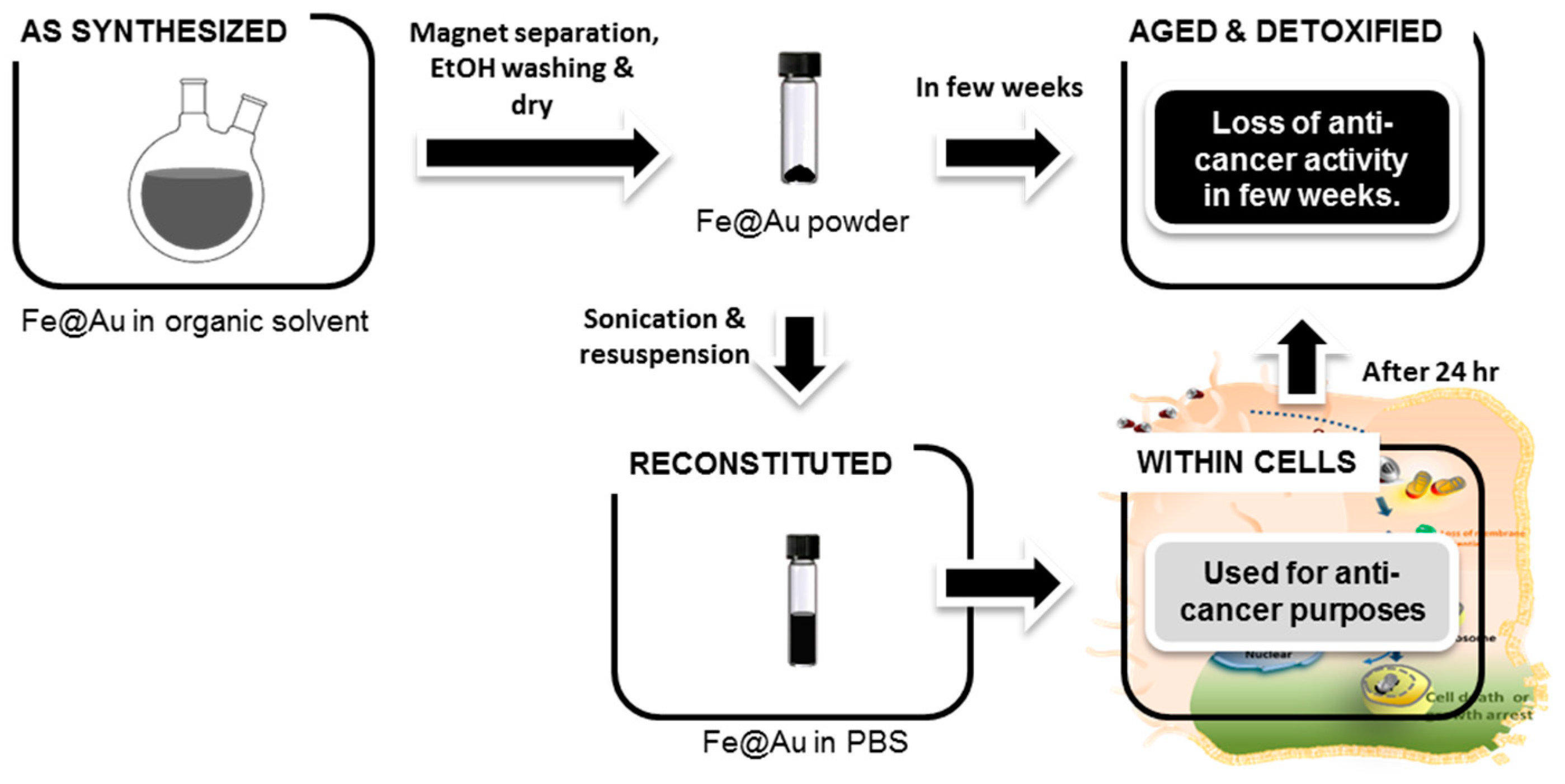
3.2. Aging of Fe@Au Nanoparticles and Their Diminished Anti-Cancer Activity

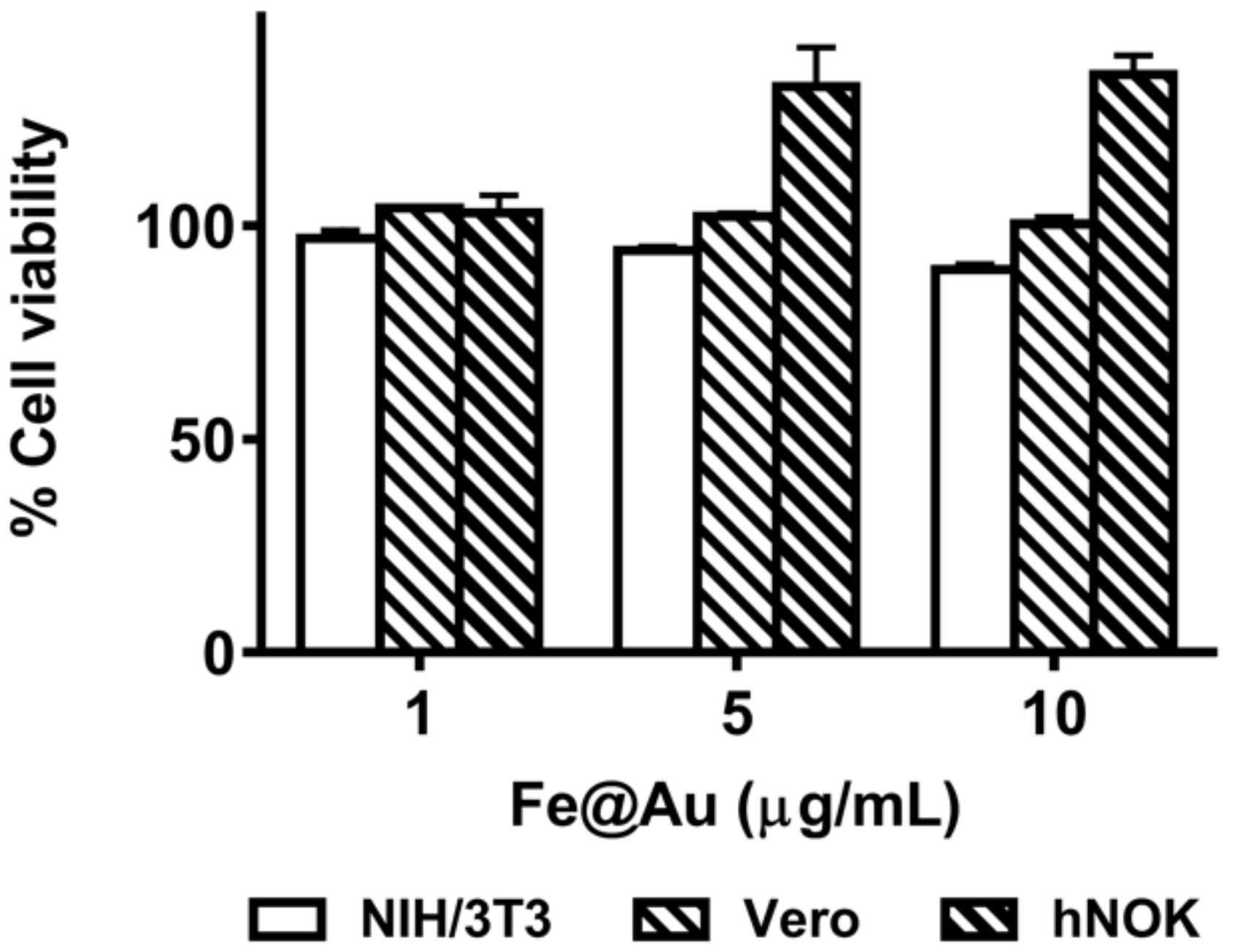
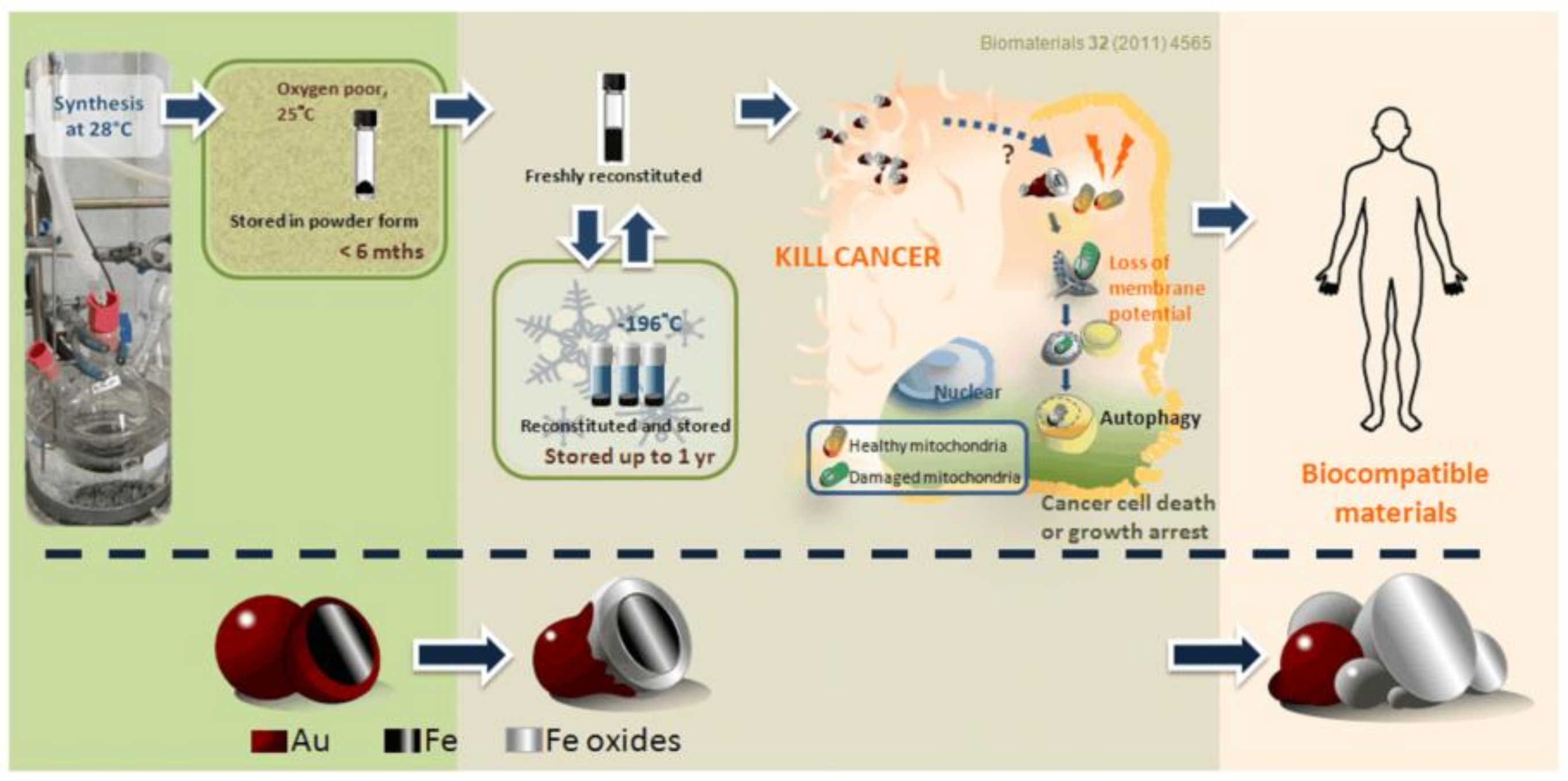
3.3. Long-Term Storage of Fe@Au Nanoparticles and Preservation of Their Anti-Cancer Properties
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Ervik, M.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. GLOBOCAN 2012, Cancer Incidence and Mortality Worldwide: IARC. Available online: http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx (accessed on 8 November 2018).
- Beger, R.D.; Sun, J.; Schnackenberg, L.K. Metabolomics approaches for discovering biomarkers of drug-induced hepatotoxicity and nephrotoxicity. Toxicol. Appl. Pharmacol. 2010, 243, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Superfin, D.; Iannucci, A.A.; Davies, A.M. Commentary: Oncologic Drugs in Patients with Organ Dysfunction: A Summary. Oncologist 2007, 12, 1070–1083. [Google Scholar] [CrossRef] [Green Version]
- Baratelli, C.; Zichi, C.; Di Maio, M.; Brizzi, M.P.; Sonetto, C.; Scagliotti, G.V.; Tampellini, M. A systematic review of the safety profile of the different combinations of fluoropyrimidines and oxaliplatin in the treatment of colorectal cancer patients. Crit. Rev. Oncol. Hematol. 2018, 122, 21–29. [Google Scholar] [CrossRef]
- Jain, T.K.; Reddy, M.K.; Morales, M.A.; Leslie-Pelecky, D.L.; Labhasetwar, V. Biodistribution, clearance and biocompatibility of iron oxide magnetic nanoparticles in rats. Mol. Pharmacol. 2008, 5, 316–327. [Google Scholar] [CrossRef]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef]
- Li, Z.; Greden, K.; Alvarez, P.J.; Gregory, K.B.; Lowry, G.V. Adsorbed polymer and NOM limits adhesion and toxicity of nano scale zerovalent iron to E. coli. Environ. Sci. Technol. 2010, 44, 3462–3467. [Google Scholar] [CrossRef]
- Lee, C.; Kim, J.Y.; Lee, W.I.; Nelson, K.L.; Yoon, J.; Sedlak, D.L. Bactericidal effect of zero-valent iron nanoparticles on Escherichia coli. Environ. Sci. Technol. 2008, 42, 4927–4933. [Google Scholar] [CrossRef] [PubMed]
- Karn, B.; Kuiken, T.; Otto, M. Nanotechnology and in situ remediation: A review of the benefits and potential risks. Environ. Health Perspect. 2009, 117, 1813–1831. [Google Scholar] [CrossRef] [PubMed]
- Cundy, A.B.; Hopkinson, L.; Whitby, R.L. Use of iron-based technologies in contaminated land and groundwater remediation: A review. Sci. Total Environ. 2008, 400, 42–51. [Google Scholar] [CrossRef]
- Jiang, D.; Zeng, G.; Huang, D.; Chen, M.; Zhang, C.; Huang, C.; Wan, J. Remediation of contaminated soils by enhanced nanoscale zero valent iron. Environ. Res. 2018, 163, 217–227. [Google Scholar] [CrossRef]
- Asl, B.A.; Mogharizadeh, L.; Khomjani, N.; Rasti, B.; Pishva, S.P.; Akhtari, K.; Attar, F.; Falahati, M. Probing the interaction of zero valent iron nanoparticles with blood system by biophysical, docking, cellular and molecular studies. Int. J. Biol. Macromol. 2018, 109, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, S.; Hadadi, Z.; Attar, F.; Mousavi, S.E.; Zargar, S.S.; Tajik, A.; Saboury, A.A.; Rezayat, S.M.; Falahati, M. ROS-mediated heme degradation and cytotoxicity induced by iron nanoparticles: Hemoglobin and lymphocyte cells as targets. J. Biomol. Struct. Dyn. 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Keenan, C.R.; Goth-Goldstein, R.; Lucas, D.; Sedlak, D.L. Oxidative stress induced by zero-valent iron nanoparticles and Fe(II) in human bronchial epithelial cells. Environ. Sci. Technol. 2009, 43, 4555–4560. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.L.; Carpenter, E.E.; Lin, J.; Kumbhar, A.; Sims, J.; O’Connor, C.J. Nanostructures of gold coated iron core-shell nanoparticles and the nanobands assembled under magnetic field. Eur. Phys. J. D 2001, 16, 289–292. [Google Scholar] [CrossRef]
- Cho, S.J.; Idrobo, J.C.; Olamit, J.; Liu, K.; Browning, N.D.; Kauzlarich, S.M. Growth mechanisms and oxidation resistance of gold-coated iron nanoparticles. Chem. Mater. 2005, 17, 3181–3186. [Google Scholar] [CrossRef]
- Kayal, S.; Ramanujan, R.V. Anti-cancer drug loaded iron-gold core-shell nanoparticles (Fe@Au) for magnetic drug targeting. J. Nanosci. Nanotechnol. 2010, 10, 5527–5539. [Google Scholar] [CrossRef]
- Ho, D.; Sun, X.; Sun, S. Monodisperse magnetic nanoparticles for theranostic applications. Acc. Chem. Res. 2011, 44, 875–882. [Google Scholar] [CrossRef]
- Wu, Y.N.; Chen, D.H.; Shi, X.Y.; Lian, C.C.; Wang, T.Y.; Yeh, C.S.; Ratinac, K.R.; Thordarson, P.; Braet, F.; Shieh, D.B. Cancer-cell-specific cytotoxicity of non-oxidized iron elements in iron core-gold shell NPs. Nanomed. NBM 2011, 7, 420–427. [Google Scholar] [CrossRef]
- Wu, Y.N.; Wu, P.C.; Yang, L.X.; Ratinac, K.R.; Thordarson, P.; Jahn, K.A.; Chen, D.H.; Shieh, D.B.; Braet, F. The anticancer properties of iron core-gold shell nanoparticles in colorectal cancer cells. Int. J. Nanomed. 2013, 8, 3321–3331. [Google Scholar] [CrossRef]
- Wu, Y.N.; Yang, L.X.; Shi, X.Y.; Li, I.C.; Biazik, J.M.; Ratinac, K.R.; Chen, D.H.; Thordarson, P.; Shieh, D.B.; Braet, F. The selective growth inhibition of oral cancer by iron core-gold shell nanoparticles through mitochondria-mediated autophagy. Biomaterials 2011, 32, 4565–4573. [Google Scholar] [CrossRef]
- Liu, Y.C.; Leu, C.M.; Wong, F.H.; Fong, W.S.; Chen, S.C.; Chang, C.; Hu, C. Autocrine Stimulation by Insulin-Like Growth Factor I Is Involved in the Growth, Tumorigenicity and Chemoresistance of Human Esophageal Carcinoma Cells. J. Biomed. Sci. 2002, 9, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.S.; Kaga, K.; Tsuzuku, T.; Uno, A. Effects of primary auditory cortex lesions on middle latency responses in awake cats. Auris Nasus Larynx 1993, 20, 155–165. [Google Scholar] [CrossRef]
- Alkilany, A.M.; Nagaria, P.K.; Hexel, C.R.; Shaw, T.J.; Murphy, C.J.; Wyatt, M.D. Cellular Uptake and Cytotoxicity of Gold Nanorods: Molecular Origin of Cytotoxicity and Surface Effects. Small 2009, 5, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Petosa, A.R.; Jaisi, D.P.; Quevedo, I.R.; Elimelech, M.; Tufenkji, N. Aggregation and deposition of engineered nanomaterials in aquatic environments: Role of physicochemical interactions. Environ. Sci. Technol. 2010, 44, 6532–6549. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Zhou, W.L.; Kumbhar, A.; Wiemann, J.; Fang, J.Y.; Carpenter, E.E.; O’Connor, C.J. Gold-coated iron (Fe@Au) nanoparticles: Synthesis, characterization and magnetic field-induced self-assembly. J. Solid State Chem. 2001, 159, 26–31. [Google Scholar] [CrossRef]
- Coffey, H.T.; Keller, E.L.; Patterson, A.; Autler, S.H. Effect of Low-Temperature Deuteron Irradiation on Some Type-2 Superconductors. Phys. Rev. 1967, 155, 355–363. [Google Scholar] [CrossRef]
- Patterson, A.L. The Scherrer Formula for X-Ray Particle Size Determination. Phys. Rev. 1939, 56, 978–982. [Google Scholar] [CrossRef]
- Scherrer, P. Bestimmung der Grosse und Derinneren Struktur von Kolloidteilchen Mittels Rontgenstrahlen. Nachrichten von der Gesellschaft der Wissenschaften zu Gottingen, Mathematisch-Physikalische Klasse aus der Jahre 1918, 26, 98–100. [Google Scholar]
- Mindyuk, A.K. Temperature dependence of the rate of acid corrosion of iron and steel. Mater. Sci. 1977, 12, 164–166. [Google Scholar] [CrossRef]
- Lease, N.; Vasilevski, V.; Carreira, M.; De Almeida, A.; Sanau, M.; Hirva, P.; Casini, A.; Contel, M. Potential anticancer heterometallic Fe-Au and Fe-Pd agents: initial mechanistic insights. J. Med. Chem. 2013, 56, 5806–5818. [Google Scholar] [CrossRef]
- Wang, C.M.; Baer, D.R.; Thomas, L.E.; Amonette, J.E.; Antony, J.; Qiang, Y.; Duscher, G. Void formation during early stages of passivation: Initial oxidation of iron nanoparticles at room temperature. J. Appl. Phys. 2005, 98, 094308. [Google Scholar] [CrossRef]
- Cabot, A.; Puntes, V.F.; Shevchenko, E.; Yin, Y.; Balcells, L.; Marcus, M.A.; Hughes, S.M.; Alivisatos, A.P. Vacancy coalescence during oxidation of iron nanoparticles. J. Am. Chem. Soc. 2007, 129, 10358–10360. [Google Scholar] [CrossRef] [PubMed]
- Augustine, R.; Dominic, E.A.; Reju, I.; Kaimal, B.; Kalarikkal, N.; Thomas, S. Electrospun polycaprolactone membranes incorporated with ZnO nanoparticles as skin substitutes with enhanced fibroblast proliferation and wound healing. RSC Adv. 2014, 4, 24777–24785. [Google Scholar] [CrossRef]
- Lu, Z.; Gao, J.; He, Q.; Wu, J.; Liang, D.; Yang, H.; Chen, R. Enhanced antibacterial and wound healing activities of microporous chitosan-Ag/ZnO composite dressing. Carbohydr. Polym. 2017, 156, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Chen, K.; He, X.; Li, N.; Huang, J.; Tang, K.; Li, Y.; Wang, F. Nano-TiO2/collagen-chitosan porous scaffold for wound repairing. Int. J. Biol. Macromol. 2016, 91, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Augustine, R.; Mathew, A.P.; Sosnik, A. Metal Oxide Nanoparticles as Versatile Therapeutic Agents Modulating Cell Signaling Pathways: Linking Nanotechnology with Molecular Medicine. Appl. Mater. Today 2017, 7, 91–103. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Weight (%) ± Standard Dev. | Atomic (%) |
|---|---|---|
| (a) Freshly prepared Fe@Au, as shown in Figure 1a | ||
| Fe K | 20.6 ± 0.4 | 47.9 |
| Au L | 79.4 ± 0.4 | 52.1 |
| (b) The dense area of reconstituted Fe@Au, as marked with white asterisk in Figure 1b | ||
| Fe K | 3.7 ± 0.3 | 12.0 |
| Au L | 96.3 ± 0.3 | 88.0 |
| (c) A less-dense area of reconstituted Fe@Au, as marked with black asterisk in Figure 1b | ||
| Fe K | 61.2 ± 1.7 | 37.0 |
| Au L | 13.8 ± 1.6 | 1.7 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.-N.; Shieh, D.-B.; Yang, L.-X.; Sheu, H.-S.; Zheng, R.; Thordarson, P.; Chen, D.-H.; Braet, F. Characterization of Iron Core–Gold Shell Nanoparticles for Anti-Cancer Treatments: Chemical and Structural Transformations During Storage and Use. Materials 2018, 11, 2572. https://doi.org/10.3390/ma11122572
Wu Y-N, Shieh D-B, Yang L-X, Sheu H-S, Zheng R, Thordarson P, Chen D-H, Braet F. Characterization of Iron Core–Gold Shell Nanoparticles for Anti-Cancer Treatments: Chemical and Structural Transformations During Storage and Use. Materials. 2018; 11(12):2572. https://doi.org/10.3390/ma11122572
Chicago/Turabian StyleWu, Ya-Na, Dar-Bin Shieh, Li-Xing Yang, Hwo-Shuenn Sheu, Rongkun Zheng, Pall Thordarson, Dong-Hwang Chen, and Filip Braet. 2018. "Characterization of Iron Core–Gold Shell Nanoparticles for Anti-Cancer Treatments: Chemical and Structural Transformations During Storage and Use" Materials 11, no. 12: 2572. https://doi.org/10.3390/ma11122572