
Effects of Six Weeks of Hypoxia Exposure on Hepatic Fatty Acid Metabolism in ApoE Knockout Mice Fed a High-Fat Diet

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
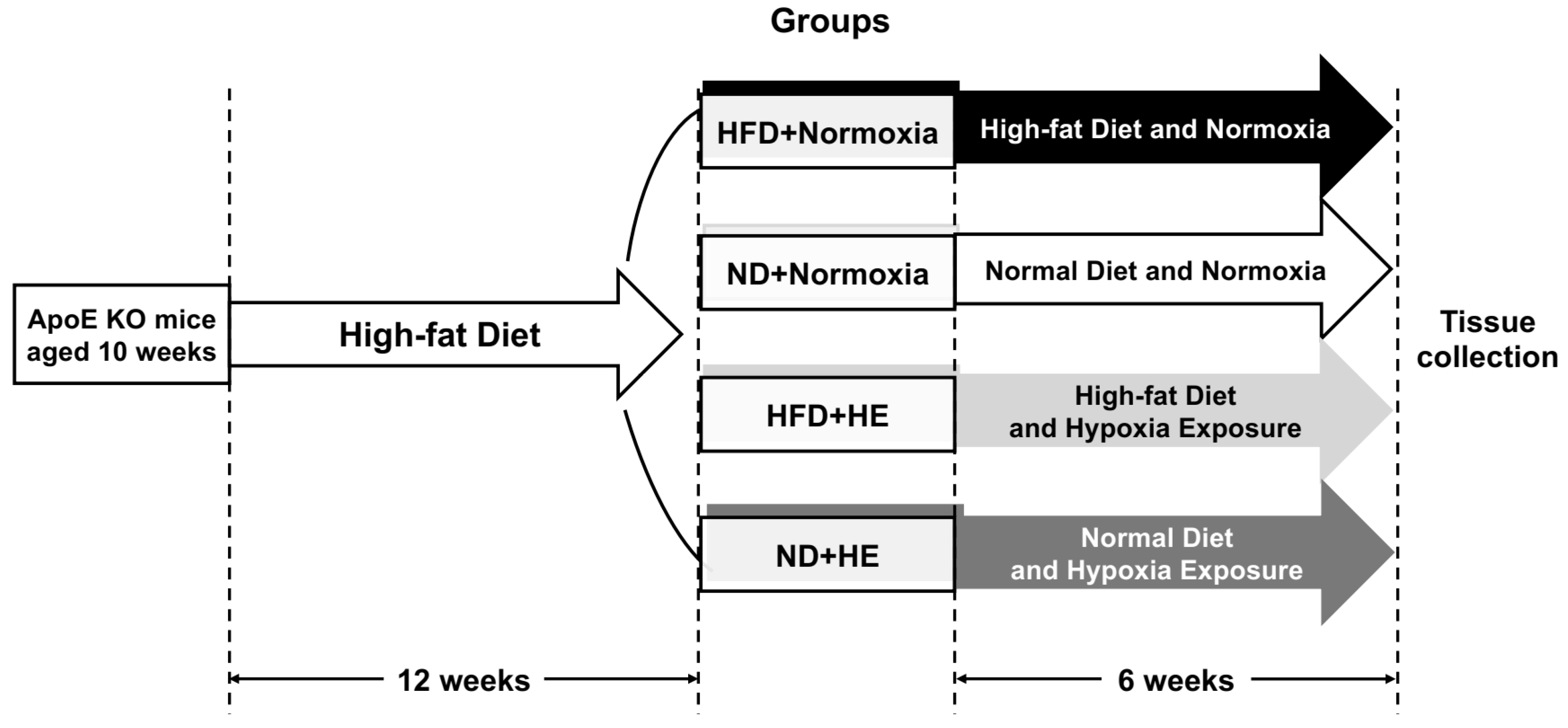
2.1. Animals
2.2. Body Weight, Food and Energy Intake
2.3. Plasma Lipid Profiles
2.4. Hepatic Lipid Contents, Plasma ALT and AST Levels
2.5. Real-Time Quantitative PCR Analysis
2.6. Western Blotting
2.7. Activities of FASN and ACC
2.8. Statistical Analysis
3. Results
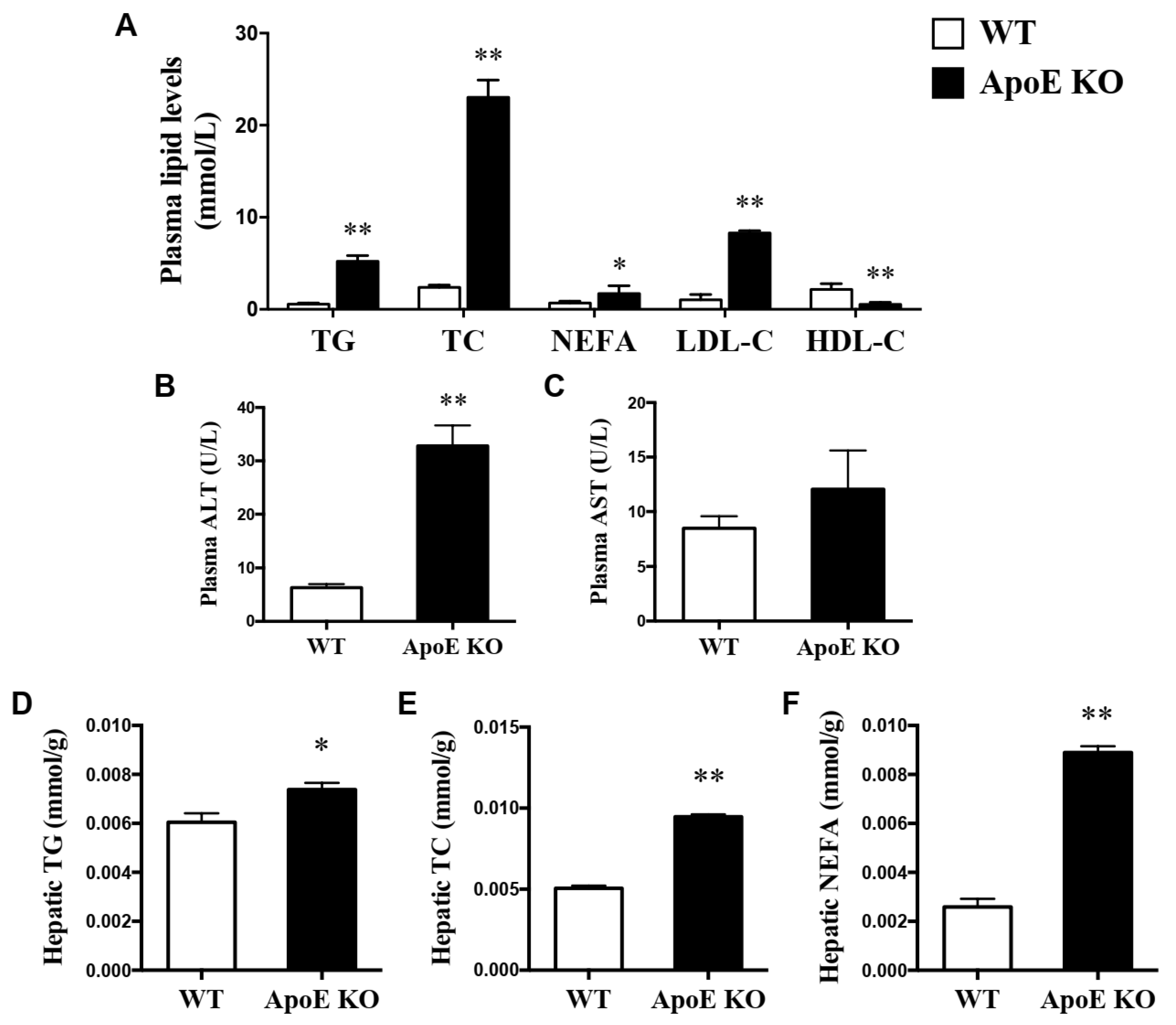
3.1. Changes in Plasma Lipid Profiles, ALT, AST Levels and Hepatic Lipid Profiles between WT and NAFLD Model
3.2. Changes in Body Weight, Food and Energy Intake
3.3. Changes in Plasma Lipid Profiles, ALT, AST Levels and Hepatic Lipid Profiles
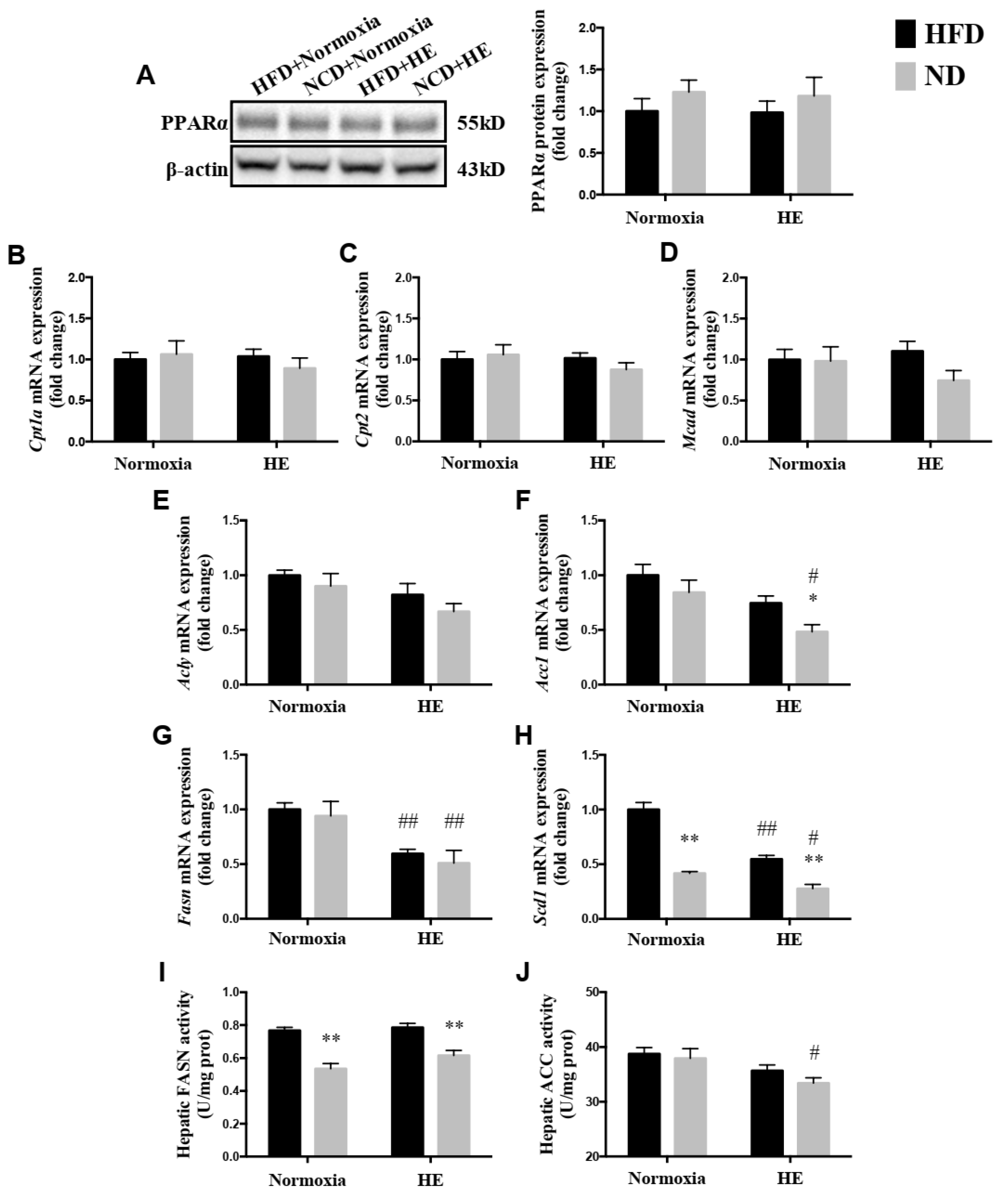
3.4. Changes in Hepatic PPARα Protein Expression, mRNA Expression Levels of Genes Involved in Mitochondrial Fatty Acid Oxidation and Synthesis, and Activities of FASN and ACC
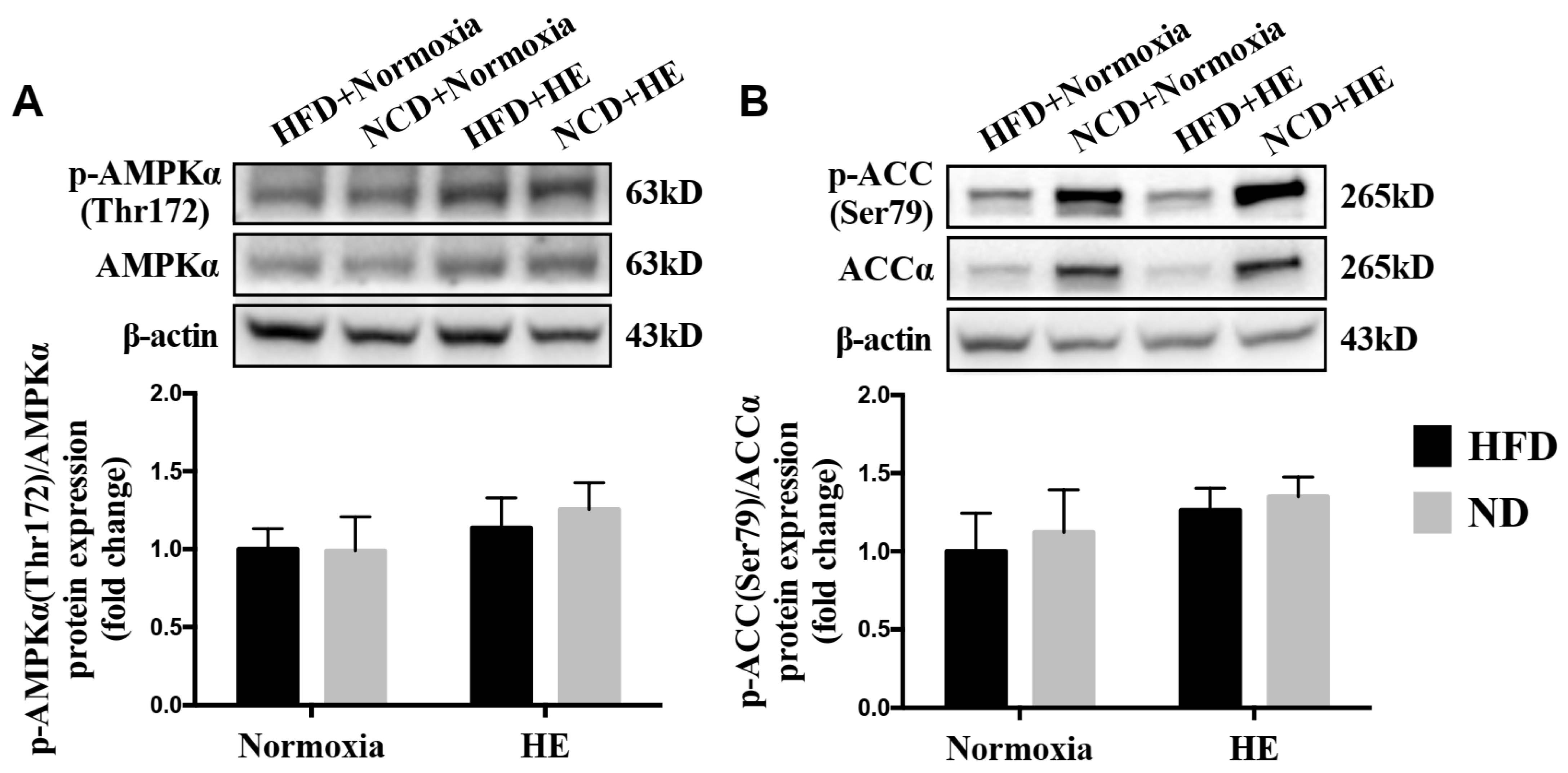
3.5. Changes in Hepatic p-AMPKα(Thr172)/AMPKα and p-ACC(Ser79)/ACCα Ratios
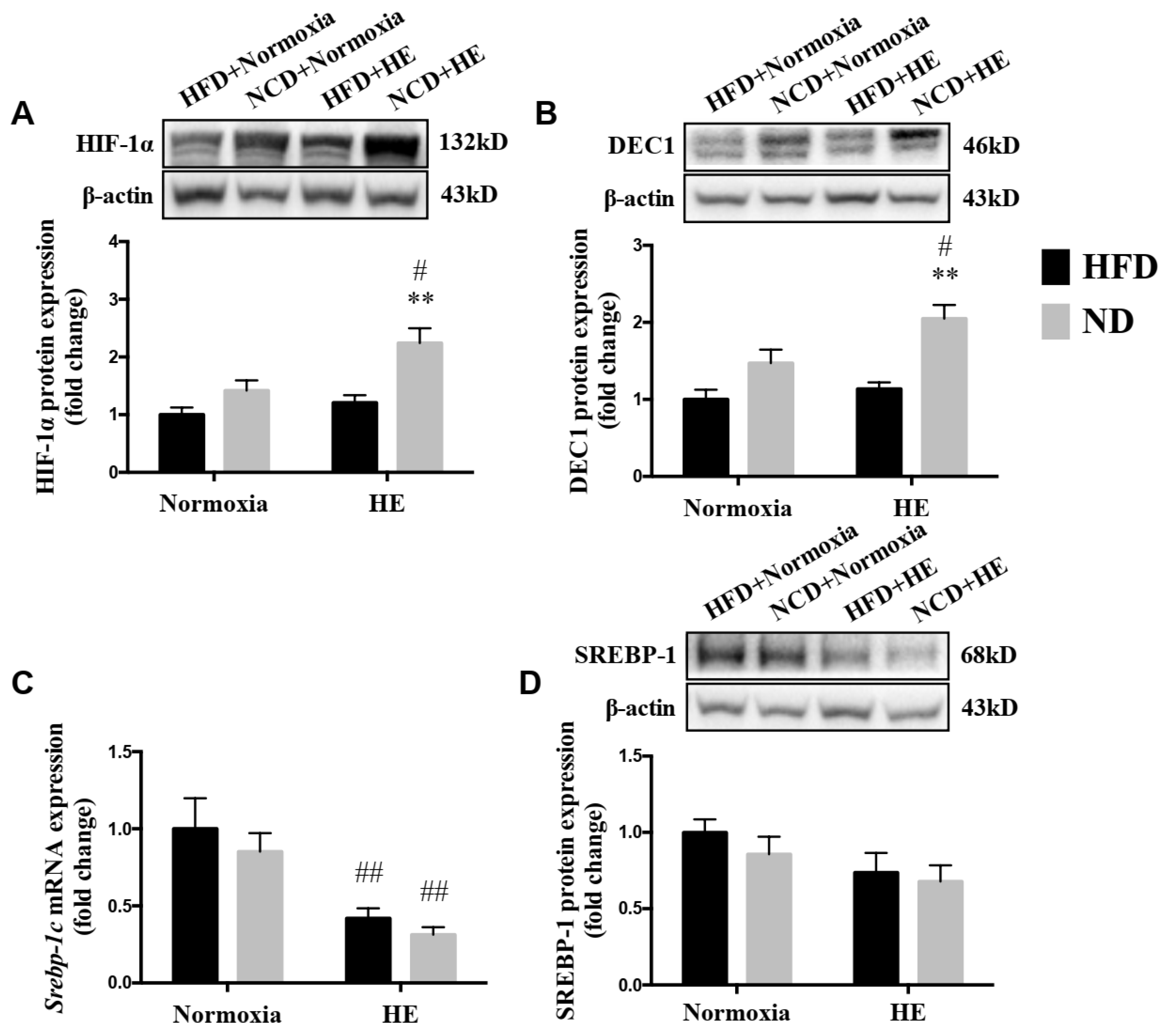
3.6. Changes in Protein Expression of HIF-1α, DEC1 and SREBP-1, and mRNA Expression of Srebp-1c
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Li, Z.; Xue, J.; Chen, P.; Chen, L.; Yan, S.; Liu, L. Prevalence of nonalcoholic fatty liver disease in mainland of China: A meta-analysis of published studies. J. Gastroenterol. Hepatol. 2014, 29, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Wong, R.J.; Harrison, S.A. Nonalcoholic Fatty Liver Disease Review: Diagnosis, Treatment, and Outcomes. Clin. Gastroenterol. Hepatol. 2015, 13, 2062–2070. [Google Scholar] [CrossRef] [PubMed]
- Meex, R.C.R.; Watt, M.J. Hepatokines: Linking nonalcoholic fatty liver disease and insulin resistance. Nat. Rev. Endocrinol. 2017, 13, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Byrne, C.D.; Lonardo, A.; Zoppini, G.; Barbui, C. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: A meta-analysis. J. Hepatol. 2016, 65, 589–600. [Google Scholar] [CrossRef]
- Abd El-Kader, S.M.; El-Den Ashmawy, E.M.S. Non-alcoholic fatty liver disease: The diagnosis and management. World J. Hepatol. 2015, 7, 846–858. [Google Scholar] [CrossRef]
- Montagner, A.; Polizzi, A.; Fouché, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Régnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef]
- Nakamura, M.T.; Yudell, B.E.; Loor, J.J. Regulation of energy metabolism by long-chain fatty acids. Prog. Lipid Res. 2014, 53, 124–144. [Google Scholar] [CrossRef]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef]
- Ohashi, T.; Nakade, Y.; Ibusuki, M.; Kitano, R.; Yamauchi, T.; Kimoto, S.; Inoue, T.; Kobayashi, Y.; Sumida, Y.; Ito, K.; et al. Conophylline inhibits high fat diet-induced non-alcoholic fatty liver disease in mice. PLoS ONE 2019, 14, e0210068. [Google Scholar] [CrossRef]
- Xu, X.; So, J.-S.; Park, J.-G.; Lee, A.-H. Transcriptional Control of Hepatic Lipid Metabolism by SREBP and ChREBP. Semin. Liver Dis. 2013, 33, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, I.; Bashmakov, Y.; Horton, J.D. Increased Levels of Nuclear SREBP-1c Associated with Fatty Livers in Two Mouse Models of Diabetes Mellitus. J. Biol. Chem. 1999, 274, 30028–30032. [Google Scholar] [CrossRef] [PubMed]
- Fang, K.; Wu, F.; Chen, G.; Dong, H.; Li, J.; Zhao, Y.; Xu, L.; Zou, X.; Lu, F. Diosgenin ameliorates palmitic acid-induced lipid accumulation via AMPK/ACC/CPT-1A and SREBP-1c/FAS signaling pathways in LO2 cells. BMC Complement. Altern. Med. 2019, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.; Daniel, S.; Broyles, S.S.; Kim, K.H. Critical phosphorylation sites for acetyl-CoA carboxylase activity. J. Biol. Chem. 1994, 269, 22162–22168. [Google Scholar] [CrossRef]
- Zhu, X.; Bian, H.; Wang, L.; Sun, X.; Xu, X.; Yan, H.; Xia, M.-F.; Chang, X.; Lu, Y.; Li, Y.; et al. Berberine attenuates nonalcoholic hepatic steatosis through the AMPK-SREBP-1c-SCD1 pathway. Free Radic. Biol. Med. 2019, 141, 192–204. [Google Scholar] [CrossRef]
- Marais, A.D. Apolipoprotein E in lipoprotein metabolism, health and cardiovascular disease. Pathology 2019, 51, 165–176. [Google Scholar] [CrossRef]
- Vasquez, E.C.; A Peotta, V.; Gava, A.L.; MC Pereira, T.; Meyrelles, S.S. Cardiac and vascular phenotypes in the apolipoprotein E-deficient mouse. J. Biomed. Sci. 2012, 19, 22. [Google Scholar] [CrossRef]
- Sfyri, P.; Matsakas, A. Crossroads between peripheral atherosclerosis, western-type diet and skeletal muscle pathophysiology: Emphasis on apolipoprotein E deficiency and peripheral arterial disease. J. Biomed. Sci. 2017, 24, 42. [Google Scholar] [CrossRef]
- Tous, M.; Ferre, N.; Camps, J.; Riu, F.; Joven, J. Feeding apolipoprotein E-knockout mice with cholesterol and fat enriched diets may be a model of non-alcoholic steatohepatitis. Mol. Cell. Biochem. 2005, 268, 53–58. [Google Scholar] [CrossRef]
- Schierwagen, R.; Maybüchen, L.; Zimmer, S.; Hittatiya, K.; Bäck, C.; Klein, S.L.; Uschner, F.E.; Reul, W.; Boor, P.; Nickenig, G.; et al. Seven weeks of Western diet in apolipoprotein-E-deficient mice induce metabolic syndrome and non-alcoholic steatohepatitis with liver fibrosis. Sci. Rep. 2015, 5, 12931. [Google Scholar] [CrossRef]
- Urdampilleta, A.; González-Muniesa, P.; Portillo, M.P.; Martínez, J.A. Usefulness of combining intermittent hypoxia and physical exercise in the treatment of obesity. J. Physiol. Biochem. 2011, 68, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Hara, Y.; Watanabe, N. Changes in expression of genes related to glucose metabolism in liver and skeletal muscle of rats exposed to acute hypoxia. Heliyon 2020, 6, e04334. [Google Scholar] [CrossRef] [PubMed]
- Cartee, G.; Douen, A.G.; Ramlal, T.; Klip, A.; Holloszy, J.O. Stimulation of glucose transport in skeletal muscle by hypoxia. J. Appl. Physiol. 1991, 70, 1593–1600. [Google Scholar] [CrossRef]
- Chiu, L.-L.; Chou, S.-W.; Cho, Y.-M.; Ho, H.-Y.; Ivy, J.L.; Hunt, D.; Wang, P.S.; Kuo, C.-H. Effect of prolonged intermittent hypoxia and exercise training on glucose tolerance and muscle GLUT4 protein expression in rats. J. Biomed. Sci. 2004, 11, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, J.L.; O Carey, J.; Pories, W.J.; Morris, P.G.; Dohm, G.L. Hypoxia Stimulates Glucose Transport in Insulin-Resistant Human Skeletal Muscle. Diabetes 1995, 44, 695–698. [Google Scholar] [CrossRef]
- Mackenzie, R.; Maxwell, N.; Castle, P.; Brickley, G.; Watt, P. Acute hypoxia and exercise improve insulin sensitivity (SI2*) in individuals with type 2 diabetes. Diabetes/Metab. Res. Rev. 2010, 27, 94–101. [Google Scholar] [CrossRef]
- Song, K.; Zhang, Y.; Ga, Q.; Bai, Z.; Ge, R.-L. High-altitude chronic hypoxia ameliorates obesity-induced non-alcoholic fatty liver disease in mice by regulating mitochondrial and AMPK signaling. Life Sci. 2020, 252, 117633. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Olivetti, C.; Rosina, F.; Carbone, G.; Gambino, R. Association of obstructive sleep apnoea with the presence and severity of non-alcoholic fatty liver disease. A systematic review and meta-analysis. Obes. Rev. 2013, 14, 417–431. [Google Scholar] [CrossRef]
- Kuvat, N.; Tanriverdi, H.; Armutcu, F. The relationship between obstructive sleep apnea syndrome and obesity: A new perspective on the pathogenesis in terms of organ crosstalk. Clin. Respir. J. 2020, 14, 595–604. [Google Scholar] [CrossRef]
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef] [Green Version]
- Ji, W.; Wang, L.; He, S.; Yan, L.; Li, T.; Wang, J.; Kong, A.-N.T.; Yu, S.; Zhang, Y. Effects of acute hypoxia exposure with different durations on activation of Nrf2-ARE pathway in mouse skeletal muscle. PLoS ONE 2018, 13, e0208474. [Google Scholar] [CrossRef]
- Wang, L.; Lavier, J.; Hua, W.; Wang, Y.; Gong, L.; Wei, H.; Wang, J.; Pellegrin, M.; Millet, G.; Zhang, Y. High-Intensity Interval Training and Moderate-Intensity Continuous Training Attenuate Oxidative Damage and Promote Myokine Response in the Skeletal Muscle of ApoE KO Mice on High-Fat Diet. Antioxidants 2021, 10, 992. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; He, S.; Liu, S.; Kong, Z.; Wang, J.; Zhang, Y. Effects of different exercise durations on Keap1-Nrf2-ARE pathway activation in mouse skeletal muscle. Free Radic. Res. 2015, 49, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Sehayek, E.; Shefer, S.; Nguyen, L.B.; Ono, J.G.; Merkel, M.; Breslow, J.L. Apolipoprotein E regulates dietary cholesterol absorption and biliary cholesterol excretion: Studies in C57BL/6 apolipoprotein E knockout mice. Proc. Natl. Acad. Sci. USA 2000, 97, 3433–3437. [Google Scholar] [CrossRef] [PubMed]
- Woolcott, O.O.; Gutierrez, C.; Castillo, O.A.; Elashoff, R.M.; Stefanovski, D.; Bergman, R.N. Inverse association between altitude and obesity: A prevalence study among andean and low-altitude adult individuals of Peru. Obesity 2016, 24, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.C.; Liu, L.; Barnhart, B.C.; Young, R.M. Hypoxia-Induced Signaling in the Cardiovascular System. Annu. Rev. Physiol. 2008, 70, 51–71. [Google Scholar] [CrossRef]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and Inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef]
- Nath, B.D.; Szabo, G. Hypoxia and hypoxia inducible factors: Diverse roles in liver diseases. Hepatology 2011, 55, 622–633. [Google Scholar] [CrossRef] [Green Version]
- Parathath, S.; Mick, S.L.; Feig, J.E.; Joaquin, V.; Grauer, L.; Habiel, D.M.; Gassmann, M.; Gardner, L.B.; Fisher, E.A. Hypoxia Is Present in Murine Atherosclerotic Plaques and Has Multiple Adverse Effects on Macrophage Lipid Metabolism. Circ. Res. 2011, 109, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Furuta, E.; Pai, S.K.; Zhan, R.; Bandyopadhyay, S.; Watabe, M.; Mo, Y.-Y.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; et al. Fatty Acid Synthase Gene Is Up-regulated by Hypoxia via Activation of Akt and Sterol Regulatory Element Binding Protein-1. Cancer Res. 2008, 68, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Burtscher, J.; Millet, G.P.; Burtscher, M. Does living at moderate altitudes in Austria affect mortality rates of various causes? An ecological study. BMJ Open 2021, 11, e048520. [Google Scholar] [CrossRef] [PubMed]
- Burtscher, J.; Millet, G.P.; Renner-Sattler, K.; Klimont, J.; Hackl, M.; Burtscher, M. Moderate Altitude Residence Reduces Male Colorectal and Female Breast Cancer Mortality More Than Incidence: Therapeutic Implications? Cancers 2021, 13, 4420. [Google Scholar] [CrossRef]
- Bailey, D.P.; Smith, L.R.; Chrismas, B.C.; Taylor, L.; Stensel, D.; Deighton, K.; Douglas, J.A.; Kerr, C.J. Appetite and gut hormone responses to moderate-intensity continuous exercise versus high-intensity interval exercise, in normoxic and hypoxic conditions. Appetite 2015, 89, 237–245. [Google Scholar] [CrossRef]
- Debevec, T. Hypoxia-Related Hormonal Appetite Modulation in Humans during Rest and Exercise: Mini Review. Front. Physiol. 2017, 8, 366. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Goda, N.; Kanai, M.; Niwa, D.; Osanai, K.; Yamamoto, Y.; Senoo-Matsuda, N.; Johnson, R.S.; Miura, S.; Kabe, Y.; et al. HIF-1α induction suppresses excessive lipid accumulation in alcoholic fatty liver in mice. J. Hepatol. 2012, 56, 441–447. [Google Scholar] [CrossRef]
- Wang, X.L.; Suzuki, R.; Lee, K.; Tran, T.; Gunton, J.E.; Saha, A.K.; Patti, M.-E.; Goldfine, A.; Ruderman, N.B.; Gonzalez, F.J.; et al. Ablation of ARNT/HIF1β in Liver Alters Gluconeogenesis, Lipogenic Gene Expression, and Serum Ketones. Cell Metab. 2009, 9, 428–439. [Google Scholar] [CrossRef]
- Carabelli, J.; Burgueño, A.L.; Rosselli, M.S.; Gianotti, T.F.; Lago, N.R.; Pirola, C.J.; Sookoian, S. High fat diet-induced liver steatosis promotes an increase in liver mitochondrial biogenesis in response to hypoxia. J. Cell. Mol. Med. 2011, 15, 1329–1338. [Google Scholar] [CrossRef]
- Palmer, B.F.; Clegg, D.J. Ascent to altitude as a weight loss method: The good and bad of hypoxia inducible factor activation. Obesity 2013, 22, 311–317. [Google Scholar] [CrossRef]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Am. J. Dig. Dis. 2016, 61, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Jensen, V.S.; Hvid, H.; Damgaard, J.; Nygaard, H.; Ingvorsen, C.; Wulff, E.M.; Lykkesfeldt, J.; Fledelius, C. Dietary fat stimulates development of NAFLD more potently than dietary fructose in Sprague–Dawley rats. Diabetol. Metab. Syndr. 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Nomura, K.; Yamanouchi, T. The role of fructose-enriched diets in mechanisms of nonalcoholic fatty liver disease. J. Nutr. Biochem. 2012, 23, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Liu, L.; Li, F. Acetate alters the process of lipid metabolism in rabbits. Animal 2018, 12, 1895–1902. [Google Scholar] [CrossRef]
- Bradbury, M.W. Lipid Metabolism and Liver Inflammation. I. Hepatic fatty acid uptake: Possible role in steatosis. Am. J. Physiol. Liver Physiol. 2006, 290, G194–G198. [Google Scholar] [CrossRef]
- Raghow, R.; Yellaturu, C.; Deng, X.; Park, E.A.; Elam, M.B. SREBPs: The crossroads of physiological and pathological lipid homeostasis. Trends Endocrinol. Metab. 2008, 19, 65–73. [Google Scholar] [CrossRef]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J. The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef]
- Yuan, F.; Teng, X.; Guo, Z.; Zhou, J.-J.; Zhang, Y.; Wang, S. Chronic intermittent hypobaric hypoxia ameliorates endoplasmic reticulum stress mediated liver damage induced by fructose in rats. Life Sci. 2014, 121, 40–45. [Google Scholar] [CrossRef]
- Tian, Y.-M.; Liu, Y.; Wang, S.; Dong, Y.; Su, T.; Ma, H.-J.; Zhang, Y. Anti-diabetes effect of chronic intermittent hypobaric hypoxia through improving liver insulin resistance in diabetic rats. Life Sci. 2016, 150, 1–7. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Gene ID | Forward Primer | Reverse Primer |
|---|---|---|---|
| Cpt1a | 12894 | CTCCGCCTGAGCCATGAAG | CACCAGTGATGATGCCATTCT |
| Cpt2 | 12896 | CAGCACAGCATCGTACCCA | TCCCAATGCCGTTCTCAAAAT |
| Acc1 | 107476 | ATGGGCGGAATGGTCTCTTTC | TGGGGACCTTGTCTTCATCAT |
| Acly | 104112 | ACCCTTTCACTGGGGATCACA | GACAGGGATCAGGATTTCCTTG |
| Fasn | 14104 | GGAGGTGGTGATAGCCGGTAT | TGGGTAATCCATAGAGCCCAG |
| Scd1 | 20249 | TTCTTGCGATACACTCTGGTGC | CGGGATTGAATGTTCTTGTCGT |
| Srebp-1c | 20787 | GTGAGCCTGACAAGCAATCA | GGTGCCTACAGAGCAAGAG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Lavier, J.; Hua, W.; Gong, L.; Wei, H.; Wang, J.; Pellegrin, M.; Millet, G.P.; Zhang, Y. Effects of Six Weeks of Hypoxia Exposure on Hepatic Fatty Acid Metabolism in ApoE Knockout Mice Fed a High-Fat Diet. Life 2022, 12, 1535. https://doi.org/10.3390/life12101535
Wang Y, Lavier J, Hua W, Gong L, Wei H, Wang J, Pellegrin M, Millet GP, Zhang Y. Effects of Six Weeks of Hypoxia Exposure on Hepatic Fatty Acid Metabolism in ApoE Knockout Mice Fed a High-Fat Diet. Life. 2022; 12(10):1535. https://doi.org/10.3390/life12101535
Chicago/Turabian StyleWang, Yangwenjie, Jessica Lavier, Weicheng Hua, Lijing Gong, Hao Wei, Jianxiong Wang, Maxime Pellegrin, Grégoire P. Millet, and Ying Zhang. 2022. "Effects of Six Weeks of Hypoxia Exposure on Hepatic Fatty Acid Metabolism in ApoE Knockout Mice Fed a High-Fat Diet" Life 12, no. 10: 1535. https://doi.org/10.3390/life12101535