
Abdominal Aortic Aneurysm Formation with a Focus on Vascular Smooth Muscle Cells


Department of Cellular and Molecular Biology, The University of Texas Health Science Center at Tyler, Tyler, TX 75708, USA
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Life 2022, 12(2), 191; https://doi.org/10.3390/life12020191
Submission received: 12 January 2022
/
Revised: 24 January 2022
/
Accepted: 25 January 2022
/
Published: 27 January 2022
(This article belongs to the Special Issue Vascular Smooth Muscle Cell (VSMC) Differentiation)
{kind=link}
{kind=link}
Abstract
:Abdominal aortic aneurysm (AAA) is a lethal degenerative vascular disease that affects, mostly, the elder population, with a high mortality rate (>80%) upon rupture. It features a dilation of the aortic diameter to larger than 30 mm or more than 50%. Diverse pathological processes are involved in the development of AAA, including aortic wall inflammation, elastin breakdown, oxidative stress, smooth muscle cell (SMC) phenotypic switching and dysfunction, and extracellular matrix degradation. With open surgery being the only therapeutic option up to date, the lack of pharmaceutical treatment approach calls for identifying novel and effective targets and further understanding the pathological process of AAA. Both lifestyle and genetic predisposition have an important role in increasing the risk of AAA. Several cell types are closely related to the pathogenesis of AAA. Among them, vascular SMCs (VSMCs) are gaining much attention as a critical contributor for AAA initiation and/or progression. In this review, we summarize what is known about AAA, including the risk factors, the pathophysiology, and the established animal models of AAA. In particular, we focus on the VSMC phenotypic switching and dysfunction in AAA formation. Further understanding the regulation of VSMC phenotypic changes may provide novel therapeutic targets for the treatment or prevention of AAA.
1. Introduction
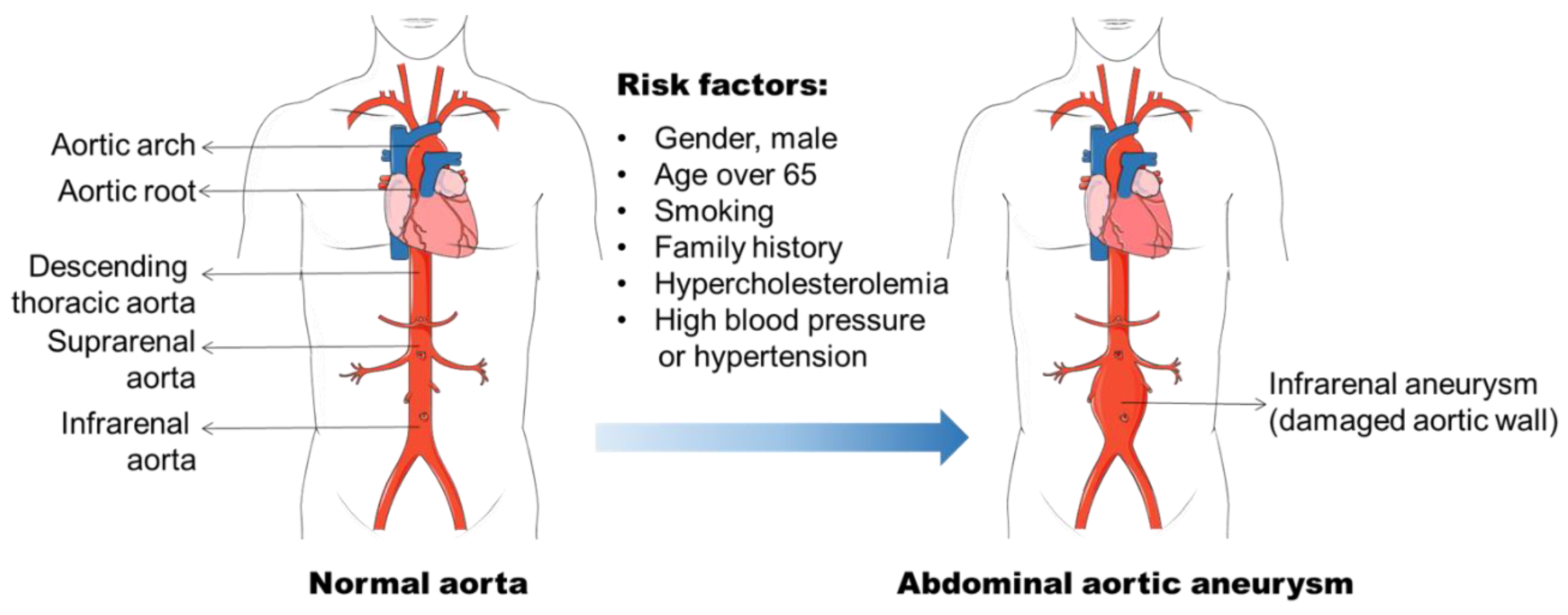
Aneurysm is the term for a dilated blood vessel with a diameter at least 1.5 times its normal size [1]. According to the locations, aneurysms can be divided into three major types, i.e., cerebral aneurysm (brain), aortic aneurysm (AA, aorta), and popliteal aneurysm (popliteal artery) [2,3,4]. The aorta is the largest blood vessel in the body and the AA is categorized into two main types: thoracic aortic aneurysm (TAA, in the chest) and abdominal aortic aneurysm (AAA, in the abdomen) [3]. AAA usually occurs in the infra-renal segment with a diameter exceeding 3.0 cm (Figure 1) [3]. AAA is the most common aneurysm and predominantly affects men aged 65 years and older [5,6].
AAA diagnosis remains a challenge because it does not present with any symptoms, nor can it be detected by a mere physical examination [7]. With the growing of the aortic diameter, the risk of AAA rupture increases [8]. The rupture of AAA results in profound internal bleeding with a mortality around 80% [9]. The ultrasound screening of the high-risk populations (men of 65-years and older) has been demonstrated to be an effective approach to prevent the AAA related mortality [10,11]. However, it is costly and not appropriate for the assessment of AAA progression. The treatment option for patients with large (≥55 mm), rapidly growing (>10 mm), or symptomatic AAAs remains endovascular exclusion or open surgery [5], although the postsurgical mortality for emergency operations stays at around 50% [12]. Patients who have small AAAs (<55 mm) are not beneficial from surgical repair [7]. Currently, there is no specific drug available to prevent or reverse AAA progression.
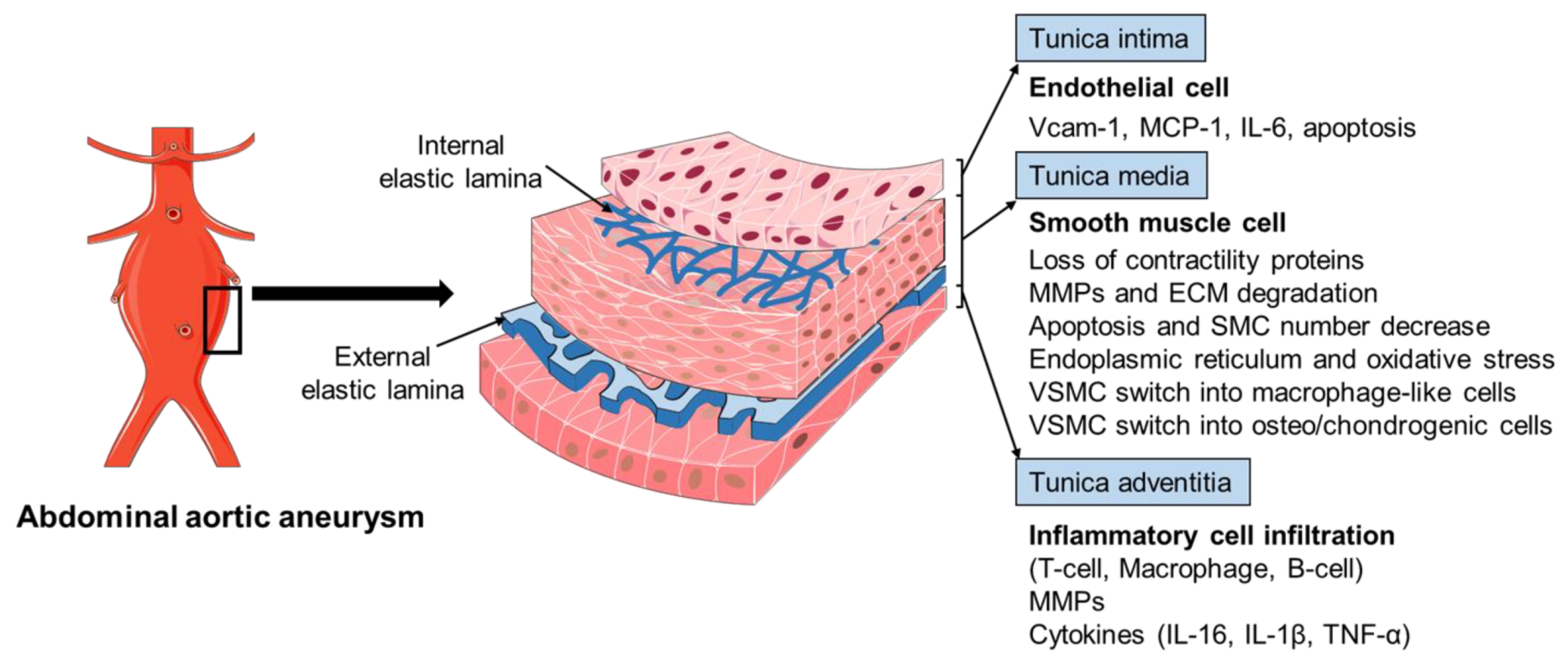
The pathophysiology of AAA is complex, involving the increased expression of endothelial cell (EC) adhesion molecules and chemokines, the inflammatory cell infiltration into the aortic wall, vascular smooth muscle cell (VSMC) dysfunction, aortic extracellular matrix (ECM) remodeling, oxidative stress, and the formation of intraluminal thrombus (Figure 2) [3,6,13]. The mechanisms of the AAA initiation and progression remain incompletely understood. The current review discusses the formation and progression of AAA with a focus on VSMC phenotypic switching and dysfunction.
2. AAA Formation
2.1. Risk Factors for AAA
The identification of risk factors for AAA formation provides strategies for AAA prevention and therapy. In the past few decades, various factors including male gender, aging, cigarette smoking, and elevated blood cholesterol level, etc., have been found to be related to AAA initiation and progression (Figure 1) [6,14,15]. The incidence of AAA is directly proportional to an increase in age, and AAA is a prominent cause of death in older people from 65 years of age [5]. It is often seen in older men, and the ratio of male and female patients ranges 3.5–6:1 based on various screening studies [16,17]. Therefore, the men with an age of more than 65 years old are the high-risk population for AAA development.
Smoking has been recognized as a strong risk factor for AAA in various epidemiological studies [14,16,18]. Approximately 87% of the men with AAA were reported to be current or ex-smokers in a screening study of AAA among 65-year-old Swedish men with an odd ratio of 3.5 (95%CI: 2.4–5.1) [19]. The nicotine in plasma contributes to AAA progression, which has been confirmed in Apolipoprotein E deficient (ApoE−/−) mouse model and elastase-perfusion model of AAA [20,21,22]. The mechanisms underlying smoking induced AAA involve VSMC dysfunction and inflammatory cell function that have been discussed in Norman, P. E. and Curci, J. A. published review [20]. Hypercholesterolemia is a common facilitating cause of AAA, although there is limited knowledge as to its role in the development of AAA [15]. The abdominal aortic region is prone to the atherosclerotic lesion, which is found in virtually all cases of human AAA patients and that atherosclerosis is involved in the process of aortic dilation. Additional evidence to support the important role of hypercholesterolemia in aneurysm formation is the increased AAA formation in angiotensin II (Ang II) infused ApoE−/− mice (hypercholesterolemia mouse model) compared with wild-type (WT) C57BL/6 mice [23]. Genetic analyses of AAA from population screening and gene-associated studies show that different genetic risk factors are involved in the pathogenesis of AAA [24]. A heritability of 70% was reported in a large twin study by Jesper Swedenborg’s research group [25]. Several other studies have found that the risk of AAA in first-degree relatives of affected individuals was almost doubled [26,27,28]. Two loci for AAA have been identified and mapped on chromosome 19q13 and 4q31 [29,30]. The genome wide association studies (GWAS) have identified associations of single nucleotide polymorphisms (SNPs) with AAAs, such as CDKN2BAS1 rs10757274, the low-density lipoprotein receptor (LDLR) rs6511720, and DAB2IP rs10985349 [31,32]. In addition, other factors including DNA methylation, hypertension, and viral infections (e.g., cytomegalovirus, CMV) are also associated with AAA pathogenesis [33,34,35,36,37].
2.2. Histopathology of AAA
The histopathology of AAA is characterized by aortic wall inflammation, EC alteration, SMC dysfunction, oxidative stress, and ECM degradation, which cause a progressive luminal dilation, and finally a rupture (Figure 2) [6,13,38,39]. The aortic wall inflammation with the infiltration of inflammatory cells, including T-cells, B-cells, and macrophages, are essential features of AAA (Figure 2) [40,41,42]. In clinical and experimental AAA, a number of pro-inflammatory cytokines are increased, such as monocyte chemoattractant protein 1 (MCP-1), interleukin-1β (IL-1β), IL-6, and tumor necrosis factor-α (TNF-α). Polymorphisms in inflammatory cytokines may affect the production of these cytokines and, therefore, influence the pathogenesis of AAA [23,43,44]. SNPs (rs1800795 and rs1800796) in the IL-6 promoter have been linked with the development of AAA [44,45]. These inflammatory mediators play a critical role in inflammatory cell infiltration, which promotes an inflammatory response and subsequent SMC dysfunction, ECM degradation (mainly through matrix metalloproteinases, MMPs), and eventual AAA formation [42,46,47,48]. In addition, fragments of the aortic wall degradation serve as attracting agents for macrophage infiltration into the aortic wall to initiate the immune responses and AAA formation [49]. Dysregulation of EC function is another important factor implicated in AAA initiation and/or progression [50,51]. The increased EC expression of MCP-1 and vascular cell adhesion molecule 1 (Vcam-1) recruits macrophages into the aortic wall and leads to ECM degradation and, finally, aneurysm formation (Figure 2). In addition, EC apoptosis with the reduced expression of endothelial nitric oxide synthase (eNOS) also facilitates AAA formation by affecting the activity of NO, which is important in the stability of vascular tone, blood pressure, and SMC relaxation.
VSMCs in the tunica media represent the most abundant cells in aorta arteries. The important role and phenotypic modulation of VSMCs in AAA have been explored extensively, including the SMC genetic modulation, SMC-mediated ECM production and degradation, SMC-upregulated inflammatory response, and SMC-modulated oxidative stress and apoptosis (Figure 2) [3,13,52,53,54,55]. The cellular and molecular mechanisms of SMCs mediated AAA development will be discussed in more detail in the following section.
3. Vascular Smooth Muscle Cells in AAA Formation
3.1. VSMC Phenotypic Plasticity
The artery wall is made up of three layers, including the tunica intima (ECs), tunica media (mainly VSMCs), and tunica adventitia (fibroblasts and extracellular matrix) (Figure 2) [56]. During vascular development, precursor VSMCs are recruited to the endothelial vascular network and further differentiate into mature SMCs through various signaling pathways, such as transforming growth factor-β (TGF-β)/Smads, platelet-derived growth factor-BB (PDGF-BB), Wnt/Notch, histone deacetylases (HDACs)/epigenetics, micro-RNAs, etc. [57,58,59]. In a healthy vessel wall, VSMCs play a major role in maintaining vascular tone to mediate blood pressure and blood flow through their contraction and dilatation capability [56,60]. Accumulating evidence shows that SMCs’ genetic and epigenetic modulation in vasculogenesis during embryo development is a major mechanism in AAA formation. The embryological origins of SMCs in the aortic arch and descending aorta are different [58,60,61]. Compared with the aortic arch SMCs derived from the neural crest, those descending aortic SMCs instead are derived from the mesoderm with less elastic lamellae formed during vascular development. There are also differences in the cellular content and genetic activity, which renders the infra-renal region an area prone to aneurysm. For example, the mesoderm derived SMCs are more responsive to the cytokines IL-1β, which can upregulate MMPs’ expression and further ECM degradation. In addition, TGF-β induction increased DNA synthesis and collagen production in neural crest-derived SMCs but not in mesoderm-derived SMCs.
SMCs are highly plastic and undergo significant changes between two phenotypes, i.e., a rather ‘dormant’ one with differentiated SMCs, and a proliferating/synthetic one with dedifferentiated SMCs in response to stress signals [56,60,62]. Differentiated SMCs are spindle shaped and express high levels of contractile proteins, such as α-smooth muscle actin (α-SMA), SM myosin heavy chain (SMMHC), smooth muscle 22α (SM22α), SM-calponin (CNN), and smoothelin-B [59]. Under pathological conditions, SMCs could be induced by TGF-β, PDGF-BB, Ang II, etc., and change into a dedifferentiated phenotype with low levels of contractile proteins but high levels of molecules associated with proliferation, migration, fibrosis, and inflammation [60,63,64,65]. Mechanical injury, atherosclerosis, hypertension, as well as aneurysm are all related with the SMC phenotypic changes [56,64,65,66,67].
3.2. VSMC Phenotypic Modulation in AAA
3.2.1. VSMC Contractility and TGF-β
VSMCs play a central role in aneurysm formation. VSMCs in the healthy vessel wall display a contractile phenotype to maintain the vascular tone. The loss of SMC contractile function may alter the vascular tone and increase the aortic wall stress to promote aneurysm formation [52,64,68]. The mutations in genes encoding SMC contractile proteins have been reported to be associated with AAA development. The TGF-β pathway components, including the receptors and Smad proteins, are involved in SMC contractility through upregulating the expression of SMC contractile proteins (α-SMA, SMMHC, and CNN, etc.) [68,69,70,71]. TGF-β neutralization has been shown to augment Ang II induced aneurysms in both thoracic and abdominal regions [72,73]. Consistently, the disruption of the TGF-β receptors in SMCs impairs their contractile ability and results in aneurysm [70]. The genetic deletion of Smad3 causes extensive aneurysm formation with elastic fiber fragmentation, collagen fiber reorganization, and vessel inflammation in the calcium chloride (CaCl2)-induced mouse AAA model [71]. In addition, a previous study showed a remarkable downregulation of TGF-β receptor 2 (TGFBR2) in human AAA biopsies. Eleven of twelve AAA biopsies demonstrated TGFBR2 exon 8 deletion with a marked downregulation of TGFBR2 in AAA biopsies [74]. Further, an AAA case-control study from a Dutch population also reported that SNPs in both TGFBR1 (rs1626340) and TGFBR2 (rs1036095 and rs4522809) are associated with AAA prevalence [32,75]. These studies demonstrate that TGF-β signaling is necessary to sustain the structural integrity of SMCs and prevent aortic dilatation during AAA. A recent lineage tracing study using SMC-specific deletion of TGFBR2 provides consistent and convincing in vivo evidence. Those mice lacking TGF-β signaling in their SMCs on an ApoE−/− background developed AAAs after 4 months’ feeding with a high cholesterol high fat diet, while the wild-type counterparts did not. The trans-differentiation of a subset of contractile SMCs into mesenchymal stem cell-like cells might account for such phenomenon since the latter can further give rise to other cell types, including osteoblasts/chondrocytes, adipocytes, and macrophages [76].
Another layer of control in VSMC phenotypic changes lies in micro-RNA (miRNA). micro-RNAs are small and non-coding RNAs, which function to repress gene expression by degrading messenger RNAs or mimicking small interfering RNAs (siRNAs) to inhibit translation. The miR-143/145 cluster highly expressed in VSMCs has been shown to be most abundantly expressed in the heart and the aorta [77]. Serum response factor (SRF), myocardin, and Nkx2.5 are reported to induce the miR-143/145 expression in SMCs [78]. Overexpression of miR-145 upregulates the levels of SMC contractile proteins, including α-SMA, SMMHC, and CNN. Such a function of miR-145 in increasing SMCs’ contractility primarily acts through suppressing Kruppel-like transcription factor 4 (KLF4) and KLF5, which represses myocardin to downregulate VSMC differentiation marker genes [78]. In addition, the deregulation of miR-143/145 has also been implicated in human aneurysm development [79].
3.2.2. SMCs Express Proteolytic Enzymes to Induce ECM Disorganization
Aortic aneurysm is a matrix degenerative disease with dilated blood vessels. Multiple studies have demonstrated that ECM degradation during AAA formation is actively mediated by VSMCs [6,13,52,64]. The ECM consists of elastin, collagen, fibronectin, and fibrillin. VSMCs produce elastin and collagen to resist vasodilation and rupture. On the other hand, VSMCs control the integrity and degradation of ECM by the release and maturation of MMPs and the tissue inhibitor of metalloproteinases (TIMPs) [54,56,80,81,82,83,84,85]. During the aneurysm formation, the breakdown of elastin results in SMC phenotypic changes characterized by an increased expression of MMPs, such as MMP-1, -2, -9, -13, and -14 [54,80]. These MMP proteins exert proteolytic activity towards elastin and collagen, which are essential in ECM degradation and disorganization during AAA formation. Increased expression/activity of MMP-2 and MMP-9 are of particular significance in degrading the extracellular matrix and weakening the aortic vascular wall in the context of AAA formation [81]. The gene polymorphic sites have been recognized in the promoter of a number of MMPs [82]. The substitution of cytosine with thymidine in the promoter region of MMP-9 increases its promoter activity [86], leading to upregulated MMP-9 in AAA patients. A similar substitution occurs in the polymorphic site in the promoter regions of MMPs -2, -3, -9, and -12 genes [87,88,89].
VSMCs modulate the MMP activities and ECM degradation through inhibiting the expression of TIMPs (TIMP-1, -2, and -3) [83,84,85]. The decreased TIMP expression or activity has been shown to mediate aneurysm development through resulting upregulated MMP activities and increased ECM degradation [90]. The TIMP1-deficient mice developed larger aneurysms compared to the WT mice after porcine pancreatic elastase (PPE) perfusion [91]. Additionally, the MMP/TIMP ratio is controlled by plasminogen activator inhibitors (PAIs) [92]. VSMCs also express a number of miRNAs to mediate ECM degradation and promote AAA [93,94,95]. The murine miR-712 or human/murine homolog miR205 have been shown to repress TIMP translation and, consequently, increase MMP activity and promote AAA development [95]. The upregulated expression of miRNAs, including miR-21, -133, and -378, have been found in aortic aneurysm tissues from mouse AAA models and human AAA patients [94]. HDACs, regulators of gene transcription, have been shown to modulate the expression of VSMC genes involved in AAA [96]. Consistently, the HDAC inhibitor MCT-1 decreases MMP2 expression and activity in VSMCs in vitro [97]. An increased expression of the HDAC profile has also been noted in aneurysm samples from AAA animal models and human patients [96]. Further, the HDAC inhibitors including MS-275, MC-1568, and MCT-1 have been found to improve ECM disorganization and inhibit aneurysm development in mouse AAA models [93,98]. In a study exploring the effects of calorie restriction on AAA development, Sirtuin 1 (SIRT1) expression in VSMCs was found critical for mediating the protective effects of calorie restriction against aortic aneurysm formation. The VSMC-specification knockout of SIRT1 abolished the protective effect of calorie restriction. Such an effect was attributed partly to the SIRT1–dependent deacetylation of histone H3 lysine 9 on the MMP2 promoter [99]. The results suggest SIRT1 as a novel regulator of VSMC ECM remodeling during energy restriction in the context of AAA development.
3.2.3. Endoplasmic Reticulum Stress and Oxidative Stress
The increased endoplasmic reticulum (ER) stress has been implicated in aortic aneurysm formation [100,101,102]. The inhibition of ER stress successfully decreases aneurysm formation in Ang II-induced mouse AAA models [102]. In the absence of ER stress, the transcription factor unspliced X box protein 1(XBP1u) is expressed to maintain VSMC contractile phenotype. XBP1u deficiency induced AAA formation in vivo with VSMC dedifferentiation and the increase of proinflammatory and proteolytic VSMCs [103]. ER stress triggers a thoracic aortic aneurysm and dissection (TAAD) formation through the C/EBP homologous protein (CHOP), which controls ER stress-induced apoptosis. CHOP deletion, therefore, has been shown to prevent SMC apoptosis and TAAD development [104].
Oxidative stress, as defined by the excess production of reactive oxygen species (ROS), promotes AAA development through multiple mechanisms, such as VSMC apoptosis induction, MMP activation, and pro-inflammatory cytokine production [13,53,93,105,106,107,108,109]. Antioxidant treatment via vitamin E administration lowered oxidative stress and inhibited AAA formation and rupture [105]. The ROS has also been found increased in SMCs and human AAA tissues [53,109,110,111]. Generally, scavenging ROS reduces the formation of AAA in mice and safeguards against aortic aneurysm development [110]. Elevated ROS has been correlated with increased HAT activity and augmented histone acetylation in SMCs, which promotes inflammatory changes in SMCs [109]. A previous in vivo study also showed that ROS contributes to inflammatory SMCs via upregulating cyclophilin, a pro-inflammatory mediator that promotes ERK1/2 phosphorylation and MMP-2 activation [112]. NADPH (nicotinamide adenine dinucleotide phosphate) oxidase, NOX, is a major source of ROS in AAA development [111]. The enhanced expression of NOX1, NOX2, NOX4, and NOX5 have been observed in human SMCs [111]. The mechanical stress is one of the inducers to activate NADPH oxidase to enhance ROS production in VSMCs, in addition to Ang II, PDGF, oxidized low-density lipoprotein (ox-LDL), and certain cytokines such as TNF-α. NOX enzymes have been well studied in AAA pathology [108]. The iNOS deficiency and NADPH oxidase inhibition by apocynin decreases NO(x) levels and suppresses aneurysm formation [110]. The genetic removal of p47phox, a subunit of NADPH oxidase, reduces AAA incidence in the Ang II induced mouse AAA model [113]. NOX1-mediated ROS generation has been reported to lower contractile protein expression in aneurysms [114]. NOX4-mediated oxidative stress has been shown to be implicated in SMC apoptosis, which is a vital process in AAA formation [111]. Taken together, SMC oxidative stress is an important pathological event in the development of AAA; its effects diverge depending on the activated components and the context of aneurysmal induction.
3.2.4. Apoptosis and SMC Loss
A fundamental difference between the healthy vessel wall and the aneurysmal wall lies in the decreased number of VSMCs [6,13,52]. The reduced VSMC number in the aortic wall attenuates their abilities in producing connective tissue and repairing elastin breaks, which induces AAA formation. SMC apoptosis is a major cause of SMC number reduction in AAA development [52,55,115]. The apoptotic SMCs have been frequently observed in the medial aortic wall of AAAs [52,115]. Apoptosis is associated with the generation of apoptotic bodies, which promotes calcium deposition if not cleared by phagocytosis. Calcium deposition in the aortic wall increases vessel wall stiffness and promotes AAA development [116]. SMC apoptosis could be triggered by inflammatory mediators, growth factors such as PDGF, cell stretch, hypoxia, and DNA damage [55,117]. In addition, SMC aging can ultimately progress into cell death [55,118]. The death mediator Fas/Fas ligand (FasL) signaling activates a caspase cascade (caspase-3 and -7) to induce the degradation of chromosomal DNA and apoptosis [119,120]. Further, the activation of Fas/FasL has been reported in SMCs of aneurysm tissues [121,122]. The serpin proteinase inhibitor B9 (serpinb9) has shown success in inhibiting VSMC cell apoptosis and ECM degradation induced by elastase [123]. In accord, the decreased expression of serpin proteinase inhibitors has been reported in AAA [124]. Growing evidence shows that ER stress and oxidative stress induce SMC apoptosis [104,111]. Autophagy related apoptosis is also implicated in the development of AAA. Recently, Lu et al. shows that the transcription factor EB (TFEB), a master regulator of autophagy, is critical for AAA development via regulating VSMC apoptosis. The VSMC-specific knockout of TFEB enhances VSMC apoptosis and promotes AAA formation in different preclinical models of AAA [125]. Consistently, the knockout of ATG7, a key regulator of autophagy, also aggravates angiotensin II-associated aortic remodeling [126].
A number of SNPs have been found through GWAS to be associated with SMC apoptosis in aneurysms, such as CDKN2BAS, DAB2IP, and LDL receptor-related protein 1 (LRP1). Knockdown or knockout of Cdkn2b, DAB2IP, and LRP1 in SMCs are capable of inducing SMC apoptosis and AAA formation [93].
Recent studies have demonstrated the critical roles of noncoding RNAs, including long noncoding RNA (lncRNA) and micro-RNA, in regulating VSMC apoptosis and aneurysm development [94,127,128]. miR-21 and miR-26a have been shown to protect against AAA formation through inhibiting VSMC apoptosis [22,94]. The overexpression of miR-21 through lentiviral transduction results in the decreased expression of phosphatase and tensin homolog (PTEN) and leads to the phosphorylation and activation of AKT, thus preventing VSMC apoptosis [22]. The inhibition of miR-26a via anti-miR transfection promoted H2O2-induced apoptosis of the human aortic SMCs [126]. In addition, lncRNA H19 has been found to be upregulated in Ang II and PPE induced AAA animal models, and knockdown of H19 inhibited the aneurysm formation in both AAA models. Mechanistically, H19 increases the expression of HIF1α, leading to increased VSMC apoptosis in aneurysm [129]. The overexpression of the lncRNA PVT1 has been found to induce VSMC apoptosis, elevate MMP-2 and MMP-9, and decrease TIMP-1 in Ang II-induced mouse AAA models. Conversely, blocking PVT1 reverses these effects in in vitro and in vivo settings [130]. Together, these findings emphasize apoptosis of SMCs as an important pathological event in the development of AAA.
In addition to apoptosis, key mediators of necroptosis, including the receptor-interacting protein kinase 1 (RIPK1) and 3 (RIPK3), have been found to be increased in human AAA samples (especially in VSMCs) and in the elastase-induced mouse model of AAAs [131]. RIPK1/3 are inducers of aortic SMC necroptosis, silent mutation or inhibitors of RIPK1/3 could attenuate AAA expansion in elastase perfusion induced AAA models [131,132,133,134]. Increasing evidence also suggests a role of other cell death types such as ferroptosis and pyroptosis in the development of AAA [135]. In spite of limited knowledge, there is growing interest in targeting cell death pathways as a novel approach for AAA treatment.
3.2.5. VSMCs Inflammatory Phenotypic Change and Transdifferentiation into Macrophage-like Cells
The aortic wall inflammation and inflammatory cell infiltration is an important component of AAA development [13,82,116,136,137]. Lineage tracing techniques have been employed to confirm that VSMCs can transdifferentiate into macrophage-like cells. The phenotypically transdifferentiated VSMCs comprised of approximately 30% of the macrophage population within the atherosclerosis lesions, as shown by lineage tracing in the ApoE−/− mice model [138]. The infiltrated macrophages are responsible for the upregulated cytokines in aneurysm tissues such as IL-1α, IL-1β, IL-6, and TNF-α, which could induce SMC phenotypic change. Interestingly, the inflammatory VSMCs also produce these cytokines, implying that the cytokine upregulation in aneurysms is likely also related to autocrine mechanism. The cytokine upregulation in the aortic wall could further induce MMP activation, VSMC apoptosis, and EC dysfunction to promote AAA formation [6,13]. The SET and MYND domain-containing 2 (SMYD2) methylation in aortic SMCs correlates with suppressed gene expression. The SMYD promoter region of SMCs is drastically hypomethylated in AAAs [139]. SMYD2 can methylate TNF-α and IL-6 promoters to suppress their transcription and inhibit NF-κB and ERK signaling pathways [140]. Therefore, hypomethylation of SMYD may result in increased inflammation and promote AAA development. In addition, miR-24 was shown to target chitinase 3-like 1 (Chi3l1, an inflammation marker of AAA disease progression) and decrease its expression. It suppresses inflammation by blocking IL-8 and MCP-1/CCL2 production by VSMCs. In accord, decreased plasma levels of miR-24 have been observed in AAA patients and murine AAA models [141].
3.2.6. VSMC Phenotypic Switch into Osteo/Chondrogenic VSMCs
Vascular calcification, closely related with arterial stiffening, is characterized by the deposition of calcium phosphate crystals in the vessel wall [142,143,144]. It has been implicated in the development of a number of clinical diseases including chronic kidney disease, atherosclerosis, and hypertension. Recent studies show that vascular calcification is involved in AAA initiation and progression, and even rupture [144,145,146]. The detection of microcalcification assists the evaluation of the risk for AAA events [147,148]. Further, the active mineralization in AAA, as determined by the 18F-sodium fluoride uptake, has been shown to correlate with AAA progression [149,150]. The switching from contractile VSMCs to osteo/chondrogenic VSMCs is a key process in vessel wall calcification [151,152]. The osteo/chondrogenic VSMCs are featured by the increased expression of mineralization regulators, including osteocalcin, alkaline phosphatase, osteopontin, osteoprotegerin, etc., which are controlled by transcription factors such as Runt related transcription factor 2, Osterix, and SRY-box transcription factor 9 [151]. In bovine aortic SMCs, β-glycerophosphate (β-GP) stimulated calcium deposition and, meanwhile, decreased the expression of the elastic fibers [153]. These dysregulations of elastic fibers further cause calcification and decrease vascular elasticity, and finally affect the aneurysm development. The MMP-mediated elastin disorganization has also been shown to correlate with the calcium deposition on elastin fibers and the calcification during AAA [154]. The subsequent detachment of VSMCs from the elastic fibers, VSMC apoptosis, and the loss of matrix Gla protein (a calcification inhibitor), together contribute to the stimulation of the VSMC calcification event.
4. Animal Models Used to Investigate SMC Phenotypic Change and AAA
To study the pathogenesis of AAA and test the potential of prospective therapies, a range of animal models have been developed in different species, such as mouse, rat, rabbit, and pig, etc. [155]. Rodent models, especially mouse models, of AAA have been widely used to explore the underlying mechanisms and therapies of aneurysm. The mouse model of Ang II infusion represents the most widely used animal model of AAA. Daugherty et al. firstly reported AAA formation in ApoE−/− mice when an osmotic mini pump with Ang II was implanted subcutaneously for 4 weeks [23]. The Ang II-induced mouse AAA model is established based on the hyperlipidemia mice, such as ApoE−/− or LDLR−/− male mice, that are susceptible to AAA formation with Ang II infusion, which yields an AAA incidence of approximately 80%, while in WT C57BL/6 mice the incidence is only 25%. The Ang II mouse model is technically easy and takes about one month for aneurysm formation. A common feature of the Ang II infusion model is the induction of arterial inflammation, with massive infiltration of leucocytes into the aorta, the degradation of the extracellular matrix, and smooth muscle phenotypic changes. Differing from the human AAA that occurs predominantly in the infrarenal aorta, the Ang II-induced aneurysms are present in suprarenal region and ascending aorta. However, the Ang II infusion model remains of great interest in the study of pharmaceutical intervention and genetic mutant effects in the development of AAA as it captures several important features of human AAA, including leukocyte infiltration, dissection, medial degeneration, and a close association with atherosclerosis [155].
Unlike the Ang II-induced mouse AAA model, the elastase-induced AAA model does not require hyperlipidemia. Thus, this model could be used to induce AAA in different species with results mimicking that seen in humans. In this model, a segment of surgically isolated aorta is exposed to PPE for 5 to 10 min, which causes cleaved aortic elastic fibers, and triggers an inflammatory response, SMC phenotypic changes, and, ultimately, aneurysm formation within two weeks. A simplified model has also been established by applying PPE at a higher concentration to the outside adventitia to cause acute inflammatory infiltration, ECM degradation, and aortic dilation [156,157]. The loss of VSMCs is directly proportional to the concentration of PPE used, further supporting the essential role of SMCs in AAA development. The PPE model is a frequently used AAA model because it is fast, genetic background independent, and feasible in various species. The weakness of this model includes failure of dissection and intraluminal thrombus induction in the aneurysm tissues [158]. In the CaCl2-induced AAA model, the aneurysm is achieved by applying cotton gauze soaked with CaCl2 to the peri-aortic wall for 15 to 20 min, followed by adding PBS on the aorta [116]. This model is easy to handle and feasible for different species without genetic background requirements. The pathology of this model involves calcification, inflammation, ECM degradation, and VSMC loss [159]. However, the dissection, rupture, and intraluminal thrombus do not develop in this model. A surgery-induced AAA model is also very useful, particularly in large animals, which could be utilized to test endovascular devices or surgical procedures. The frequently used approaches of the surgical model include an aortic patch, artificial aneurysm graft, intra-aortic PPE infusion, and aortic dissection with endovascular treatment [155]. The surgery-induced AAA model is technically challenging and more costly compared to small animal models, which limits the usage of this model.
β-Aminopropionitrile, a lysyl oxidase (LOX) inhibitor, is capable of blocking LOX function in crosslinking elastin and collagen. The combination of β-Aminopropionitrile with the Ang II model or PPE model increases the incidence of AAA and the aneurysm diameter, even the rupture [160,161]. A high-fat diet combined with the Ang II model or PPE model has also been employed to enhance the aneurysm formation [161,162]. The PPE-induced aneurysm could be enhanced through a combined exposure to cigarette smoking, the most potent environmental risk factor for AAA. In addition, the combinations of the different AAA models have also been reported but will not be discussed in this review. It is evident that each AAA model has its own advantages and inherent limitations. Therefore, the selection of AAA animal model should take into account their respective features and the aim and objectives of the research.
5. Application of Single-Cell RNA-Sequencing (scRNA Seq) in AAA Studies
scRNA seq emerges as a powerful approach to study transcriptome profile changes that are useful for identifying cellular clusters and exploring cellular responses in AAA. During AAA development, a myriad of cell types is involved, ranging from circulating immune cells to vascular resident cells (e.g., SMC). Recently, studies using scRNA seq have shed light on the heterogeneity and cellular responses of vascular cells in AAA progression. One recent study identified 17 clusters representing nine-cell lineages. Further Seurat clustering analysis identified four SMC subpopulations and five monocyte/macrophage subpopulations. During AAA progression, three major SMC subpopulations were proportionally decreased, whereas a small subpopulation was increased with downregulated SMC contractile markers and increased pro-inflammatory genes [163], suggesting phenotypic changes. Interestingly, scRNA seq analysis of lesioned aortas has identified macrophage-derived Netrin-1 as a robust inducer of the intracellular calcium flux and MMP3 activity by VSMCs, thereby it mediates the dynamic crosstalk between inflammation and ECM remodeling in AAA [164]. Single-cell analysis of the clinical aortic specimens from Marfan syndrome patients also revealed defective TGF-β signaling, i.e., downregulated TGFBR2 and Smad in a subset of SMCs [165]. Furthermore, an altered subpopulation of dedifferentiated proliferative SMCs was noted in the aortic tissues from Marfan syndrome patients but not from control subjects. These studies underscore the importance of the selective targeting of subgroups of VSMCs based on their transcriptome profiles. The scRNA seq analysis of AAA tissues are useful for dissecting the heterogeneity of cell subpopulations, deregulated signaling pathways, and cellular responses, as well as their interactions during AAA development. It also holds the key for identifying disease-relevant transcriptional signatures in VSMC-lineage cells, which might provide clues for disease predication, diagnosis, and prevention.
6. Conclusions
In summary, we present an overview of what is known about AAA, including the risk factors, the pathophysiology, and animal models used to explore the mechanism and therapies for AAA. AAA remains a serious threat to public health because of its high mortality after rupture. The endovascular exclusion and open surgical techniques are still the major treatment options for AAA. No drugs have been demonstrated to be effective in clinical trials. While mounting evidence from animal models and clinical research suggests inflammatory response and vascular remodeling as important pathological processes for AAA initiation, progression, and rupture, the VSMC phenotypic modulation is of particular interest as it is involved in both processes. Despite intensive research, it needs to be recognized that VSMC phenotypic switching is an evolving area, and the pathophysiology of VSMCs in AAA development remains incompletely understood. Further understanding the regulation of VSMC phenotypic changes and dysfunction in the development of AAA may help identify novel therapeutic targets for the treatment or prevention of AAA.
Author Contributions
G.Q. and O.A. reviewed the literature and drafted the manuscript. A.O. drew the figures. G.Q. and X.G. revised the manuscript. X.G. conceived the article. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Institutes of Health grant (HL141583 to X.G.) and the University of Texas Rising Star Award (to X.G.).
Conflicts of Interest
All authors declare no conflict of interest.
Abbreviations
| AAA | abdominal aortic aneurysm |
| Ang II | angiotensin II |
| α-SMA | α-smooth muscle actin |
| CNN | SM-calponin |
| EC | endothelial cell |
| ECM | extracellular matrix |
| HDACs | histone deacetylases |
| IL-1β | interleukin-1β |
| MCP-1 | monocyte chemoattractant protein 1 |
| MMP | matrix metalloproteinase |
| NOX | nicotinamide adenine dinucleotide phosphate oxidase |
| PDGF-BB | platelet-derived growth factor-BB |
| PPE | porcine pancreatic elastase |
| RIPK1/3 | receptor-interacting protein kinase 1/3 |
| ROS | reactive oxygen species |
| SMMHC | SM myosin heavy chain |
| SM22α | smooth muscle 22α |
| TAA | thoracic aortic aneurysm |
| TFEB | transcription factor EB |
| TGF-β | transforming growth factor-β |
| TGFBR1/2 | TGF-β receptor 1/2 |
| TIMPs | tissue inhibitor of metalloproteinases |
| TNF-α | tumor necrosis factor-α |
| Vcam-1 | vascular cell adhesion molecule 1 |
| VSMC | vascular smooth muscle cell |
References
- Johnston, K.W.; Rutherford, R.B.; Tilson, M.D.; Shah, D.M.; Hollier, L.; Stanley, J.C. Suggested standards for reporting on arterial aneurysms. Subcommittee on Reporting Standards for Arterial Aneurysms, Ad Hoc Committee on Reporting Standards, Society for Vascular Surgery and North American Chapter, International Society for Cardiovascular Surgery. J. Vasc. Surg. 1991, 13, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jersey, A.M.; Foster, D.M. Cerebral Aneurysm; StatPearls Publishing LLC: Treasure Island, FL, USA, 2021. [Google Scholar]
- Quintana, R.A.; Taylor, W.R. Cellular Mechanisms of Aortic Aneurysm Formation. Circ. Res. 2019, 124, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Kainth, A.; Smeds, M.R. Popliteal Aneurysm Repair; StatPearls Publishing LLC: Treasure Island, FL, USA, 2021. [Google Scholar]
- Anderson, P.L.; Arons, R.R.; Moskowitz, A.J.; Gelijns, A.; Magnell, C.; Faries, P.L.; Clair, D.; Nowygrod, R.; Kent, K.C. A statewide experience with endovascular abdominal aortic aneurysm repair: Rapid diffusion with excellent early results. J. Vasc. Surg. 2004, 39, 10–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordon, I.M.; Hinchliffe, R.J.; Loftus, I.M.; Thompson, M.M. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat. Rev. Cardiol. 2011, 8, 92–102. [Google Scholar] [CrossRef]
- Eickhoff, J. Incidence of diagnosis, operation and death from abdominal aortic aneurysms in Danish hospitals: Results from a nation-wide survey, 1977–1990. Eur. J. Surg. Acta Chir. 1993, 159, 619–623. [Google Scholar]
- Moll, F.L.; Powell, J.T.; Fraedrich, G.; Verzini, F.; Haulon, S.; Waltham, M.; van Herwaarden, J.A.; Holt, P.J.; van Keulen, J.W.; Rantner, B. Management of abdominal aortic aneurysms clinical practice guidelines of the European society for vascular surgery. Eur. J. Vasc. Endovasc. Surg. 2011, 41, S1–S58. [Google Scholar] [CrossRef] [Green Version]
- Kantonen, I.; Lepantalo, M.; Brommels, M.; Luther, M.; Salenius, J.P.; Ylonen, K. Mortality in ruptured abdominal aortic aneurysms. The Finnvasc Study Group. Eur. J. Vasc. Endovasc. Surg. 1999, 17, 208–212. [Google Scholar] [CrossRef] [Green Version]
- Earnshaw, J.J.; Lees, T. Update on Screening for Abdominal Aortic Aneurysm. Eur. J. Vasc. Endovasc. Surg. 2017, 54, 1–2. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, T.F.X.; Landon, B.E.; Schermerhorn, M.L. AAA Screening Should Be Expanded. Circulation 2019, 140, 889–890. [Google Scholar] [CrossRef]
- Hallin, A.; Bergqvist, D.; Holmberg, L. Literature review of surgical management of abdominal aortic aneurysm. Eur. J. Vasc. Endovasc. Surg. 2001, 22, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Kuivaniemi, H.; Ryer, E.J.; Elmore, J.R.; Tromp, G. Understanding the pathogenesis of abdominal aortic aneurysms. Expert Rev. Cardiovasc. 2015, 13, 975–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altobelli, E.; Rapacchietta, L.; Profeta, V.F.; Fagnano, R. Risk factors for abdominal aortic aneurysm in population-based studies: A systematic review and meta-analysis. Int. J. Environ. Res. Public Health 2018, 15, 2805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilson, M. Aortic aneurysms and atherosclerosis. Circulation 1992, 85, 378–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonamigo, T.P.; Siqueira, I. Screening for abdominal aortic aneurysms. Rev. Hosp. Clin. Fac. Med. Sao Paulo 2003, 58, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.G.; Thompson, S.G.; Marteau, T.M.; Scott, R.A.; Multicentre Aneurysm Screening Study, G. Screening for abdominal aortic aneurysms: The effects of age and social deprivation on screening uptake, prevalence and attendance at follow-up in the MASS trial. J. Med. Screen. 2004, 11, 50–53. [Google Scholar] [CrossRef] [Green Version]
- Saratzis, A.; Dattani, N.; Brown, A.; Shalhoub, J.; Bosanquet, D.; Sidloff, D.; Stather, P.; Vascular, T.; Endovascular Research, N. Multi-Centre Study on Cardiovascular Risk Management on Patients Undergoing AAA Surveillance. Eur. J. Vasc. Endovasc. Surg. 2017, 54, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Svensjo, S.; Bjorck, M.; Gurtelschmid, M.; Djavani Gidlund, K.; Hellberg, A.; Wanhainen, A. Low prevalence of abdominal aortic aneurysm among 65-year-old Swedish men indicates a change in the epidemiology of the disease. Circulation 2011, 124, 1118–1123. [Google Scholar] [CrossRef] [Green Version]
- Norman, P.E.; Curci, J.A. Understanding the effects of tobacco smoke on the pathogenesis of aortic aneurysm. Arter. Thromb. Vasc. Biol. 2013, 33, 1473–1477. [Google Scholar] [CrossRef] [Green Version]
- Stolle, K.; Berges, A.; Lietz, M.; Lebrun, S.; Wallerath, T. Cigarette smoke enhances abdominal aortic aneurysm formation in angiotensin II-treated apolipoprotein E-deficient mice. Toxicol. Lett. 2010, 199, 403–409. [Google Scholar] [CrossRef]
- Maegdefessel, L.; Azuma, J.; Toh, R.; Deng, A.; Merk, D.R.; Raiesdana, A.; Leeper, N.J.; Raaz, U.; Schoelmerich, A.M.; McConnell, M.V.; et al. MicroRNA-21 blocks abdominal aortic aneurysm development and nicotine-augmented expansion. Sci. Transl. Med. 2012, 4, 122ra122. [Google Scholar] [CrossRef] [Green Version]
- Daugherty, A.; Manning, M.W.; Cassis, L.A. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J. Clin. Investig. 2000, 105, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuivaniemi, H.; Platsoucas, C.D.; Tilson III, M.D. Aortic aneurysms: An immune disease with a strong genetic component. Circulation 2008, 117, 242–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlgren, C.M.; Larsson, E.; Magnusson, P.K.; Hultgren, R.; Swedenborg, J. Genetic and environmental contributions to abdominal aortic aneurysm development in a twin population. J. Vasc. Surg. 2010, 51, 3–7; discussion 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Vlijmen-van Keulen, C.J.; Pals, G.; Rauwerda, J.A. Familial abdominal aortic aneurysm: A systematic review of a genetic background. Eur. J. Vasc. Endovasc. Surg. 2002, 24, 105–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, C.W.; Barber, G.G.; Bouchard, A.G.; McPhail, N.V.; Roberge, C.; Waddell, W.G.; Wellington, J.L. Abdominal aortic aneurysm: Consequences of a positive family history. Can. J. Surg. 1989, 32, 117–120. [Google Scholar] [PubMed]
- Larsson, E.; Granath, F.; Swedenborg, J.; Hultgren, R. A population-based case-control study of the familial risk of abdominal aortic aneurysm. J. Vasc. Surg. 2009, 49, 47–50; discussion 51. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, C.J.; Casey, M.; He, J.; Veugelers, M.; Henderson, K.; Guo, D.; Campagna, R.; Roman, M.J.; Milewicz, D.M.; Devereux, R.B. Identification of a chromosome 11q23. 2-q24 locus for familial aortic aneurysm disease, a genetically heterogeneous disorder. Circulation 2001, 103, 2469–2475. [Google Scholar] [CrossRef] [Green Version]
- Shibamura, H.; Olson, J.M.; van Vlijmen-Van Keulen, C.; Buxbaum, S.G.; Dudek, D.M.; Tromp, G.; Ogata, T.; Skunca, M.; Sakalihasan, N.; Pals, G.; et al. Genome scan for familial abdominal aortic aneurysm using sex and family history as covariates suggests genetic heterogeneity and identifies linkage to chromosome 19q13. Circulation 2004, 109, 2103–2108. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.T.; Tromp, G.; Kuivaniemi, H.; Gretarsdottir, S.; Baas, A.F.; Giusti, B.; Strauss, E.; Van’t Hof, F.N.; Webb, T.R.; Erdman, R.; et al. Meta-Analysis of Genome-Wide Association Studies for Abdominal Aortic Aneurysm Identifies Four New Disease-Specific Risk Loci. Circ. Res. 2017, 120, 341–353. [Google Scholar] [CrossRef]
- Singh, T.P.; Field, M.A.; Bown, M.J.; Jones, G.T.; Golledge, J. Systematic review of genome-wide association studies of abdominal aortic aneurysm. Atherosclerosis 2021, 327, 39–48. [Google Scholar] [CrossRef]
- Vats, S.; Sundquist, K.; Wang, X.; Zarrouk, M.; Agren-Witteschus, S.; Sundquist, J.; Gottsater, A.; Memon, A.A. Associations of global DNA methylation and homocysteine levels with abdominal aortic aneurysm: A cohort study from a population-based screening program in Sweden. Int. J. Cardiol. 2020, 321, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Toghill, B.J.; Saratzis, A.; Harrison, S.C.; Verissimo, A.R.; Mallon, E.B.; Bown, M.J. The potential role of DNA methylation in the pathogenesis of abdominal aortic aneurysm. Atherosclerosis 2015, 241, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Umemoto, T.; Group, A. Association of Hypertension with Abdominal Aortic Aneurysm Expansion. Ann. Vasc. Surg. 2017, 39, 74–89. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, A.; Skagius, E.; Englund, E.; Nilsson, I.; Ljungh, A.; Henriksson, A.E. Abdominal aortic aneurysm and the impact of infectious burden. Eur. J. Vasc. Endovasc. Surg. 2008, 36, 292–296. [Google Scholar] [CrossRef] [Green Version]
- Jablonska, A.; Zagrapan, B.; Paradowska, E.; Neumayer, C.; Eilenberg, W.; Brostjan, C.; Klinger, M.; Nanobachvili, J.; Huk, I. Abdominal aortic aneurysm and virus infection: A potential causative role for cytomegalovirus infection? J. Med. Virol. 2021, 93, 5017–5024. [Google Scholar] [CrossRef]
- Wolinsky, H.; Glagov, S. A lamellar unit of aortic medial structure and function in mammals. Circ. Res. 1967, 20, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Crawford, E.S.; Cohen, E.S. Aortic aneurysm: A multifocal disease: Presidential address. Arch. Surg. 1982, 117, 1393–1400. [Google Scholar] [CrossRef]
- Koch, A.E.; Haines, G.K.; Rizzo, R.J.; Radosevich, J.A.; Pope, R.M.; Robinson, P.G.; Pearce, W.H. Human abdominal aortic aneurysms. Immunophenotypic analysis suggesting an immune-mediated response. Am. J. Pathol. 1990, 137, 1199. [Google Scholar]
- Gregory, A.K.; Yin, N.X.; Capella, J.; Xia, S.; Newman, K.M.; Tilson, M.D. Features of autoimmunity in the abdominal aortic aneurysm. Arch. Surg. 1996, 131, 85–88. [Google Scholar] [CrossRef]
- Yuan, Z.; Lu, Y.; Wei, J.; Wu, J.; Yang, J.; Cai, Z. Abdominal Aortic Aneurysm: Roles of Inflammatory Cells. Front. Immunol. 2020, 11, 609161. [Google Scholar] [CrossRef]
- Marculescu, R.; Sodeck, G.; Domanovits, H.; Hobusch, G.; Exner, M.; Heinzl, H.; Huber, K.; Mannhalter, C.; Minar, E.; Wagner, O. Interleukin-1 gene cluster variants and abdominal aortic aneurysms. Thromb. Haemost. 2005, 94, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Bown, M.J.; Burton, P.R.; Horsburgh, T.; Nicholson, M.L.; Bell, P.R.; Sayers, R.D. The role of cytokine gene polymorphisms in the pathogenesis of abdominal aortic aneurysms: A case-control study. J. Vasc. Surg. 2003, 37, 999–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, K.G.; Brull, D.J.; Brown, L.C.; Sian, M.; Greenhalgh, R.M.; Humphries, S.E.; Powell, J.T. Interleukin-6 (IL-6) and the prognosis of abdominal aortic aneurysms. Circulation 2001, 103, 2260–2265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anidjar, S.; Dobrin, P.B.; Eichorst, M.; Graham, G.P.; Chejfec, G. Correlation of inflammatory infiltrate with the enlargement of experimental aortic aneurysms. J. Vasc. Surg. 1992, 16, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Juvonen, J.; Surcel, H.-M.; Satta, J.; Teppo, A.-M.; Bloigu, A.; Syrjälä, H.; Airaksinen, J.; Leinonen, M.; Saikku, P.; Juvonen, T. Elevated circulating levels of inflammatory cytokines in patients with abdominal aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2843–2847. [Google Scholar] [CrossRef]
- Colonnello, J.S.; Hance, K.A.; Shames, M.L.; Wyble, C.W.; Ziporin, S.J.; Leidenfrost, J.E.; Ennis, T.L.; Upchurch Jr, G.R.; Thompson, R.W. Transient exposure to elastase induces mouse aortic wall smooth muscle cell production of MCP-1 and RANTES during development of experimental aortic aneurysm. J. Vasc. Surg. 2003, 38, 138–146. [Google Scholar] [CrossRef] [Green Version]
- Hance, K.A.; Tataria, M.; Ziporin, S.J.; Lee, J.K.; Thompson, R.W. Monocyte chemotactic activity in human abdominal aortic aneurysms: Role of elastin degradation peptides and the 67–kD cell surface elastin receptor. J. Vasc. Surg. 2002, 35, 254–261. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Deng, H.; Zhou, Z.; Xiong, X.; Gao, L. Endothelium as a Potential Target for Treatment of Abdominal Aortic Aneurysm. Oxid. Med. Cell. Longev. 2018, 2018, 6306542. [Google Scholar] [CrossRef] [Green Version]
- Spartalis, E.; Spartalis, M.; Athanasiou, A.; Paschou, S.A.; Patelis, N.; Voudris, V.; Iliopoulos, D.C. Endothelium in Aortic Aneurysm Disease: New Insights. Curr. Med. Chem. 2020, 27, 1081–1088. [Google Scholar] [CrossRef]
- Lopez-Candales, A.; Holmes, D.R.; Liao, S.; Scott, M.J.; Wickline, S.A.; Thompson, R.W. Decreased vascular smooth muscle cell density in medial degeneration of human abdominal aortic aneurysms. Am. J. Pathol. 1997, 150, 993–1007. [Google Scholar]
- Miller, F.J., Jr.; Sharp, W.J.; Fang, X.; Oberley, L.W.; Oberley, T.D.; Weintraub, N.L. Oxidative stress in human abdominal aortic aneurysms: A potential mediator of aneurysmal remodeling. Arter. Thromb. Vasc. Biol. 2002, 22, 560–565. [Google Scholar] [CrossRef] [Green Version]
- Airhart, N.; Arif, B.; Curci, J. Vascular smooth muscle cells from abdominal aortic aneurysms have uniquely high elastolytic potential associated with activation of MMP-2. J. Surg. Res. 2013, 179, 282. [Google Scholar] [CrossRef]
- Thompson, R.W.; Liao, S.; Curci, J.A. Vascular smooth muscle cell apoptosis in abdominal aortic aneurysms. Coron. Artery Dis. 1997, 8, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, R.; Dave, J.M.; Chandran, R.R.; Misra, A.; Sheikh, A.Q.; Greif, D.M. Vascular Cells in Blood Vessel Wall Development and Disease. Adv. Pharm. 2017, 78, 323–350. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Huang, H.; Sun, X.; Guo, Y.; Hamblin, M.; Ritchie, R.P.; Garcia-Barrio, M.T.; Zhang, J.; Chen, Y.E. MicroRNA-1 regulates smooth muscle cell differentiation by repressing Kruppel-like factor 4. Stem Cells Dev. 2011, 20, 205–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Jacquet, L.; Karamariti, E.; Xu, Q. Origin and differentiation of vascular smooth muscle cells. J. Physiol. 2015, 593, 3013–3030. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Chen, S.Y. Transforming growth factor-beta and smooth muscle differentiation. World J. Biol. Chem. 2012, 3, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef]
- Wasteson, P.; Johansson, B.R.; Jukkola, T.; Breuer, S.; Akyürek, L.M.; Partanen, J.; Lindahl, P. Developmental origin of smooth muscle cells in the descending aorta in mice. Development 2008, 135, 1823–1832. [Google Scholar] [CrossRef] [Green Version]
- Alexander, M.R.; Owens, G.K. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu. Rev. Physiol. 2012, 74, 13–40. [Google Scholar] [CrossRef]
- Coll-Bonfill, N.; De La Cruz-Thea, B.; Pisano, M.; Musri, M. Noncoding RNAs in smooth muscle cell homeostasis: Implications in phenotypic switch and vascular disorders. Pflügers Arch.-Eur. J. Physiol. 2016, 468, 1071–1087. [Google Scholar] [CrossRef] [PubMed]
- Ailawadi, G.; Moehle, C.W.; Pei, H.; Walton, S.P.; Yang, Z.; Kron, I.L.; Lau, C.L.; Owens, G.K. Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J. Thorac. Cardiovasc. Surg. 2009, 138, 1392–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, T.; Kaestner, K.H.; Owens, G.K. Conditional deletion of Kruppel-like factor 4 delays downregulation of smooth muscle cell differentiation markers but accelerates neointimal formation following vascular injury. Circ. Res. 2008, 102, 1548–1557. [Google Scholar] [CrossRef]
- Chen, P.Y.; Qin, L.; Li, G.; Tellides, G.; Simons, M. Smooth muscle FGF/TGF β cross talk regulates atherosclerosis progression. EMBO Mol. Med. 2016, 8, 712–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Shi, N.; Cui, X.B.; Wang, J.N.; Fukui, Y.; Chen, S.Y. Dedicator of cytokinesis 2, a novel regulator for smooth muscle phenotypic modulation and vascular remodeling. Circ. Res. 2015, 116, e71–e80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crosas-Molist, E.; Meirelles, T.; López-Luque, J.; Serra-Peinado, C.; Selva, J.; Caja, L.; Gorbenko del Blanco, D.; Uriarte, J.J.; Bertran, E.; Mendizábal, Y. Vascular smooth muscle cell phenotypic changes in patients with Marfan syndrome. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 960–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Vranckx, R.; Van Kien, P.K.; Lalande, A.; Boisset, N.; Mathieu, F.; Wegman, M.; Glancy, L.; Gasc, J.-M.; Brunotte, F. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat. Genet. 2006, 38, 343–349. [Google Scholar] [CrossRef]
- Li, W.; Li, Q.; Jiao, Y.; Qin, L.; Ali, R.; Zhou, J.; Ferruzzi, J.; Kim, R.W.; Geirsson, A.; Dietz, H.C.; et al. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J. Clin. Investig. 2014, 124, 755–767. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Shen, J.; Annam, N.P.; Jiang, H.; Levi, E.; Schworer, C.M.; Tromp, G.; Arora, A.; Higgins, M.; Wang, X.F.; et al. SMAD3 deficiency promotes vessel wall remodeling, collagen fiber reorganization and leukocyte infiltration in an inflammatory abdominal aortic aneurysm mouse model. Sci. Rep. 2015, 5, 10180. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ait-Oufella, H.; Herbin, O.; Bonnin, P.; Ramkhelawon, B.; Taleb, S.; Huang, J.; Offenstadt, G.; Combadiere, C.; Renia, L.; et al. TGF-beta activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. J. Clin. Investig. 2010, 120, 422–432. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Rateri, D.L.; Howatt, D.A.; Balakrishnan, A.; Moorleghen, J.J.; Cassis, L.A.; Daugherty, A. TGF-beta Neutralization Enhances AngII-Induced Aortic Rupture and Aneurysm in Both Thoracic and Abdominal Regions. PLoS ONE 2016, 11, e0153811. [Google Scholar] [CrossRef]
- Biros, E.; Walker, P.J.; Nataatmadja, M.; West, M.; Golledge, J. Downregulation of transforming growth factor, beta receptor 2 and Notch signaling pathway in human abdominal aortic aneurysm. Atherosclerosis 2012, 221, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Baas, A.F.; Medic, J.; van’t Slot, R.; de Kovel, C.G.; Zhernakova, A.; Geelkerken, R.H.; Kranendonk, S.E.; van Sterkenburg, S.M.; Grobbee, D.E.; Boll, A.P.; et al. Association of the TGF-beta receptor genes with abdominal aortic aneurysm. Eur. J. Hum. Genet. 2010, 18, 240–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.Y.; Qin, L.; Li, G.; Malagon-Lopez, J.; Wang, Z.; Bergaya, S.; Gujja, S.; Caulk, A.W.; Murtada, S.I.; Zhang, X.; et al. Smooth Muscle Cell Reprogramming in Aortic Aneurysms. Cell Stem Cell 2020, 26, 542–557.e511. [Google Scholar] [CrossRef]
- Rangrez, A.Y.; Massy, Z.A.; Metzinger-Le Meuth, V.; Metzinger, L. miR-143 and miR-145: Molecular keys to switch the phenotype of vascular smooth muscle cells. Circ. Cardiovasc. Genet. 2011, 4, 197–205. [Google Scholar] [CrossRef]
- Xin, M.; Small, E.M.; Sutherland, L.B.; Qi, X.; McAnally, J.; Plato, C.F.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 2009, 23, 2166–2178. [Google Scholar] [CrossRef] [Green Version]
- Elia, L.; Quintavalle, M.; Zhang, J.; Contu, R.; Cossu, L.; Latronico, M.V.; Peterson, K.L.; Indolfi, C.; Catalucci, D.; Chen, J.; et al. The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: Correlates with human disease. Cell Death Differ. 2009, 16, 1590–1598. [Google Scholar] [CrossRef]
- Van Varik, B.; Rennenberg, R.; Reutelingsperger, C.; Kroon, A.; de Leeuw, P.; Schurgers, L.J. Mechanisms of arterial remodeling: Lessons from genetic diseases. Front. Genet. 2012, 3, 290. [Google Scholar] [CrossRef] [Green Version]
- Bendeck, M.P.; Irvin, C.; Reidy, M.A. Inhibition of matrix metalloproteinase activity inhibits smooth muscle cell migration but not neointimal thickening after arterial injury. Circ. Res. 1996, 78, 38–43. [Google Scholar] [CrossRef]
- Sandford, R.; Bown, M.; London, N.; Sayers, R. The genetic basis of abdominal aortic aneurysms: A review. Eur. J. Vasc. Endovasc. Surg. 2007, 33, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Tilson, M.D.; Reilly, J.M.; Brophy, C.M.; Webster, E.L.; Barnett, T.R. Expression and sequence of the gene for tissue inhibitor of metalloproteinases in patients with abdominal aortic aneurysms. J. Vasc. Surg. 1993, 18, 266–270. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Khalil, R.A. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv. Pharm. 2018, 81, 241–330. [Google Scholar] [CrossRef]
- Raffetto, J.D.; Khalil, R.A. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem. Pharm. 2008, 75, 346–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, G.T.; Phillips, V.L.; Harris, E.L.; Rossaak, J.I.; van Rij, A.M. Functional matrix metalloproteinase-9 polymorphism (C-1562T) associated with abdominal aortic aneurysm. J. Vasc. Surg. 2003, 38, 1363–1367. [Google Scholar] [CrossRef] [Green Version]
- Lamblin, N.; Bauters, C.; Hermant, X.; Lablanche, J.-M.; Helbecque, N.; Amouyel, P. Polymorphisms in the promoter regions of MMP-2, MMP-3, MMP-9 and MMP-12 genes as determinants of aneurysmal coronary artery disease. J. Am. Coll. Cardiol. 2002, 40, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, P.; Jormsjö-Pettersson, S.; Brady, A.; Deguchi, H.; Hamsten, A.; Powell, J. Genotype–phenotype relationships in an investigation of the role of proteases in abdominal aortic aneurysm expansion. J. Br. Surg. 2005, 92, 1372–1376. [Google Scholar] [CrossRef]
- Longo, G.M.; Buda, S.J.; Fiotta, N.; Xiong, W.; Griener, T.; Shapiro, S.; Baxter, B.T. MMP-12 has a role in abdominal aortic aneurysms in mice. Surgery 2005, 137, 457–462. [Google Scholar] [CrossRef]
- Brophy, C.M.; Marks, W.H.; Reilly, J.M.; Tilson, M.D. Decreased tissue inhibitor of metalloproteinases (TIMP) in abdominal aortic aneurysm tissue: A preliminary report. J. Surg. Res. 1991, 50, 653–657. [Google Scholar] [CrossRef]
- Eskandari, M.K.; Vijungco, J.D.; Flores, A.; Borensztajn, J.; Shively, V.; Pearce, W.H. Enhanced abdominal aortic aneurysm in TIMP-1-deficient mice. J. Surg. Res. 2005, 123, 289–293. [Google Scholar] [CrossRef]
- Rossaak, J.I.; van Rij, A.M.; Jones, G.T.; Harris, E.L. Association of the 4G/5G polymorphism in the promoter region of plasminogen activator inhibitor-1 with abdominal aortic aneurysms. J. Vasc. Surg. 2000, 31, 1026–1032. [Google Scholar] [CrossRef]
- Gurung, R.; Choong, A.M.; Woo, C.C.; Foo, R.; Sorokin, V. Genetic and Epigenetic Mechanisms Underlying Vascular Smooth Muscle Cell Phenotypic Modulation in Abdominal Aortic Aneurysm. Int. J. Mol. Sci. 2020, 21, 6334. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Dimmeler, S. MicroRNAs and aneurysm formation. Trends Cardiovasc. Med. 2011, 21, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Kumar, S.; Son, D.J.; Jang, I.H.; Griendling, K.K.; Jo, H. Prevention of abdominal aortic aneurysm by anti-microRNA-712 or anti-microRNA-205 in angiotensin II-infused mice. Arter. Thromb. Vasc. Biol. 2014, 34, 1412–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galan, M.; Varona, S.; Orriols, M.; Rodriguez, J.A.; Aguilo, S.; Dilme, J.; Camacho, M.; Martinez-Gonzalez, J.; Rodriguez, C. Induction of histone deacetylases (HDACs) in human abdominal aortic aneurysm: Therapeutic potential of HDAC inhibitors. Dis. Model. Mech. 2016, 9, 541–552. [Google Scholar] [CrossRef] [Green Version]
- Vinh, A.; Gaspari, T.A.; Liu, H.B.; Dousha, L.F.; Widdop, R.E.; Dear, A.E. A novel histone deacetylase inhibitor reduces abdominal aortic aneurysm formation in angiotensin II-infused apolipoprotein E-deficient mice. J. Vasc. Res. 2008, 45, 143–152. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Z.; Xie, N.; Huang, C.; Li, Z.; Yu, F.; Fu, Y.; Cui, Q.; Kong, W. Pan-HDAC (Histone Deacetylase) Inhibitors Increase Susceptibility of Thoracic Aortic Aneurysm and Dissection in Mice. Arter. Thromb. Vasc. Biol. 2021, 41, 2848–2850. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, T.T.; Zhang, R.; Fu, W.Y.; Wang, X.; Wang, F.; Gao, P.; Ding, Y.N.; Xie, Y.; Hao, D.L.; et al. Calorie restriction protects against experimental abdominal aortic aneurysms in mice. J. Exp. Med. 2016, 213, 2473–2488. [Google Scholar] [CrossRef]
- Furmanik, M.; M Shanahan, C. Endoplasmic reticulum stress in arterial smooth muscle cells: A novel regulator of vascular disease. Curr. Cardiol. Rev. 2017, 13, 94–105. [Google Scholar]
- Navas-Madronal, M.; Rodriguez, C.; Kassan, M.; Fite, J.; Escudero, J.R.; Canes, L.; Martinez-Gonzalez, J.; Camacho, M.; Galan, M. Enhanced endoplasmic reticulum and mitochondrial stress in abdominal aortic aneurysm. Clin. Sci. 2019, 133, 1421–1438. [Google Scholar] [CrossRef]
- Qin, Y.; Wang, Y.; Liu, O.; Jia, L.; Fang, W.; Du, J.; Wei, Y. Tauroursodeoxycholic Acid Attenuates Angiotensin II Induced Abdominal Aortic Aneurysm Formation in Apolipoprotein E-deficient Mice by Inhibiting Endoplasmic Reticulum Stress. Eur. J. Vasc. Endovasc. Surg. 2017, 53, 337–345. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Fu, Y.; Cai, Z.; Yu, F.; Gong, Z.; Dai, R.; Hu, Y.; Zeng, L.; Xu, Q.; Kong, W. Unspliced XBP1 confers VSMC homeostasis and prevents aortic aneurysm formation via FoxO4 interaction. Circ. Res. 2017, 121, 1331–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, L.X.; Zhang, W.M.; Zhang, H.J.; Li, T.T.; Wang, Y.L.; Qin, Y.W.; Gu, H.; Du, J. Mechanical stretch-induced endoplasmic reticulum stress, apoptosis and inflammation contribute to thoracic aortic aneurysm and dissection. J. Pathol. 2015, 236, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavrila, D.; Li, W.G.; McCormick, M.L.; Thomas, M.; Daugherty, A.; Cassis, L.A.; Miller, F.J., Jr.; Oberley, L.W.; Dellsperger, K.C.; Weintraub, N.L. Vitamin E inhibits abdominal aortic aneurysm formation in angiotensin II-infused apolipoprotein E-deficient mice. Arter. Thromb. Vasc. Biol. 2005, 25, 1671–1677. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.-W.; Jia, L.-X.; Ni, X.-Q.; Zhao, L.; Chang, J.-R.; Zhang, J.-S.; Hou, Y.-L.; Zhu, Y.; Guan, Y.-F.; Yu, Y.-R. Intermedin1− 53 Attenuates Abdominal Aortic Aneurysm by Inhibiting Oxidative Stress. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2176–2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branchetti, E.; Poggio, P.; Sainger, R.; Shang, E.; Grau, J.B.; Jackson, B.M.; Lai, E.K.; Parmacek, M.S.; Gorman, R.C.; Gorman, J.H. Oxidative stress modulates vascular smooth muscle cell phenotype via CTGF in thoracic aortic aneurysm. Cardiovasc. Res. 2013, 100, 316–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siu, K.L.; Li, Q.; Zhang, Y.; Guo, J.; Youn, J.Y.; Du, J.; Cai, H. NOX isoforms in the development of abdominal aortic aneurysm. Redox Biol. 2017, 11, 118–125. [Google Scholar] [CrossRef] [Green Version]
- McCormick, M.L.; Gavrila, D.; Weintraub, N.L. Role of oxidative stress in the pathogenesis of abdominal aortic aneurysms. Arter. Thromb. Vasc. Biol. 2007, 27, 461–469. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.; Mactaggart, J.; Knispel, R.; Worth, J.; Zhu, Z.; Li, Y.; Sun, Y.; Baxter, B.T.; Johanning, J. Inhibition of reactive oxygen species attenuates aneurysm formation in a murine model. Atherosclerosis 2009, 202, 128–134. [Google Scholar] [CrossRef] [Green Version]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH oxidases in vascular pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [Green Version]
- Soe, N.N.; Sowden, M.; Baskaran, P.; Kim, Y.; Nigro, P.; Smolock, E.M.; Berk, B.C. Acetylation of cyclophilin A is required for its secretion and vascular cell activation. Cardiovasc. Res. 2014, 101, 444–453. [Google Scholar] [CrossRef]
- Thomas, M.; Gavrila, D.; McCormick, M.L.; Miller, F.J., Jr.; Daugherty, A.; Cassis, L.A.; Dellsperger, K.C.; Weintraub, N.L. Deletion of p47phox attenuates angiotensin II-induced abdominal aortic aneurysm formation in apolipoprotein E-deficient mice. Circulation 2006, 114, 404–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starke, R.M.; Thompson, J.W.; Ali, M.S.; Pascale, C.L.; Martinez Lege, A.; Ding, D.; Chalouhi, N.; Hasan, D.M.; Jabbour, P.; Owens, G.K. Cigarette smoke initiates oxidative stress-induced cellular phenotypic modulation leading to cerebral aneurysm pathogenesis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 610–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, V.L.; Stevens, S.L.; Reddick, T.T.; Freeman, M.B.; Donnell, R.; Carroll, R.C.; Goldman, M.H. Vascular smooth muscle cell apoptosis in aneurysmal, occlusive, and normal human aortas. J. Vasc. Surg. 2000, 31, 567–576. [Google Scholar] [CrossRef]
- Yamanouchi, D.; Morgan, S.; Stair, C.; Seedial, S.; Lengfeld, J.; Kent, K.C.; Liu, B. Accelerated aneurysmal dilation associated with apoptosis and inflammation in a newly developed calcium phosphate rodent abdominal aortic aneurysm model. J. Vasc. Surg. 2012, 56, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopalan, S.; Meng, X.P.; Ramasamy, S.; Harrison, D.G.; Galis, Z.S. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J. Clin. Investig. 1996, 98, 2572–2579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, L.-X.; Zhang, W.-M.; Li, T.-T.; Liu, Y.; Piao, C.-M.; Ma, Y.-C.; Lu, Y.; Wang, Y.; Liu, T.-T.; Qi, Y.-F. ER stress dependent microparticles derived from smooth muscle cells promote endothelial dysfunction during thoracic aortic aneurysm and dissection. Clin. Sci. 2017, 131, 1287–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, S. Fas ligand-induced apoptosis. Annu. Rev. Genet. 1999, 33, 29–55. [Google Scholar] [CrossRef]
- Waring, P.; Mullbacher, A. Cell death induced by the Fas/Fas ligand pathway and its role in pathology. Immunol. Cell. Biol. 1999, 77, 312–317. [Google Scholar] [CrossRef]
- Henderson, E.L.; Geng, Y.J.; Sukhova, G.K.; Whittemore, A.D.; Knox, J.; Libby, P. Death of smooth muscle cells and expression of mediators of apoptosis by T lymphocytes in human abdominal aortic aneurysms. Circulation 1999, 99, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.; Chau, L. Fas/Fas ligand-mediated death pathway is involved in oxLDL-induced apoptosis in vascular smooth muscle cells. Am J. Physiol. Cell Physiol. 2001, 280, C709–C718. [Google Scholar] [CrossRef]
- Kuiper, J.; Quax, P.H.; Bot, I. Anti-apoptotic serpins as therapeutics in cardiovascular diseases. Cardiovasc. Hematol. Disord. Drug Targets 2013, 13, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Ang, L.S.; Boivin, W.A.; Williams, S.J.; Zhao, H.; Abraham, T.; Carmine-Simmen, K.; McManus, B.M.; Bleackley, R.C.; Granville, D.J. Serpina3n attenuates granzyme B-mediated decorin cleavage and rupture in a murine model of aortic aneurysm. Cell Death Dis. 2011, 2, e209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Sun, J.; Liang, W.; Chang, Z.; Rom, O.; Zhao, Y.; Zhao, G.; Xiong, W.; Wang, H.; Zhu, T.; et al. Cyclodextrin Prevents Abdominal Aortic Aneurysm via Activation of Vascular Smooth Muscle Cell Transcription Factor EB. Circulation 2020, 142, 483–498. [Google Scholar] [CrossRef] [PubMed]
- Leeper, N.J.; Raiesdana, A.; Kojima, Y.; Chun, H.J.; Azuma, J.; Maegdefessel, L.; Kundu, R.K.; Quertermous, T.; Tsao, P.S.; Spin, J.M. MicroRNA-26a is a novel regulator of vascular smooth muscle cell function. J. Cell. Physiol. 2011, 226, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.Y.; Trenner, M.; Boon, R.A.; Spin, J.M.; Maegdefessel, L. Long noncoding RNAs in key cellular processes involved in aortic aneurysms. Atherosclerosis 2020, 292, 112–118. [Google Scholar] [CrossRef] [Green Version]
- Kalayinia, S.; Arjmand, F.; Maleki, M.; Malakootian, M.; Singh, C.P. MicroRNAs: Roles in cardiovascular development and disease. Cardiovasc. Pathol. 2021, 50, 107296. [Google Scholar] [CrossRef]
- Li, D.Y.; Busch, A.; Jin, H.; Chernogubova, E.; Pelisek, J.; Karlsson, J.; Sennblad, B.; Liu, S.; Lao, S.; Hofmann, P.; et al. H19 Induces Abdominal Aortic Aneurysm Development and Progression. Circulation 2018, 138, 1551–1568. [Google Scholar] [CrossRef]
- Zhang, Z.; Zou, G.; Chen, X.; Lu, W.; Liu, J.; Zhai, S.; Qiao, G. Knockdown of lncRNA PVT1 Inhibits Vascular Smooth Muscle Cell Apoptosis and Extracellular Matrix Disruption in a Murine Abdominal Aortic Aneurysm Model. Mol. Cells 2019, 42, 218–227. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Z.; Ren, J.; Morgan, S.; Assa, C.; Liu, B. Receptor-interacting protein kinase 3 contributes to abdominal aortic aneurysms via smooth muscle cell necrosis and inflammation. Circ. Res. 2015, 116, 600–611. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Wang, Q.; Phan, N.; Ren, J.; Yang, H.; Feldman, C.C.; Feltenberger, J.B.; Ye, Z.; Wildman, S.A.; Tang, W.; et al. Identification of a novel class of RIP1/RIP3 dual inhibitors that impede cell death and inflammation in mouse abdominal aortic aneurysm models. Cell Death Dis. 2019, 10, 226. [Google Scholar] [CrossRef]
- Khoury, M.K.; Zhou, T.; Yang, H.; Prince, S.R.; Gupta, K.; Stranz, A.R.; Wang, Q.; Liu, B. GSK2593074A blocks progression of existing abdominal aortic dilation. JVS Vasc. Sci. 2020, 1, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; DeRoo, E.; Yang, H.; Stranz, A.; Wang, Q.; Ginnan, R.; Singer, H.A.; Liu, B. MLKL and CaMKII Are Involved in RIPK3-Mediated Smooth Muscle Cell Necroptosis. Cells 2021, 10, 2397. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; He, Y.; Wei, X.; Jiang, D.S. Targeting regulated cell death in aortic aneurysm and dissection therapy. Pharm. Res 2021, 176, 106048. [Google Scholar] [CrossRef]
- Abdul-Hussien, H.; Hanemaaijer, R.; Kleemann, R.; Verhaaren, B.F.; van Bockel, J.H.; Lindeman, J.H. The pathophysiology of abdominal aortic aneurysm growth: Corresponding and discordant inflammatory and proteolytic processes in abdominal aortic and popliteal artery aneurysms. J. Vasc. Surg. 2010, 51, 1479–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizas, K.D.; Ippagunta, N.; Tilson, M.D., 3rd. Immune cells and molecular mediators in the pathogenesis of the abdominal aortic aneurysm. Cardiol. Rev. 2009, 17, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef] [Green Version]
- Toghill, B.J.; Saratzis, A.; Freeman, P.J.; Sylvius, N.; Collaborators, U.; Bown, M.J. SMYD2 promoter DNA methylation is associated with abdominal aortic aneurysm (AAA) and SMYD2 expression in vascular smooth muscle cells. Clin. Epigenetics 2018, 10, 29. [Google Scholar] [CrossRef]
- Xu, G.; Liu, G.; Xiong, S.; Liu, H.; Chen, X.; Zheng, B. The histone methyltransferase Smyd2 is a negative regulator of macrophage activation by suppressing interleukin 6 (IL-6) and tumor necrosis factor alpha (TNF-alpha) production. J. Biol. Chem. 2015, 290, 5414–5423. [Google Scholar] [CrossRef] [Green Version]
- Maegdefessel, L.; Spin, J.M.; Raaz, U.; Eken, S.M.; Toh, R.; Azuma, J.; Adam, M.; Nakagami, F.; Heymann, H.M.; Chernogubova, E.; et al. miR-24 limits aortic vascular inflammation and murine abdominal aneurysm development. Nat. Commun. 2014, 5, 5214. [Google Scholar] [CrossRef] [Green Version]
- Villa-Bellosta, R.; Millan, A.; Sorribas, V. Role of calcium-phosphate deposition in vascular smooth muscle cell calcification. Am. J. Physiol. Cell. Physiol. 2011, 300, C210–C220. [Google Scholar] [CrossRef] [Green Version]
- Duer, M.J.; Friscic, T.; Proudfoot, D.; Reid, D.G.; Schoppet, M.; Shanahan, C.M.; Skepper, J.N.; Wise, E.R. Mineral surface in calcified plaque is like that of bone: Further evidence for regulated mineralization. Arter. Thromb. Vasc. Biol. 2008, 28, 2030–2034. [Google Scholar] [CrossRef] [PubMed]
- Strauss, H.W.; Nakahara, T.; Narula, N.; Narula, J. Vascular Calcification: The Evolving Relationship of Vascular Calcification to Major Acute Coronary Events. J. Nucl. Med. 2019, 60, 1207–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buijs, R.V.; Willems, T.P.; Tio, R.A.; Boersma, H.H.; Tielliu, I.F.; Slart, R.H.; Zeebregts, C.J. Calcification as a risk factor for rupture of abdominal aortic aneurysm. Eur. J. Vasc. Endovasc. Surg. 2013, 46, 542–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petsophonsakul, P.; Furmanik, M.; Forsythe, R.; Dweck, M.; Schurink, G.W.; Natour, E.; Reutelingsperger, C.; Jacobs, M.; Mees, B.; Schurgers, L. Role of Vascular Smooth Muscle Cell Phenotypic Switching and Calcification in Aortic Aneurysm Formation. Arter. Thromb. Vasc. Biol. 2019, 39, 1351–1368. [Google Scholar] [CrossRef]
- Forsythe, R.O.; Newby, D.E.; Robson, J.M. Monitoring the biological activity of abdominal aortic aneurysms Beyond Ultrasound. Heart 2016, 102, 817–824. [Google Scholar] [CrossRef] [Green Version]
- Lindholt, J.S. Aneurysmal wall calcification predicts natural history of small abdominal aortic aneurysms. Atherosclerosis 2008, 197, 673–678. [Google Scholar] [CrossRef]
- Forsythe, R.O.; Dweck, M.R.; McBride, O.M.B.; Vesey, A.T.; Semple, S.I.; Shah, A.S.V.; Adamson, P.D.; Wallace, W.A.; Kaczynski, J.; Ho, W.; et al. (18)F-Sodium Fluoride Uptake in Abdominal Aortic Aneurysms: The SoFIA(3) Study. J. Am. Coll. Cardiol. 2018, 71, 513–523. [Google Scholar] [CrossRef]
- Reeps, C.; Essler, M.; Pelisek, J.; Seidl, S.; Eckstein, H.H.; Krause, B.J. Increased 18F-fluorodeoxyglucose uptake in abdominal aortic aneurysms in positron emission/computed tomography is associated with inflammation, aortic wall instability, and acute symptoms. J. Vasc. Surg. 2008, 48, 417–423; discussion 424. [Google Scholar] [CrossRef] [Green Version]
- Iyemere, V.P.; Proudfoot, D.; Weissberg, P.L.; Shanahan, C.M. Vascular smooth muscle cell phenotypic plasticity and the regulation of vascular calcification. J. Intern. Med. 2006, 260, 192–210. [Google Scholar] [CrossRef]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef] [Green Version]
- Shioi, A.; Nishizawa, Y.; Jono, S.; Koyama, H.; Hosoi, M.; Morii, H. Beta-glycerophosphate accelerates calcification in cultured bovine vascular smooth muscle cells. Arter. Thromb. Vasc. Biol. 1995, 15, 2003–2009. [Google Scholar] [CrossRef] [PubMed]
- Basalyga, D.M.; Simionescu, D.T.; Xiong, W.; Baxter, B.T.; Starcher, B.C.; Vyavahare, N.R. Elastin degradation and calcification in an abdominal aorta injury model: Role of matrix metalloproteinases. Circulation 2004, 110, 3480–3487. [Google Scholar] [CrossRef] [PubMed]
- Patelis, N.; Moris, D.; Schizas, D.; Damaskos, C.; Perrea, D.; Bakoyiannis, C.; Liakakos, T.; Georgopoulos, S. Animal models in the research of abdominal aortic aneurysms development. Physiol. Res. 2017, 66, 899–915. [Google Scholar] [CrossRef] [PubMed]
- Busch, A.; Holm, A.; Wagner, N.; Ergun, S.; Rosenfeld, M.; Otto, C.; Baur, J.; Kellersmann, R.; Lorenz, U. Extra- and Intraluminal Elastase Induce Morphologically Distinct Abdominal Aortic Aneurysms in Mice and Thus Represent Specific Subtypes of Human Disease. J. Vasc. Res. 2016, 53, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Bhamidipati, C.M.; Mehta, G.S.; Lu, G.; Moehle, C.W.; Barbery, C.; DiMusto, P.D.; Laser, A.; Kron, I.L.; Upchurch, G.R., Jr.; Ailawadi, G. Development of a novel murine model of aortic aneurysms using peri-adventitial elastase. Surgery 2012, 152, 238–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senemaud, J.; Caligiuri, G.; Etienne, H.; Delbosc, S.; Michel, J.B.; Coscas, R. Translational Relevance and Recent Advances of Animal Models of Abdominal Aortic Aneurysm. Arter. Thromb. Vasc. Biol. 2017, 37, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Chiou, A.C.; Chiu, B.; Pearce, W.H. Murine aortic aneurysm produced by periarterial application of calcium chloride. J. Surg. Res. 2001, 99, 371–376. [Google Scholar] [CrossRef]
- Lu, G.; Su, G.; Davis, J.P.; Schaheen, B.; Downs, E.; Roy, R.J.; Ailawadi, G.; Upchurch, G.R., Jr. A novel chronic advanced stage abdominal aortic aneurysm murine model. J. Vasc. Surg. 2017, 66, 232–242.e234. [Google Scholar] [CrossRef] [Green Version]
- Fashandi, A.Z.; Hawkins, R.B.; Salmon, M.D.; Spinosa, M.D.; Montgomery, W.G.; Cullen, J.M.; Lu, G.; Su, G.; Ailawadi, G.; Upchurch, G.R., Jr. A novel reproducible model of aortic aneurysm rupture. Surgery 2018, 163, 397–403. [Google Scholar] [CrossRef]
- Gopal, K.; Kumar, K.; Nandini, R.; Jahan, P.; Kumar, M.J. High fat diet containing cholesterol induce aortic aneurysm through recruitment and proliferation of circulating agranulocytes in apoE knock out mice model. J. Thromb. Thrombolysis 2010, 30, 154–163. [Google Scholar] [CrossRef]
- Zhao, G.; Lu, H.; Chang, Z.; Zhao, Y.; Zhu, T.; Chang, L.; Guo, Y.; Garcia-Barrio, M.T.; Chen, Y.E.; Zhang, J. Single-cell RNA sequencing reveals the cellular heterogeneity of aneurysmal infrarenal abdominal aorta. Cardiovasc. Res. 2021, 117, 1402–1416. [Google Scholar] [CrossRef] [PubMed]
- Hadi, T.; Boytard, L.; Silvestro, M.; Alebrahim, D.; Jacob, S.; Feinstein, J.; Barone, K.; Spiro, W.; Hutchison, S.; Simon, R.; et al. Macrophage-derived netrin-1 promotes abdominal aortic aneurysm formation by activating MMP3 in vascular smooth muscle cells. Nat. Commun. 2018, 9, 5022. [Google Scholar] [CrossRef] [PubMed]
- Dawson, A.; Li, Y.; Li, Y.; Ren, P.; Vasquez, H.G.; Zhang, C.; Rebello, K.R.; Ageedi, W.; Azares, A.R.; Mattar, A.B.; et al. Single-Cell Analysis of Aneurysmal Aortic Tissue in Patients with Marfan Syndrome Reveals Dysfunctional TGF-β Signaling. Genes 2021, 13, 95. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Abdominal aortic aneurysm formation and its risk factors. Abdominal aortic aneurysm (AAA) occurs in the infra-renal segment with a diameter exceeding 3.0 cm. The risk factors, including male gender, aging, smoking, and hypercholesterolemia, etc., have been found to be related to AAA initiation and progression.
Figure 1.
Abdominal aortic aneurysm formation and its risk factors. Abdominal aortic aneurysm (AAA) occurs in the infra-renal segment with a diameter exceeding 3.0 cm. The risk factors, including male gender, aging, smoking, and hypercholesterolemia, etc., have been found to be related to AAA initiation and progression.

Figure 2.
The pathophysiology of abdominal aortic aneurysm. The pathophysiology of abdominal aortic aneurysm (AAA) is a complicated process, involving the endothelial cell (EC) dysfunction with increased expression of adhesion molecules and chemokines, vascular smooth muscle cell (VSMC) phenotypic changes and dysfunction, inflammatory cell infiltration into the aortic wall, oxidative stress, and extracellular matrix (ECM) remodeling. Various mediators are involved in this process, including vascular cell adhesion molecule 1 (Vcam-1), monocyte chemoattractant protein 1 (MCP-1), interleukin-6 (IL-6), interleukin-1β (IL-1β), matrix metalloproteinases (MMPs), and tumor necrosis factor-α (TNF-α).
Figure 2.
The pathophysiology of abdominal aortic aneurysm. The pathophysiology of abdominal aortic aneurysm (AAA) is a complicated process, involving the endothelial cell (EC) dysfunction with increased expression of adhesion molecules and chemokines, vascular smooth muscle cell (VSMC) phenotypic changes and dysfunction, inflammatory cell infiltration into the aortic wall, oxidative stress, and extracellular matrix (ECM) remodeling. Various mediators are involved in this process, including vascular cell adhesion molecule 1 (Vcam-1), monocyte chemoattractant protein 1 (MCP-1), interleukin-6 (IL-6), interleukin-1β (IL-1β), matrix metalloproteinases (MMPs), and tumor necrosis factor-α (TNF-α).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Qian, G.; Adeyanju, O.; Olajuyin, A.; Guo, X. Abdominal Aortic Aneurysm Formation with a Focus on Vascular Smooth Muscle Cells. Life 2022, 12, 191. https://doi.org/10.3390/life12020191
AMA Style
Qian G, Adeyanju O, Olajuyin A, Guo X. Abdominal Aortic Aneurysm Formation with a Focus on Vascular Smooth Muscle Cells. Life. 2022; 12(2):191. https://doi.org/10.3390/life12020191
Chicago/Turabian StyleQian, Guoqing, Oluwaseun Adeyanju, Ayobami Olajuyin, and Xia Guo. 2022. "Abdominal Aortic Aneurysm Formation with a Focus on Vascular Smooth Muscle Cells" Life 12, no. 2: 191. https://doi.org/10.3390/life12020191
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.