
Tumor Stimulus-Responsive Biodegradable Diblock Copolymer Conjugates as Efficient Anti-Cancer Nanomedicines

 ,
,  ,
, 
Abstract
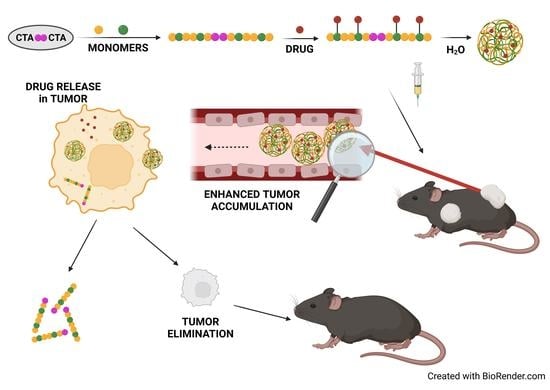
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Monomers, Oligopeptide and Chain Transfer Agents
2.2.1. Synthesis of PVGLIGK Peptide
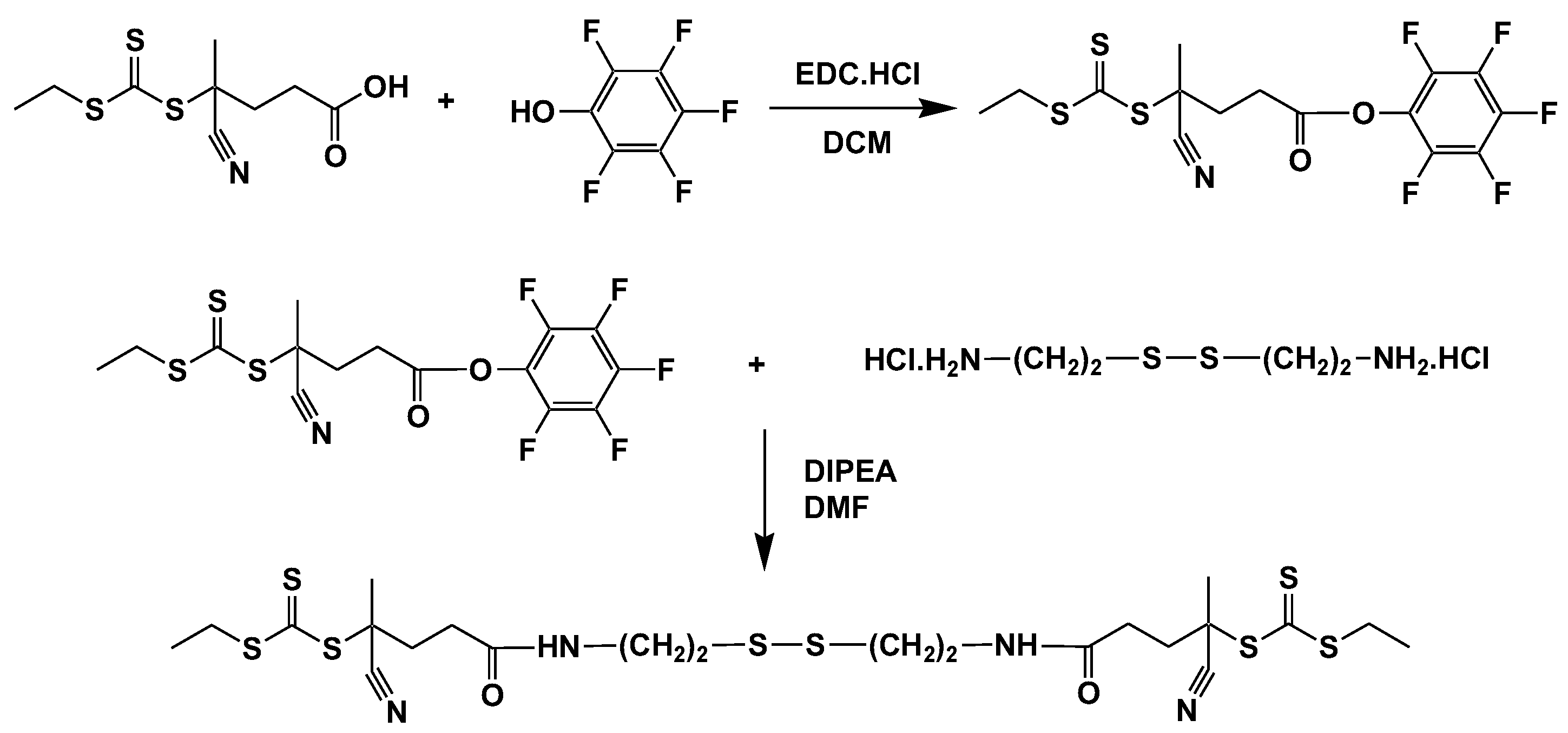
2.2.2. Synthesis of Pentafluoro Phenyl Functional Chain Transfer Agent (2,3,4,5,6-Pentafluorophenyl) 4-Cyano-4-Ethylsulfanylcarbothioylsulfanyl-Pentanoate (CTA-PFP)
2.2.3. Synthesis of 4-Cyano-N-[2-[2-[(4-Cyano-4-Ethylsulfanylcarbothioylsulfanyl-Pentanoyl)Amino]ethyldisulfanyl]ethyl]-4-Ethylsulfanylcarbothioylsulfanyl-Pentanamide (CTA-S-S-CTA)
2.3. Synthesis of the Polymer Precursors and Polymer Conjugates
2.4. Characterization of the Polymer Precursors and Conjugates
2.5. In Vitro Drug Release from the Polymer Conjugates
2.6. Degradation of Diblock Copolymers
2.7. In Vitro Cytotoxicity Assay
2.8. Intracellular Uptake of Conjugates in C26 Cells
2.9. Spheroid Assay for the Penetration and Uptake of Different P-THPs
2.10. In Vivo Pharmacokinetics of P-THPs
2.11. In Vivo Antitumor Effect
3. Results
3.1. Synthesis of the Polymer Precursors
3.2. Synthesis of the Polymer–Drug Conjugates
3.3. Release of Drugs from Polymer Conjugates
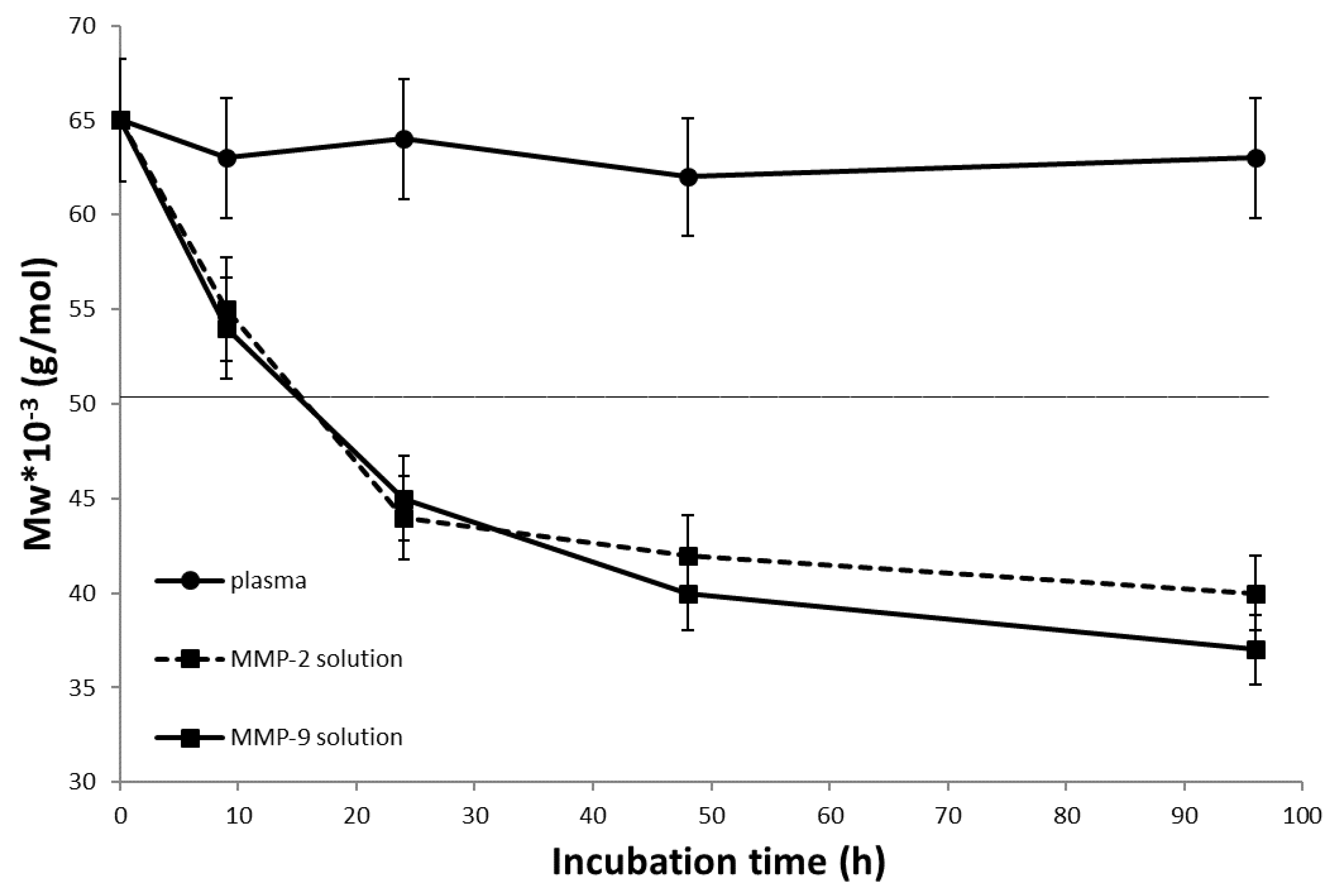
3.4. In Vitro Degradation of Diblock Conjugates
3.5. In Vitro Cytotoxicity of Diblock Conjugates
3.6. Intracellular Uptake of Diblock Conjugates
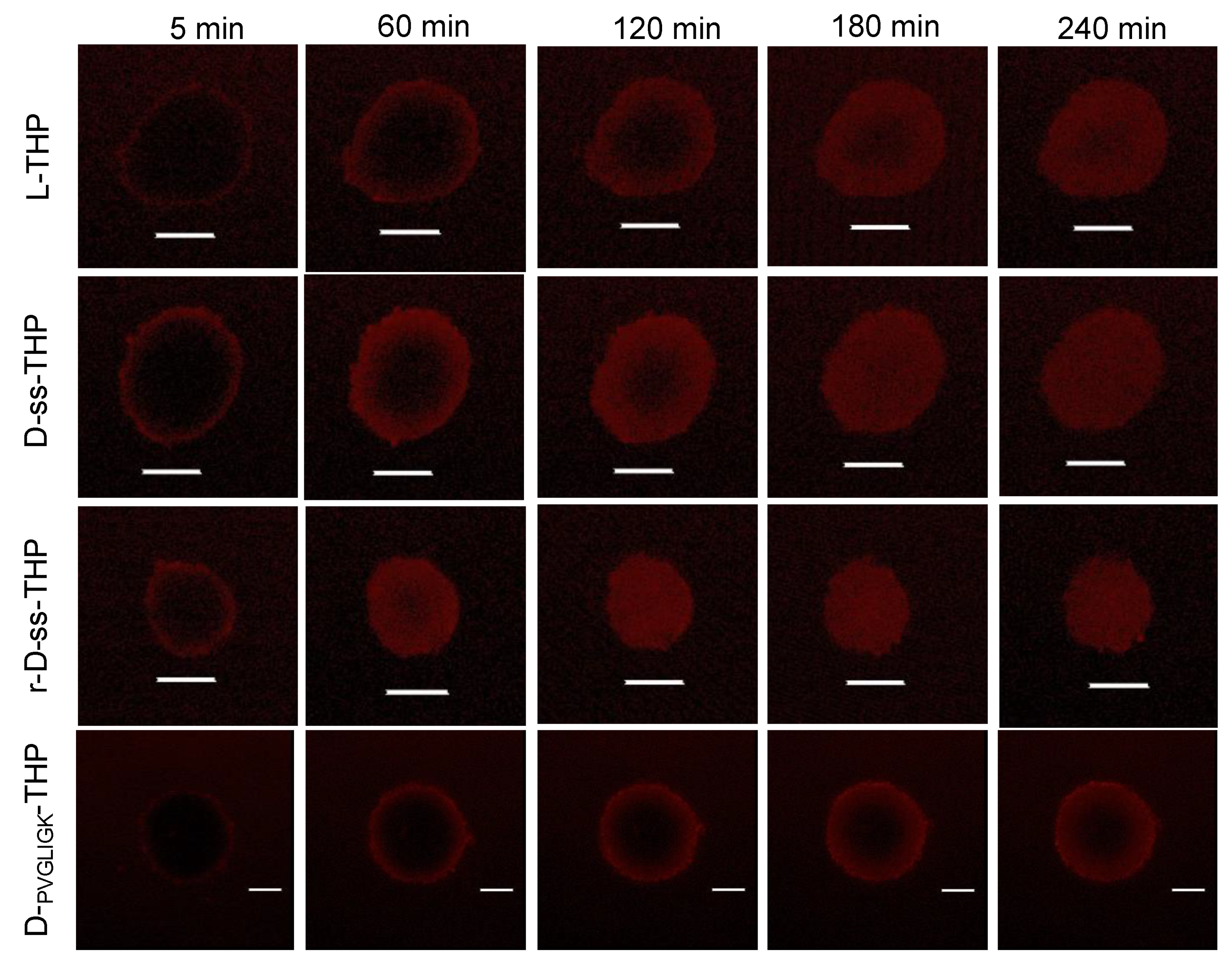
3.7. Penetration and Uptake of P-THP Conjugates in C26 Tumor Cell Spheroid
3.8. In Vivo Pharmacokinetics of P-THP Conjugates
3.9. In Vivo Antitumor Effects of P-THP Conjugates
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lammers, T.; Kiessling, F.; Hennink, W.E.; Storm, G. Nanotheranostics and Image-Guided Drug Delivery: Current Concepts and Future Directions. Mol. Pharm. 2010, 7, 1899–1912. [Google Scholar] [CrossRef] [PubMed]
- Tong, R.; Langer, R. Nanomedicines Targeting the Tumor Microenvironment. Cancer 2015, 21, 314–321. [Google Scholar] [CrossRef]
- Canal, F.; Sanchis, J.; Vicent, M.J. Polymer-drug conjugates as nano-sized medicines. Curr. Opin. Biotechnol. 2011, 22, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Dordevic, S.; Gonzalez, M.M.; Conejos-Sanchez, I.; Carreira, B.; Pozzi, S.; Acurcio, R.C.; Satchi-Fainaro, R.; Florindo, H.F.; Vicent, M.J. Current hurdles to the translation of nanomedicines from bench to the clinic. Drug Deliv. Transl. Res. 2022, 12, 500–525. [Google Scholar] [CrossRef] [PubMed]
- Kopeček, J.; Yang, J.Y. Polymer nanomedicines. Adv. Drug Deliv. Rev. 2020, 156, 40–64. [Google Scholar] [CrossRef]
- Sawant, R.R.; Torchilin, V.P. Liposomes as ‘smart’ pharmaceutical nanocarriers. Soft Matter 2010, 6, 4026–4044. [Google Scholar] [CrossRef]
- Ulbrich, K.; Holá, K.; Šubr, V.; Bakandritsos, A.; Tuček, J.; Zbořil, R. Targeted Drug Delivery with Polymers and Magnetic Nanoparticles: Covalent and Noncovalent Approaches, Release Control, and Clinical Studies. Chem. Rev. 2016, 116, 5338–5431. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P. Micellar nanocarriers: Pharmaceutical perspectives. Pharm. Res. 2007, 24, 1–16. [Google Scholar] [CrossRef]
- Oerlemans, C.; Bult, W.; Bos, M.; Storm, G.; Nijsen, J.F.W.; Hennink, W.E. Polymeric Micelles in Anticancer Therapy: Targeting, Imaging and Triggered Release. Pharm. Res. 2010, 27, 2569–2589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, J.; Teo, J.Y.; Aakalu, V.K.; Yang, Y.Y.; Kong, H. Engineering Polymersomes for Diagnostics and Therapy. Adv. Healthc. Mater. 2018, 7, 1701276. [Google Scholar] [CrossRef]
- Meng, F.H.; Zhong, Z.Y.; Feijen, J. Stimuli-Responsive Polymersomes for Programmed Drug Delivery. Biomacromolecules 2009, 10, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Kao, W.Y.J. Dendrimers for pharmaceutical and biomedical applications. J. Biomater. Sci. Polym. Ed. 2006, 17, 3–19. [Google Scholar] [CrossRef]
- Gulla, S.; Lomada, D.; Srikanth, V.; Shankar, M.V.; Reddy, K.R.; Soni, S.; Reddy, M.C. Recent advances in nanoparticles-based strategies for cancer therapeutics and antibacterial applications. Methods Microbiol. 2019, 46, 255–293. [Google Scholar]
- Arslan, F.B.; Atar, K.O.; Calis, S. Antibody-mediated drug delivery. Int. J. Pharm. 2021, 596, 120268. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, U.; Maeda, H.; Jain, R.K.; Sevick-Muraca, E.M.; Zamboni, W.; Farokhzad, O.C.; Barry, S.T.; Gabizon, A.; Grodzinski, P.; Blakey, D.C. Challenges and Key Considerations of the Enhanced Permeability and Retention Effect for Nanomedicine Drug Delivery in Oncology. Cancer Res. 2013, 73, 2412–2417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malugin, A.; Kopečková, P.; Kopeček, J. Liberation of doxorubicin from HPMA copolymer conjugate is essential for the induction of cell cycle arrest and nuclear fragmentation in ovarian carcinoma cells. J. Control. Release 2007, 124, 6–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatakeyama, H.; Akita, H.; Harashima, H. A multifunctional envelope type nano device (MEND) for gene delivery to tumours based on the EPR effect: A strategy for overcoming the PEG dilemma. Adv. Drug Deliv. Rev. 2011, 63, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Chytil, P.; Kostka, L.; Etrych, T. Structural design and synthesis of polymer prodrugs. In Polymers for Biomedicine: Synthesis, Characterization, and Applications; Scholz, C., Ed.; Wiley: Hoboken, NJ, USA, 2017; pp. 391–420. [Google Scholar]
- Pan, H.; Sima, M.; Yang, J.; Kopeček, J. Synthesis of long-circulating, backbone degradable HPMA copolymer-doxorubicin conjugates and evaluation of molecular-weight-dependent antitumor efficacy. Macromol. Biosci. 2013, 13, 155–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, H.T.; Hoang, T.X.; Kim, J.Y. All-Trans Retinoic Acid Enhances Matrix Metalloproteinase 2 Expression and Secretion in Human Myeloid Leukemia THP-1 Cells. Biomed Res. Int. 2018, 2018, 5971080. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Adhikari, N.; Banerjee, S.; Amin, S.A.; Jha, T. Matrix metalloproteinase-9 (MMP-9) and its inhibitors in cancer: A minireview. Eur. J. Med. Chem. 2020, 194, 112260. [Google Scholar] [CrossRef] [PubMed]
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011, 278, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Li, M.J.; Luo, T.; Yin, Y.C.; Jiang, Y.F. Matrix metalloproteinases in tumorigenesis: An evolving paradigm. Cell. Mol. Life Sci. 2011, 68, 3853–3868. [Google Scholar] [CrossRef] [PubMed]
- Scannevin, R.H.; Alexander, R.; Haarlander, T.M.; Burke, S.L.; Singer, M.; Huo, C.F.; Zhang, Y.M.; Maguire, D.; Spurlino, J.; Deckman, I.; et al. Discovery of a highly selective chemical inhibitor of matrix metalloproteinase-9 (MMP-9) that allosterically inhibits zymogen activation. J. Biol. Chem. 2017, 292, 17963–17974. [Google Scholar] [CrossRef] [Green Version]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [Green Version]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Cancer therapy—Matrix metalloproteinase inhibitors and cancer: Trials and tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef] [PubMed]
- Cox, G.; O’Byrne, K.J. Matrix metalloproteinases and cancer. Anticancer Res. 2001, 21, 4207–4219. [Google Scholar]
- Etrych, T.; Šubr, V.; Laga, R.; Říhová, B.; Ulbrich, K. Polymer conjugates of doxorubicin bound through an amide and hydrazone bond: Impact of the carrier structure onto synergistic action in the treatment of solid tumours. Eur. J. Pharm. Sci. 2014, 58, 1–12. [Google Scholar] [CrossRef]
- Klepac, D.; Kostková, H.; Petrova, S.; Chytil, P.; Etrych, T.; Kereiche, S.; Raška, I.; Weitz, D.A.; Filippov, S.K. Interaction of spin-labeled HPMA-based nanoparticles with human blood plasma proteins—The introduction of protein-corona-free polymer nanomedicine. Nanoscale 2018, 10, 6194–6204. [Google Scholar] [CrossRef]
- Ulbrich, K.; Šubr, V. Structural and chemical aspects of HPMA copolymers as drug carriers. Adv. Drug. Deliv. Rev. 2010, 62, 150–166. [Google Scholar] [CrossRef]
- Kopečková, P.; Rathi, R.; Takada, S.; Říhová, B.; Berenson, M.M.; Kopeček, J. Bioadhesive N-(2-hydroxypropyl) methacrylamide copolymers for colon-specific drug delivery. J. Control. Release 1994, 28, 211–222. [Google Scholar] [CrossRef]
- Říhová, B. Clinical experience with anthracycline antibiotics-HPMA copolymer-human immunoglobulin conjugates. Adv. Drug. Deliv. Rev. 2009, 61, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Seymour, L.W.; Ferry, D.R.; Kerr, D.J.; Rea, D.; Whitlock, M.; Poyner, R.; Boivin, C.; Hesslewood, S.; Twelves, C.; Blackie, R.; et al. Phase II studies of polymer-doxorubicin (PK1, FCE28068) in the treatment of breast, lung and colorectal cancer. Int. J. Oncol. 2009, 34, 1629–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dozono, H.; Yanazune, S.; Nakamura, H.; Etrych, T.; Chytil, P.; Ulbrich, K.; Fang, J.; Arimura, T.; Douchi, T.; Kobayashi, H.; et al. HPMA Copolymer-Conjugated Pirarubicin in Multimodal Treatment of a Patient with Stage IV Prostate Cancer and Extensive Lung and Bone Metastases. Target. Oncol. 2016, 11, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Etrych, T.; Chytil, P.; Ohkubo, M.; Fang, J.; Ulbrich, K.; Maeda, H. Two step mechanisms of tumor selective delivery of N-(2-hydroxypropyl)methacrylamide copolymer conjugated with pirarubicin via an acid-cleavable linkage. J. Control. Release 2014, 174, 81–87. [Google Scholar] [CrossRef]
- Etrych, T.; Jelínková, M.; Říhová, B.; Ulbrich, K. New HPMA copolymers containing doxorubicin bound via pH-sensitive linkage: Synthesis and preliminary in vitro and in vivo biological properties. J. Control. Release 2001, 73, 89–102. [Google Scholar] [CrossRef]
- Etrych, T.; Chytil, P.; Mrkvan, T.; Šírová, M.; Říhová, B.; Ulbrich, K. Conjugates of doxorubicin with graft HPMA copolymers for passive tumor targeting. J. Control. Release 2008, 132, 184–192. [Google Scholar] [CrossRef]
- Etrych, T.; Strohalm, J.; Chytil, P.; Říhová, B.; Ulbrich, K. Novel star HPMA-based polymer conjugates for passive targeting to solid tumors. J. Drug Target. 2011, 19, 874–889. [Google Scholar] [CrossRef]
- Etrych, T.; Mrkvan, T.; Chytil, P.; Koňák, Č.; Říhová, B.; Ulbrich, K. N-(2-hydroxypropyl)methacrylamide-based polymer conjugates with pH-controlled activation of doxorubicin. I. New synthesis, physicochemical characterization and preliminary biological evaluation. J. Appl. Polym. Sci. 2008, 109, 3050–3061. [Google Scholar] [CrossRef]
- Šubr, V.; Sivák, L.; Koziolová, E.; Braunová, A.; Pechar, M.; Strohalm, J.; Kabešová, M.; Říhová, B.; Ulbrich, K.; Kovář, M. Synthesis of poly[N-(2-hydroxypropyl)methacrylamide] conjugates of inhibitors of the ABC transporter that overcome multidrug resistance in doxorubicin-resistant P388 cells in vitro. Biomacromolecules 2014, 15, 3030–3043. [Google Scholar] [CrossRef]
- Chytil, P.; Etrych, T.; Kříž, J.; Šubr, V.; Ulbrich, K. N-(2-Hydroxypropyl)methacrylamide-based polymer conjugates with pH-controlled activation of doxorubicin for cell-specific or passive tumour-targeting. Synthesis by RAFT polymerization and physicochemical characterisation. Eur. J. Pharm. Sci. 2010, 41, 473–482. [Google Scholar] [CrossRef]
- Convertine, A.J.; Ayres, N.; Scales, C.W.; Lowe, A.B.; McCormick, C.L. Facile, controlled, room-temperature RAFT polymerization of N-isopropylacrylamide. Biomacromolecules 2004, 5, 1177–1180. [Google Scholar] [CrossRef] [PubMed]
- Etrych, T.; Tsukigawa, K.; Nakamura, H.; Chytil, P.; Fang, J.; Ulbrich, K.; Otagiri, M.; Maeda, H. Comparison of pharmacological and biological properties of HPMA copolymer pirarubicin conjugates: A single chain copolymer and its biodegradable tandem-diblock copolymer conjugates. Eur. J. Pharm. Sci. 2017, 106, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Koziolová, E.; Chytil, P.; Tsukigawa, K.; Fang, J.; Haratake, M.; Ulbrich, K.; Etrych, T.; Maeda, H. Pronounced cellular uptake of pirarubicin versus that of other anthracyclines: Comparison of HPMA copolymer conjugates of pirarubicin and doxorubicin. Mol. Pharm. 2016, 13, 4106–4115. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Mw (g.mol−1) | Đ | Side Chain Groups | -NHNH2 Content (mol%) | THP (wt%) | Rh (nm) a |
|---|---|---|---|---|---|---|
| L-TT | 30,100 | 1.76 | -NHNH-Boc | 5.3 | - | 4.0 |
| D-PVGLIGK-Hy | 56,800 | 1.80 | -NHNH2 | 5.3 | - | 6.9 |
| D-ss-Hy | 55,000 | 1.78 | -NHNH2 | 5.2 | - | 6.5 |
| r-D-ss-Hy | 77,000 | 1.05 | -NHNH2 | 6.5 | - | n.d. |
| L-THP | 37,400 | 1.85 | -NHNH=THP | - | 9.9 | 3.9 |
| D-ss-THP | 77,000 | 1.88 | -NHNH=THP | - | 9.3 | 7.8 |
| D-PVGLIGK-THP | 65,000 | 1.72 | -NHNH=THP | - | 10.3 | 8.2 |
| r-D-ss-THP | 135,000 | 1.49 | -NHNH=THP | - | 9.0 | 8.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šubr, V.; Pola, R.; Gao, S.; Islam, R.; Hirata, T.; Miyake, D.; Koshino, K.; Zhou, J.-R.; Yokomizo, K.; Fang, J.; et al. Tumor Stimulus-Responsive Biodegradable Diblock Copolymer Conjugates as Efficient Anti-Cancer Nanomedicines. J. Pers. Med. 2022, 12, 698. https://doi.org/10.3390/jpm12050698
Šubr V, Pola R, Gao S, Islam R, Hirata T, Miyake D, Koshino K, Zhou J-R, Yokomizo K, Fang J, et al. Tumor Stimulus-Responsive Biodegradable Diblock Copolymer Conjugates as Efficient Anti-Cancer Nanomedicines. Journal of Personalized Medicine. 2022; 12(5):698. https://doi.org/10.3390/jpm12050698
Chicago/Turabian StyleŠubr, Vladimír, Robert Pola, Shanghui Gao, Rayhanul Islam, Takuma Hirata, Daiki Miyake, Kousuke Koshino, Jian-Rong Zhou, Kazumi Yokomizo, Jun Fang, and et al. 2022. "Tumor Stimulus-Responsive Biodegradable Diblock Copolymer Conjugates as Efficient Anti-Cancer Nanomedicines" Journal of Personalized Medicine 12, no. 5: 698. https://doi.org/10.3390/jpm12050698