
Human iPSC-Derived Retinal Organoids and Retinal Pigment Epithelium for Novel Intronic RPGR Variant Assessment for Therapy Suitability

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Sample Collection and Participation
2.2. Patient Ophthalmic Investigations
2.3. Exome Sequencing, Analysis and In Silico Splice Prediction
2.4. RPGR Gene Expression Studies
2.5. Cell Culture and Generation of iPSCs
2.6. iPSC Pluripotency and Trilineage Differentiation
2.7. Differentiation of iPSCs to Retinal Cells
2.8. Preparation of Samples for Immunofluorescence, Cilia and TUNEL Assays and Immunohistochemistry
2.9. Western Blot Analysis
2.10. Image Analysis, Total Fluorescenc, and Statistics
3. Results
3.1. Patient Ophthalmic Features of RCD
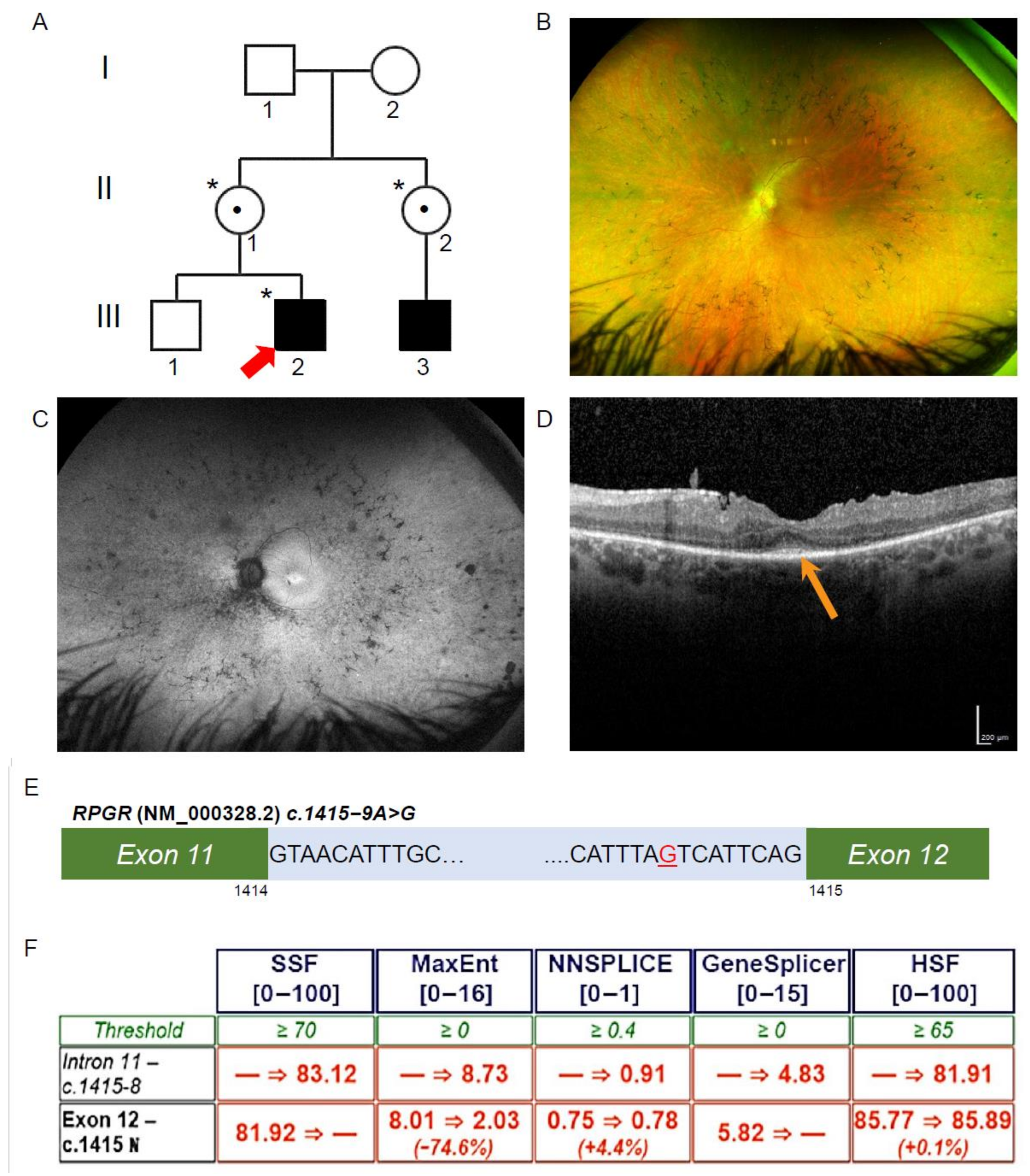
3.2. Exome Sequencing, Segregation and Bioinformatic Analysis Identified a Novel Intronic RPGR Variant
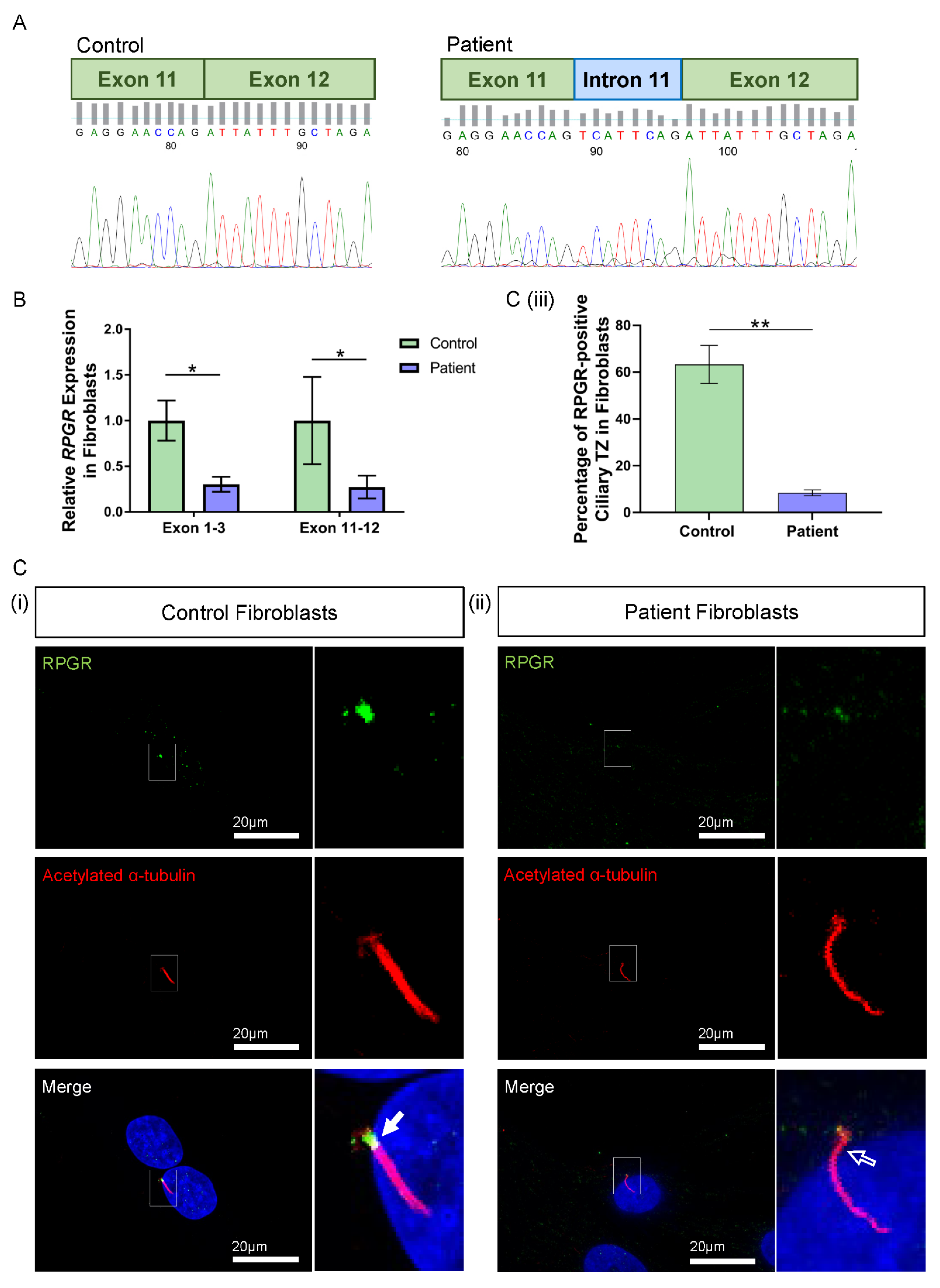
3.3. Novel RPGR Intronic Variant Creates an Alternate Splice Acceptor Site and Loss of RPGR Expression, Especially at the Transition Zone in Fibroblast Primary Cilia
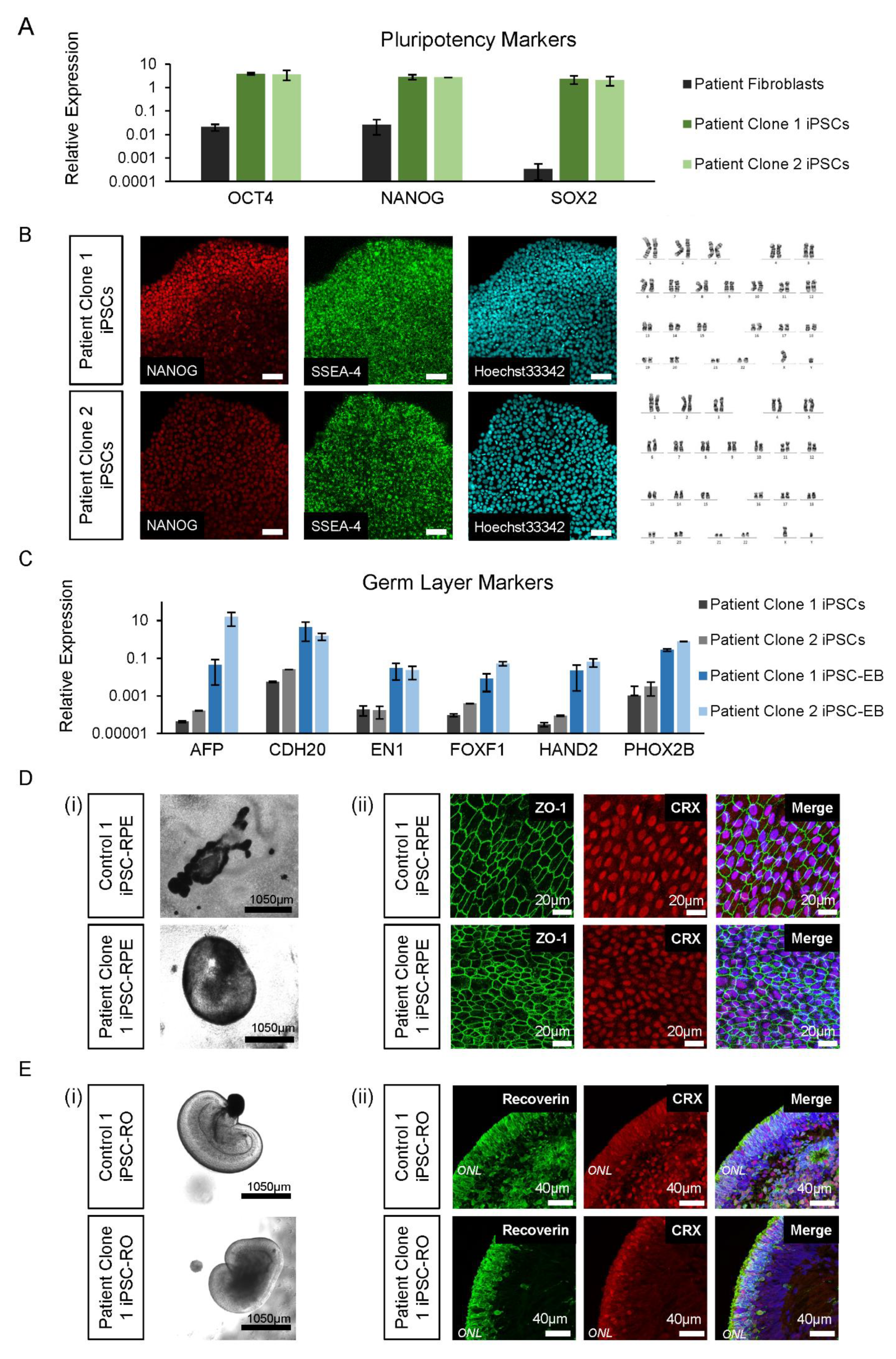
3.4. Control and Patient iPSC Lines Differentiate into iPSC-RPE and iPSC-ROs
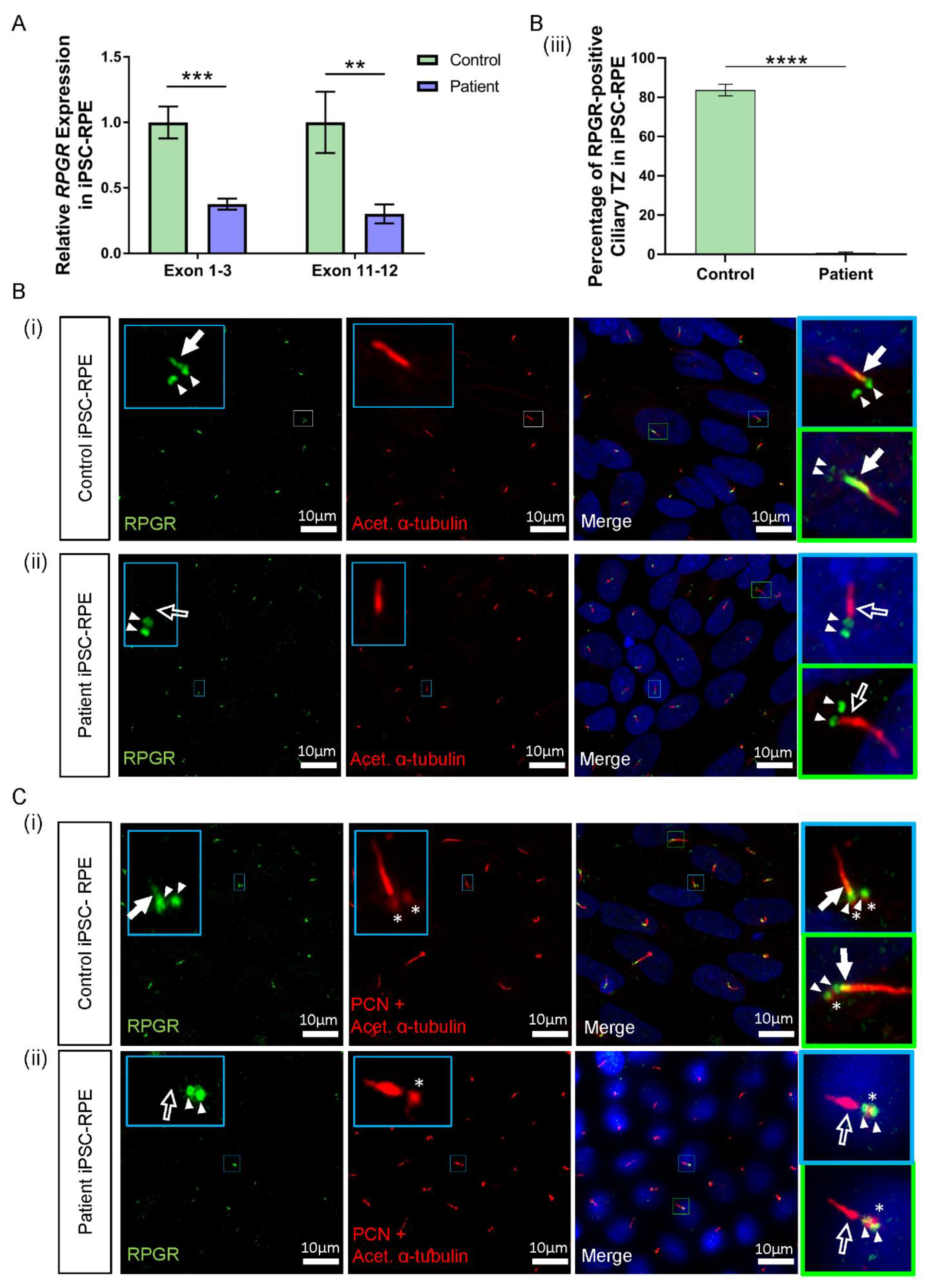
3.5. RPGR Variant iPSC-RPE Exhibits a Loss of RPGR Localisation at the Transitional Zone of Primary Cilia
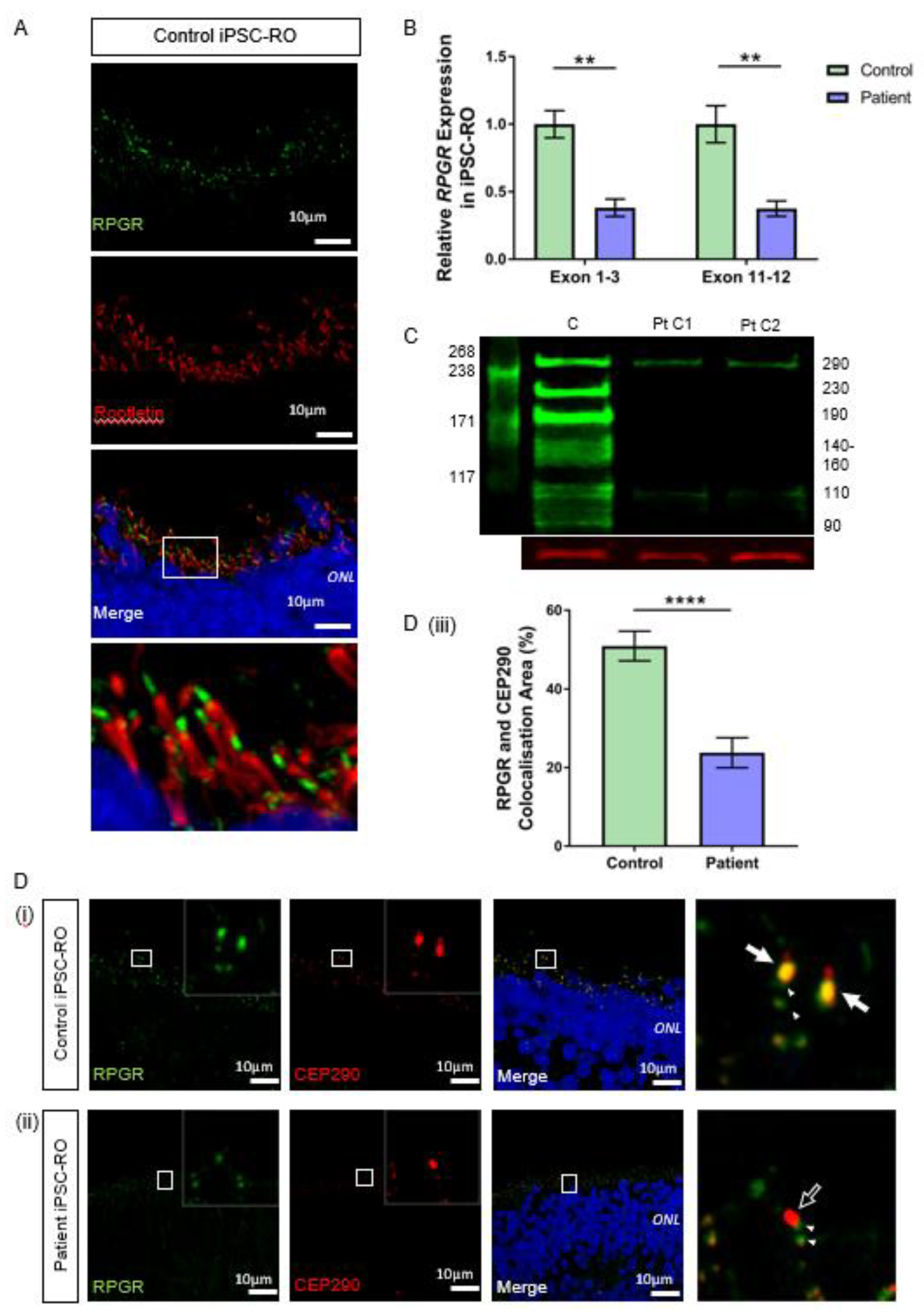
3.6. RPGR Variant iPSC-ROs Have Decreased RPGR Expression at the Ciliary TZ of the Photoreceptor Cells
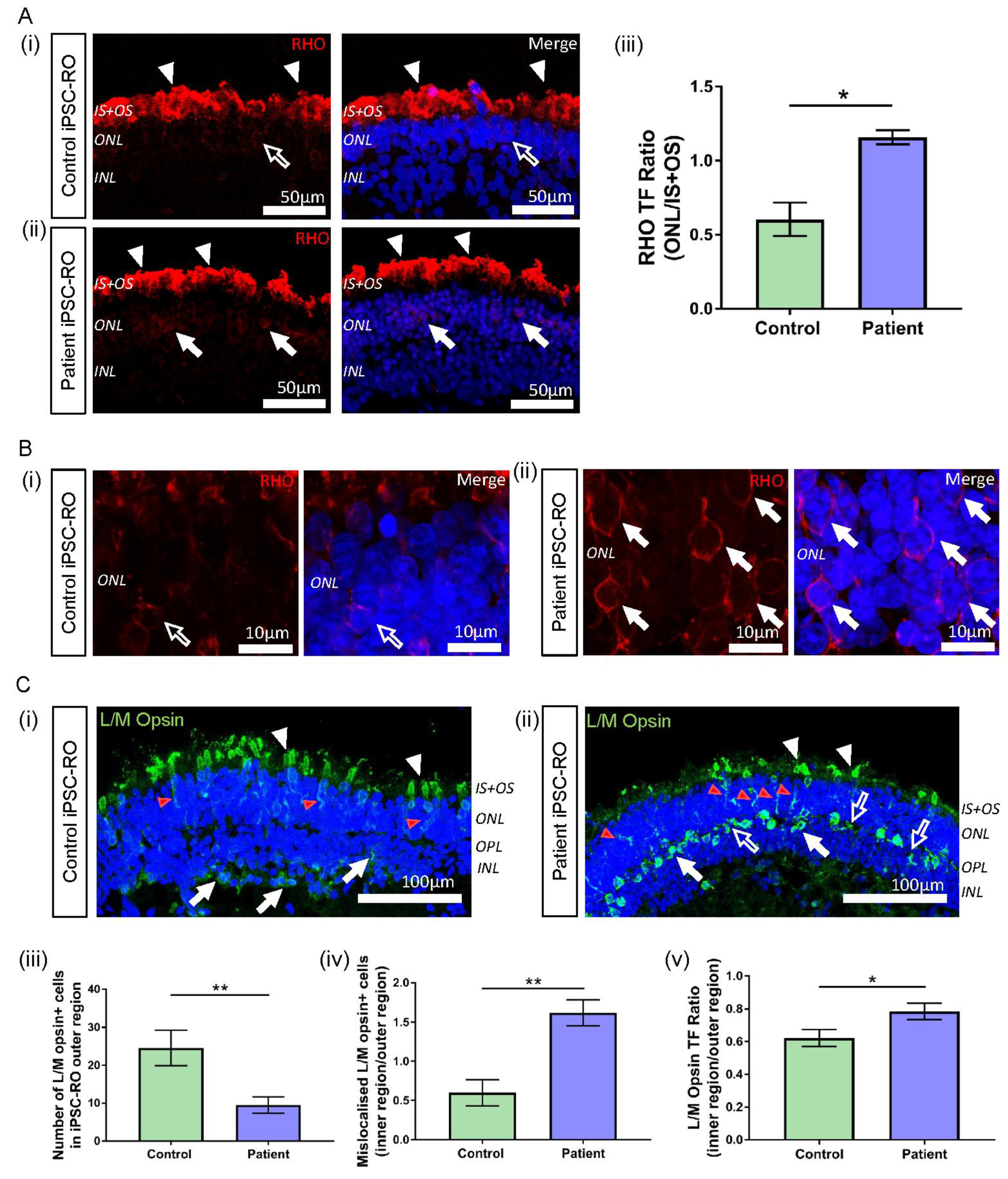
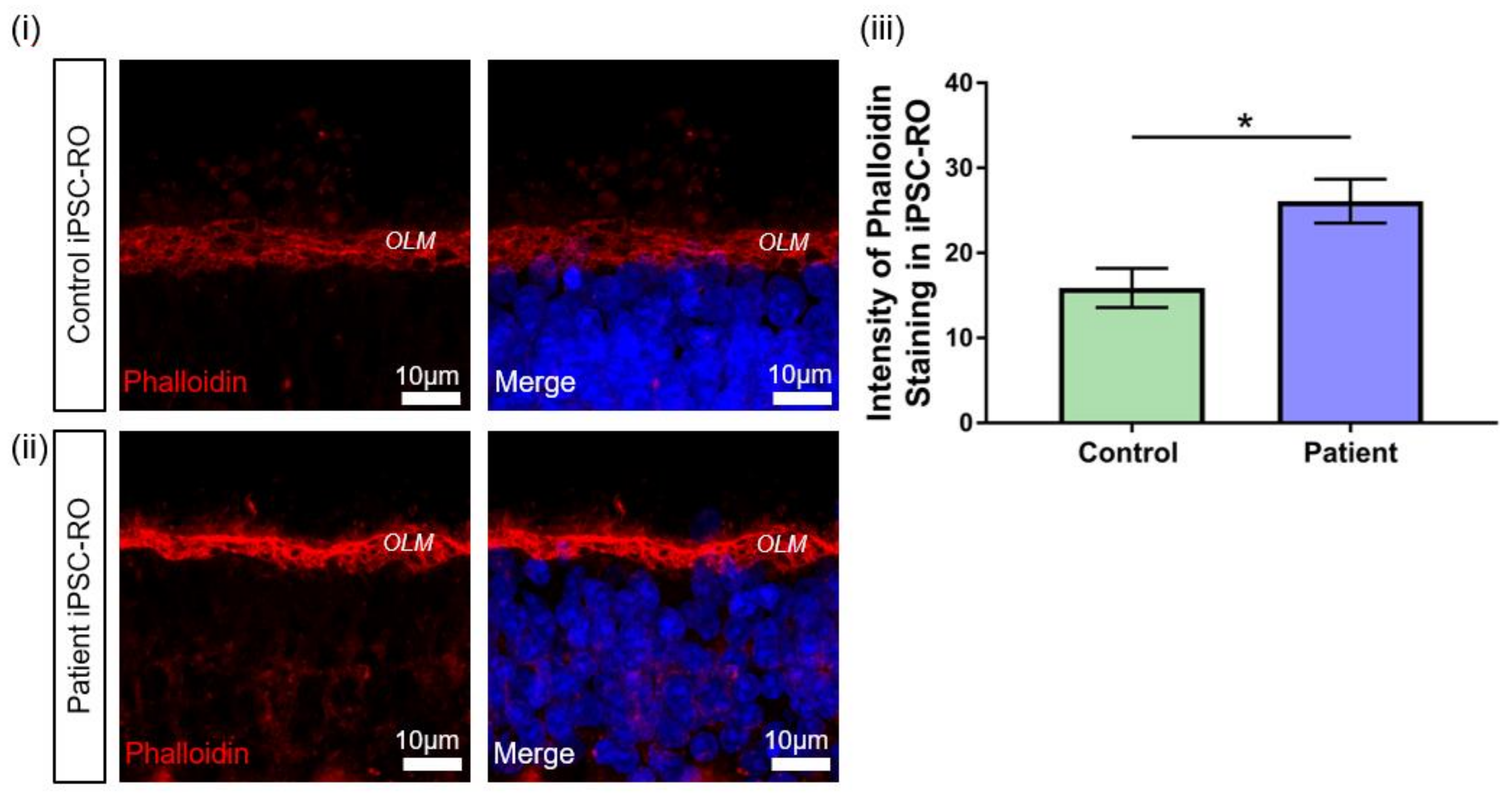
3.7. RPGR Variant iPSC-ROs Display Mislocalised Opsins, Increased Photoreceptor Apoptosis and Abnormal F-Actin Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanany, M.; Rivolta, C.; Sharon, D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl. Acad. Sci. USA 2020, 117, 2710–2716. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Gil, N.; González-Del Pozo, M.; Martín-Sánchez, M.; Méndez-Vidal, C.; Rodríguez-de la Rúa, E.; Borrego, S.; Antiñolo, G. Unravelling the genetic basis of simplex Retinitis Pigmentosa cases. Sci. Rep. 2017, 7, 41937. [Google Scholar] [CrossRef] [PubMed]
- Megaw, R.D.; Soares, D.C.; Wright, A.F. RPGR: Its role in photoreceptor physiology, human disease, and future therapies. Exp. Eye Res. 2015, 138, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhou, L.; Ouyang, J.; Xiao, X.; Sun, W.; Li, S.; Zhang, Q. Genotype-phenotype analysis of RPGR variations: Reporting of 62 chinese families and a literature review. Front. Genet. 2021, 12, 600210. [Google Scholar] [CrossRef]
- Shu, X.; Black, G.C.; Rice, J.M.; Hart-Holden, N.; Jones, A.; O’Grady, A.; Ramsden, S.; Wright, A.F. RPGR mutation analysis and disease: An update. Hum. Mutat. 2007, 28, 322–328. [Google Scholar] [CrossRef]
- Talib, M.; van Schooneveld, M.J.; Thiadens, A.A.; Fiocco, M.; Wijnholds, J.; Florijn, R.J.; Schalij-Delfos, N.E.; van Genderen, M.M.; Putter, H.; Cremers, F.P.M.; et al. Clinical and genetic characteristics of male patients with rpgr-associated retinal dystrophies: A long-term follow-up study. Retina 2019, 39, 1186–1199. [Google Scholar] [CrossRef]
- De Silva, S.R.; Arno, G.; Robson, A.G.; Fakin, A.; Pontikos, N.; Mohamed, M.D.; Bird, A.C.; Moore, A.T.; Michaelides, M.; Webster, A.R.; et al. The X-linked retinopathies: Physiological insights, pathogenic mechanisms, phenotypic features and novel therapies. Prog. Retin. Eye Res. 2021, 82, 100898. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, C.; Xiao, Y.; Liu, H. Novel splice receptor-site mutation of RPGR in a Chinese family with X-linked retinitis pigmentosa. Medicine 2018, 97, e12779. [Google Scholar] [CrossRef]
- Dry, K.L.; Manson, F.D.; Lennon, A.; Bergen, A.A.; Van Dorp, D.B.; Wright, A.F. Identification of a 5′ splice site mutation in the RPGR gene in a family with X-linked retinitis pigmentosa (RP3). Hum. Mutat. 1999, 13, 141–145. [Google Scholar] [CrossRef]
- Cehajic-Kapetanovic, J.; McClements, M.E.; Whitfield, J.; Shanks, M.; Clouston, P.; MacLaren, R.E. Association of a novel intronic variant in RPGR with hypomorphic phenotype of X-linked retinitis pigmentosa. JAMA Ophthalmol. 2020, 138, 1151. [Google Scholar] [CrossRef]
- Wright, R.N.; Hong, D.H.; Perkins, B. Misexpression of the constitutive Rpgr(ex1-19) variant leads to severe photoreceptor degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5189–5201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, N.; Seo, S. RPGR, a prenylated retinal ciliopathy protein, is targeted to cilia in a prenylation- and PDE6D-dependent manner. Biol. Open 2016, 5, 1283–1289. [Google Scholar] [CrossRef] [Green Version]
- Rao, K.N.; Li, L.; Anand, M.; Khanna, H. Ablation of retinal ciliopathy protein RPGR results in altered photoreceptor ciliary composition. Sci. Rep. 2015, 5, 11137. [Google Scholar] [CrossRef] [Green Version]
- Tsang, W.Y.; Bossard, C.; Khanna, H.; Peränen, J.; Swaroop, A.; Malhotra, V.; Dynlacht, B.D. CP110 suppresses primary cilia formation through its interaction with CEP290, a protein deficient in human ciliary disease. Dev. Cell 2008, 15, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human Gene Mutation Database (HGMD®): 2003 update. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Beltran, W.A.; Hammond, P.; Acland, G.M.; Aguirre, G.D. A frameshift mutation in RPGR exon ORF15 causes photoreceptor degeneration and inner retina remodeling in a model of X-linked retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 2006, 47, 1669–1681. [Google Scholar] [CrossRef] [Green Version]
- Ma, A.S.; Grigg, J.R.; Ho, G.; Prokudin, I.; Farnsworth, E.; Holman, K.; Cheng, A.; Billson, F.A.; Martin, F.; Fraser, C.; et al. Sporadic and familial congenital cataracts: Mutational spectrum and new diagnoses using next-generation sequencing. Hum. Mutat. 2016, 37, 371–384. [Google Scholar] [CrossRef] [Green Version]
- Nash, B.M.; Watson, C.J.G.; Hughes, E.; Hou, A.L.; Loi, T.H.; Bennetts, B.; Jelovic, D.; Polkinghorne, P.J.; Gorbatov, M.; Grigg, J.R.; et al. Heterozygous COL9A3 variants cause severe peripheral vitreoretinal degeneration and retinal detachment. Eur. J. Hum. Genet. 2021, 29, 881–886. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Nash, B.M.; Loi, T.H.; Fernando, M.; Sabri, A.; Robinson, J.; Cheng, A.; Eamegdool, S.S.; Farnsworth, E.; Bennetts, B.; Grigg, J.R.; et al. Evaluation for Retinal Therapy for RPE65 Variation Assessed in hiPSC Retinal Pigment Epithelial Cells. Stem Cells Int. 2021, 2021, 4536382. [Google Scholar] [CrossRef] [PubMed]
- Vangipuram, M.; Ting, D.; Kim, S.; Diaz, R.; Schuele, B. Skin punch biopsy explant culture for derivation of primary human fibroblasts. J. Vis. Exp. 2013, e3779. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Cordero, A.; Kruczek, K.; Naeem, A.; Fernando, M.; Kloc, M.; Ribeiro, J.; Goh, D.; Duran, Y.; Blackford, S.J.I.; Abelleira-Hervas, L.; et al. Recapitulation of human retinal development from human pluripotent stem cells generates transplantable populations of cone photoreceptors. Stem Cell Rep. 2017, 9, 820–837. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.D.; McClements, M.E.; Martinez-Fernandez de la Camara, C.; Bellingrath, J.-S.; Dauletbekov, D.; Ramsden, S.C.; Hickey, D.G.; Barnard, A.R.; MacLaren, R.E. Codon-optimized RPGR improves stability and efficacy of AAV8 gene therapy in two mouse models of X-linked retinitis pigmentosa. Mol. Ther. 2017, 25, 1854–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vössing, C.; Owczarek-Lipska, M.; Nagel-Wolfrum, K.; Reiff, C.; Jüschke, C.; Neidhardt, J. Translational read-through therapy of RPGR nonsense mutations. Int. J. Mol. Sci. 2020, 21, 8418. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Shu, X.; Fry, A.M.; Tulloch, B.; Manson, F.D.C.; Crabb, J.W.; Khanna, H.; Faragher, A.J.; Lennon, A.; He, S.; Trojan, P.; et al. RPGR ORF15 isoform co-localizes with RPGRIP1 at centrioles and basal bodies and interacts with nucleophosmin. Hum. Mol. Genet. 2005, 14, 1183–1197. [Google Scholar] [CrossRef] [Green Version]
- Khanna, H.; Hurd, T.W.; Lillo, C.; Shu, X.; Parapuram, S.K.; He, S.; Akimoto, M.; Wright, A.F.; Margolis, B.; Williams, D.S.; et al. RPGR-ORF15, which is mutated in retinitis pigmentosa, associates with SMC1, SMC3, and microtubule transport proteins. J. Biol. Chem. 2005, 280, 33580–33587. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Parapuram, S.K.; Hurd, T.W.; Behnam, B.; Margolis, B.; Swaroop, A.; Khanna, H. Retinitis Pigmentosa GTPase Regulator (RPGR) protein isoforms in mammalian retina: Insights into X-linked Retinitis Pigmentosa and associated ciliopathies. Vision Res. 2008, 48, 366–376. [Google Scholar] [CrossRef] [Green Version]
- Gakovic, M.; Shu, X.; Kasioulis, I.; Carpanini, S.; Moraga, I.; Wright, A.F. The role of RPGR in cilia formation and actin stability. Hum. Mol. Genet. 2011, 20, 4840–4850. [Google Scholar] [CrossRef] [Green Version]
- Murga-Zamalloa, C.; Swaroop, A.; Khanna, H. Multiprotein complexes of Retinitis Pigmentosa GTPase regulator (RPGR), a ciliary protein mutated in X-linked Retinitis Pigmentosa (XLRP). Adv. Exp. Med. Biol. 2010, 664, 105–114. [Google Scholar] [PubMed] [Green Version]
- Potter, V.L.; Moye, A.R.; Robichaux, M.A.; Wensel, T.G. Super-resolution microscopy reveals photoreceptor-specific subciliary location and function of ciliopathy-associated protein CEP290. JCI Insight 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.H.; Pawlyk, B.S.; Shang, J.; Sandberg, M.A.; Berson, E.L.; Li, T. A retinitis pigmentosa GTPase regulator (RPGR)-deficient mouse model for X-linked retinitis pigmentosa (RP3). Proc. Natl. Acad. Sci. USA 2000, 97, 3649–3654. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.A.; Khan, N.W.; Othman, M.I.; Chang, B.; Jia, L.; Grahek, G.; Wu, Z.; Hiriyanna, S.; Nellissery, J.; Li, T.; et al. Rd9 is a naturally occurring mouse model of a common form of retinitis pigmentosa caused by mutations in RPGR-ORF15. PLoS ONE 2012, 7, e35865. [Google Scholar] [CrossRef]
- Kallman, A.; Capowski, E.E.; Wang, J.; Kaushik, A.M.; Jansen, A.D.; Edwards, K.L.; Chen, L.; Berlinicke, C.A.; Phillips, M.J.; Pierce, E.A.; et al. Investigating cone photoreceptor development using patient-derived NRL null retinal organoids. Commun. Biol. 2020, 3, 82. [Google Scholar] [CrossRef] [Green Version]
- Lane, A.; Jovanovic, K.; Shortall, C.; Ottaviani, D.; Panes, A.B.; Schwarz, N.; Guarascio, R.; Hayes, M.J.; Palfi, A.; Chadderton, N.; et al. Modeling and rescue of RP2 retinitis pigmentosa using iPSC-derived retinal organoids. Stem Cell Rep. 2020, 15, 67–79. [Google Scholar] [CrossRef]
- Beltran, W.A.; Acland, G.M.; Aguirre, G.D. Age-dependent disease expression determines remodeling of the retinal mosaic in carriers of RPGR exon ORF15 mutations. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3985–3995. [Google Scholar] [CrossRef] [Green Version]
- Megaw, R.; Abu-Arafeh, H.; Jungnickel, M.; Mellough, C.; Gurniak, C.; Witke, W.; Zhang, W.; Khanna, H.; Mill, P.; Dhillon, B.; et al. Gelsolin dysfunction causes photoreceptor loss in induced pluripotent cell and animal retinitis pigmentosa models. Nat. Commun. 2017, 8, 271. [Google Scholar] [CrossRef]
- Patnaik, S.R.; Zhang, X.; Biswas, L.; Akhtar, S.; Zhou, X.; Kusuluri, D.K.; Reilly, J.; May-Simera, H.; Chalmers, S.; McCarron, J.G.; et al. RPGR protein complex regulates proteasome activity and mediates store-operated calcium entry. Oncotarget 2018, 9, 23183–23197. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, S.R.; Raghupathy, R.K.; Zhang, X.; Mansfield, D.; Shu, X. The Role of RPGR and Its Interacting Proteins in Ciliopathies. J. Ophthalmol. 2015, 2015, 414781. [Google Scholar] [CrossRef] [Green Version]
- Glaus, E.; Schmid, F.; Da Costa, R.; Berger, W.; Neidhardt, J. Gene therapeutic approach using mutation-adapted U1 snRNA to correct a RPGR splice defect in patient-derived cells. Mol. Ther. 2011, 19, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.N.; Zhang, W.; Li, L.; Ronquillo, C.; Baehr, W.; Khanna, H. Ciliopathy-associated protein CEP290 modifies the severity of retinal degeneration due to loss of RPGR. Hum. Mol. Genet. 2016, 25, 2005–2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.L.; Gao, M.L.; Lei, X.L.; Lv, J.N.; Zhao, H.; He, K.W.; Xia, X.X.; Li, L.Y.; Chen, Y.C.; Li, Y.P.; et al. Gene Correction Reverses Ciliopathy and Photoreceptor Loss in iPSC-Derived Retinal Organoids from Retinitis Pigmentosa Patients. Stem Cell Rep. 2018, 10, 2005. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shahani, U.; Reilly, J.; Shu, X. Disease mechanisms and neuroprotection by tauroursodeoxycholic acid in Rpgr knockout mice. J. Cell Physiol. 2019, 234, 18801–18812. [Google Scholar] [CrossRef]
- Reck-Peterson, S.L.; Redwine, W.B.; Vale, R.D.; Carter, A.P. The cytoplasmic dynein transport machinery and its many cargoes. Nat. Rev. Mol. Cell Biol. 2018, 19, 382–398. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chahine Karam, F.; Loi, T.H.; Ma, A.; Nash, B.M.; Grigg, J.R.; Parekh, D.; Riley, L.G.; Farnsworth, E.; Bennetts, B.; Gonzalez-Cordero, A.; et al. Human iPSC-Derived Retinal Organoids and Retinal Pigment Epithelium for Novel Intronic RPGR Variant Assessment for Therapy Suitability. J. Pers. Med. 2022, 12, 502. https://doi.org/10.3390/jpm12030502
Chahine Karam F, Loi TH, Ma A, Nash BM, Grigg JR, Parekh D, Riley LG, Farnsworth E, Bennetts B, Gonzalez-Cordero A, et al. Human iPSC-Derived Retinal Organoids and Retinal Pigment Epithelium for Novel Intronic RPGR Variant Assessment for Therapy Suitability. Journal of Personalized Medicine. 2022; 12(3):502. https://doi.org/10.3390/jpm12030502
Chicago/Turabian StyleChahine Karam, Fidelle, To Ha Loi, Alan Ma, Benjamin M. Nash, John R. Grigg, Darshan Parekh, Lisa G. Riley, Elizabeth Farnsworth, Bruce Bennetts, Anai Gonzalez-Cordero, and et al. 2022. "Human iPSC-Derived Retinal Organoids and Retinal Pigment Epithelium for Novel Intronic RPGR Variant Assessment for Therapy Suitability" Journal of Personalized Medicine 12, no. 3: 502. https://doi.org/10.3390/jpm12030502