Pulmonary Arterial Hypertension and Chronic Thromboembolic Pulmonary Hypertension: An Immunological Perspective

Department of Pulmonary Medicine, Erasmus MC, Doctor Molenwaterplein 40, 3015 GD Rotterdam, The Netherlands
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
J. Clin. Med. 2020, 9(2), 561; https://doi.org/10.3390/jcm9020561
Submission received: 14 December 2019
/
Revised: 10 February 2020
/
Accepted: 16 February 2020
/
Published: 19 February 2020
(This article belongs to the Special Issue Various Causes of Pulmonary Hypertension: A Multidisciplinary Approach)
Abstract
:Pulmonary hypertension (PH) is a debilitating progressive disease characterized by increased pulmonary arterial pressures, leading to right ventricular (RV) failure, heart failure and, eventually, death. Based on the underlying conditions, PH patients can be subdivided into the following five groups: (1) pulmonary arterial hypertension (PAH), (2) PH due to left heart disease, (3) PH due to lung disease, (4) chronic thromboembolic PH (CTEPH), and (5) PH with unclear and/or multifactorial mechanisms. Currently, even with PAH-specific drug treatment, prognosis for PAH and CTEPH patients remains poor, with mean five-year survival rates of 57%–59% and 53%–69% for PAH and inoperable CTEPH, respectively. Therefore, more insight into the pathogenesis of PAH and CTEPH is highly needed, so that new therapeutic strategies can be developed. Recent studies have shown increased presence and activation of innate and adaptive immune cells in both PAH and CTEPH patients. Moreover, extensive biomarker research revealed that many inflammatory and immune markers correlate with the hemodynamics and/or prognosis of PAH and CTEPH patients. Increased evidence of the pathological role of immune cells in innate and adaptive immunity has led to many promising pre-clinical interventional studies which, in turn, are leading to innovative clinical trials which are currently being performed. A combination of immunomodulatory therapies might be required besides current treatment based on vasodilatation alone, to establish an effective treatment and prevention of progression for this disease. In this review, we describe the recent progress on our understanding of the involvement of the individual cell types of the immune system in PH. We summarize the accumulating body of evidence for inflammation and immunity in the pathogenesis of PH, as well as the use of inflammatory biomarkers and immunomodulatory therapy in PAH and CTEPH.
1. Introduction
Pulmonary hypertension (PH) is a devastating condition characterized by increased pulmonary vascular resistance and elevated pulmonary arterial pressure. The definition of PH has recently been updated and is currently defined as increased mean pulmonary arterial pressures (PAP) above 20 mmHg at rest [1]. This enhanced pulmonary arterial pressure leads to elevated right ventricular (RV) hypertrophy, heart failure and, ultimately, death [2]. Structural remodeling of the vasculature, leading to diameter reduction, is linked to increased pulmonary vascular resistance and increased pulmonary pressure. PH severity is indicated by the New York Heart Association (NYHA) functional classification (FC), ranging from no symptoms (Class I) to severe symptoms at rest (Class IV).
Based on the underlying causes of PH, the WHO classification system divides PH patients into 5 groups: (1) pulmonary arterial hypertension (PAH), (2) PH due to left heart disease, (3) PH due to lung disease, (4) chronic thromboembolic PH (CTEPH), and (5) PH with unclear and/or multifactorial mechanisms [1]. WHO Group 1 (PAH), which is defined as a condition with a mean PAP >20 mmHg, normal left atrium pressure and pulmonary vascular resistance ≥3 Wood units [1], consisting of different subgroups based upon different underlying diseases or pathophysiological mechanisms. Heritable PAH (HPAH) includes patients with a family history or identified germline mutation. Such heritable susceptibility is conveyed not only through mutations in the bone morphogenetic protein receptor type II (BMPR2) gene, but also through newly identified mutations in novel causal genes [3]. PAH can also be induced by specific drugs and toxins [4]. Other causes of PAH are called APAH (associated PAH); PAH due to congenital heart disease (CHD), mainly comprising a ventricle septum defect, an atrial septum defect and patent ductus arteriosus; PAH due to liver disease (porto-pulmonary PAH), PAH due to HIV and PAH due to schistosomiasis. PAH can also be associated with auto-immune diseases, specifically with systemic sclerosis (SSc, prevalence 10%–15%), mixed connective tissue diseases (MCTD) and systemic lupus erythematosus (SLE). Two more separate identities in PAH WHO Group 1 are pulmonary veno-occlusive disease (PVOD), which can be heritable (eukaryotic translation initiation factor 2α kinase EIF2AK4 mutation), associated with autoimmune diseases or idiopathic, and persistent pulmonary hypertension of the newborn (PPHN). PPHN can be idiopathic or may be caused by several pulmonary diseases. In the largest group of PAH; idiopathic PAH (IPAH), no cause or associated disease is identified so far. WHO group 4 patients (CTEPH) can be further differentiated by whether they are operable (eligible for pulmonary endarterectomy (PEA) or balloon pulmonary angioplasty (BPA)) or inoperable CTEPH.
Currently, PAH-specific drugs focus predominantly on dilatation of the pulmonary arterial vasculature [5]. In inoperable CTEPH, PAH-specific drugs are also used to modulate the increased pulmonary vascular pressure [2]. However, even with PAH-specific drug treatment, survival for PAH patients remains poor with mean five-year survival rates of 57%–59% [6,7] and 53%–69% for PAH and inoperable CTEPH, respectively [7,8,9]. Therefore, more insight into the pathogenesis of PAH and CTEPH is urgently needed, so that new therapeutic strategies can be developed.
Especially in PAH, an increasing body of evidence shows that inflammation might play a role in its pathobiology [10]. However, recent studies have demonstrated that inflammatory cells might also contribute to disease pathology in CTEPH [11,12]. This review aims to summarize the expanding knowledge about inflammatory cells in the pathogenesis of PH, as well as the use of inflammatory biomarkers and immunomodulatory therapy in PAH and CTEPH.
2. Inflammation and Immunity in PAH and CTEPH
2.1. Histopathology in PAH and CTEPH
PAH and CTEPH are characterized by vascular remodeling as a result of increased pulmonary arterial pressures (Figure 1). Features of pulmonary vascular remodeling in PH are intima wall thickening and the formation of obliterative concentric lesions in the endothelial and/or smooth muscle cell layers. In the media, which mainly consists of smooth muscle cells, an increase in thickness is also seen. Interestingly, the combined intima and media thickness correlated significantly to the PAP and the pulmonary vascular resistance (PVR) [13]. Finally, increased adventitial thickness and remodeling were the most prominent findings in a series of 19 IPAH patient lung autopsies [14]. However, this increased adventitial thickness was not confirmed in a recent study, which might be explained by methodological differences between these studies [13].
Besides increased intimal, medial and adventitial vascular thickness, another prominent feature in PAH patients is perivascular inflammation. A feature seen specifically in IPAH patients is the formation of plexiform lesions, which are typically defined as dynamic networks of vascular channels formed by monoclonal proliferation of endothelial cells [15]. In CTEPH patients, histologic studies show neointimal, thrombotic, recanalized and atherosclerotic lesions in the pulmonary arterial vasculature. These chronic thrombotic lesions contain collagen, elastin, inflammatory cells, re-canalization vessels and calcification [16]. Current concepts state that CTEPH is a dual vascular disorder with stenosis, webs and occlusions in large- and medium-sized pulmonary arteries (Pas) at the sites of previous pulmonary emboli. Furthermore, a secondary arteriopathy affecting small resistance vessels is visible in lung biopsies of CTEPH patients [17].
2.2. Dysregulated Immune Responses in PAH and CTEPH
Immune responses can be divided into two categories—innate and adaptive immunity. Innate immunity is our swift first-line defense against microorganisms and foreign pathogens, which is very broad and nonspecific. Innate immune responses are usually initiated and executed by macrophages, neutrophils, natural killer (NK) cells and dendritic cells (DCs). In parallel, adaptive immune responses are initiated, which allows for more specific responses executed by T and B lymphocytes. Next to their innate function, DCs initiate adaptive immune responses by migrating to lymphoid organs, mostly the tissue-draining lymph nodes. There, they present parts of foreign pathogens on their cell surface through major histocompatibility complex (MHC) molecules in order to induce the clonal expansion of antigen-specific T cells. Memory T cells and B cells can ensure lifelong immunity, allowing our body to rapidly and more vigorously respond to pathogens upon a second encounter. The adaptive response relies on the ability of T and B cells to distinguish self from non-self antigens. If this capacity is disturbed, the immune system may attack the body’s own cells, which can lead to autoimmune disease. Elements hereof can be found in both PAH and CTEPH. IPAH, for instance, is characterized by perivascular inflammation, consisting of pulmonary lymphoid neogenesis with the formation of tertiary lymphoid organs (TLOs), which contain specific T and B cell zones with on-site activation and production of antibodies [18]. In addition to lymphocytes, these inflammatory lesions also contain DCs, macrophages and mast cells [18,19]. Moreover, there is a positive correlation between the degree of pulmonary perivascular inflammation and vascular intimal/medial/adventitial thickness and mean PAP, suggesting that this inflammation could be involved in pulmonary vascular remodeling and PH development [20].
In CTEPH patients, thrombotic and atherosclerotic lesions were found to contain activated B and T lymphocytes, macrophages, and neutrophils [21]. In this study, topographic analyses revealed a transmural distribution of T cells, whereas B cells were low in number and mostly localized deep within the lesion, close to internal elastic lamina and native media. Although inflammatory cells are common in thrombotic material, the accumulation of inflammatory cells may be a sign of involvement in the pathology of the non-resolution of thrombosis and atherosclerosis in CTEPH patients.
Below, we discuss several different immune cells that are part of the innate or adaptive immune system and their possible roles in the pathogenesis of PH.
2.2.1. Innate Immunity
Macrophages
Macrophages are first-line myeloid leucocytes observed in pulmonary lesions in PH patients [22,23]. Pulmonary macrophages can classically be divided into interstitial and alveolar macrophages, although recent single-cell RNA sequencing and lineage tracing studies have defined multiple pulmonary macrophage subtypes in mice [24,25,26]. In a hypoxia-driven PH mouse model, specifically interstitial macrophages—but not alveolar macrophages—were increased [27]. They might be involved in PH development through their production of cytokines [28,29,30]. Furthermore, the macrophages present in lung perivascular spaces in PAH are derived from peripheral blood monocytes [31], likely indicating their recruitment and differentiation due to pulmonary inflammation. However, chemokines, such as chemokine (C-C motif) ligand 2 (CCL2) and chemokine (C-X3-C motif) ligand 1 (CX3CL1) monocytes are recruited to the site of the inflammation and differentiate into inflammatory macrophages. In PH, patients’ expression of chemokines, such CCL1, CCL2, CCL5 and CX3CL1, was increased in circulatory monocytes, along with increased pulmonary levels of CCl1, CCL2, CCL3, CLL3 and CX3CL1 [31]. In CX3CR1 (receptor for CX3CL1)-deficient mice, both pulmonary inflammation and vascular remodeling were reduced after exposure to hypoxia when compared to control mice [31]. Interestingly, in pulmonary vascular endothelial cells from PAH patients, excessive expression of CCL2 was observed, which acts as a chemoattractant for circulating inflammatory cells and as a growth factor for pulmonary arterial smooth muscle cells (PASMCs). Moreover, PASMCs and perivascular macrophages from patients with PAH exhibited elevated CCR2 and CCR5 levels compared to controls [32,33]. Indeed, expression of CCR2 and CCL5-CCR5 was needed in both macrophages and PASMCs to initiate and amplify PASMC proliferation [33]. When circulating monocytes differentiate into interstitial macrophages in hypoxia-induced PH in mice, they express thrombospondin-1 (TSP-1), leading to Rho kinase-mediated vasoconstriction through transforming growth factor beta (TGF-β) activation [34], thereby amplifying PH pathology. All these studies suggest a potentially crucial role for chemokine-mediated macrophage recruitment in the early pathogenesis of PH.
Macrophage recruitment and activation was also shown to play an important role in the pathogenesis of hypoxia-induced PH in the mouse and in Sugen/athymic rat models [35,36,37]. A previous study has reported that hypoxia-inducible factor-1α (HIF-1α) is expressed by pulmonary macrophages in PH patients, especially in plexiform lesions [38]. When HIF-1α was absent in mice, a significant reduction in right ventricular (RV) systolic pressure (RVSP) and RV hypertrophy was observed, together with less infiltration of macrophages in the lung and RV, indicating the direct effect of on-site macrophages in inducing PH [39]. In IPAH patients, increased numbers of macrophages and monocytes were found in the lungs when compared to healthy controls [40]. Another PH-inducing factor, resistin-like molecule-α (RELMα) was increased in a hypoxia-induced mouse model for PH, and its human homologue, resistin, was also found to be upregulated in macrophage-like inflammatory cells in IPAH patients. In these patients, resistin-stimulated macrophages promoted the apoptosis-resistant proliferation of PASMCs [41].
Macrophages may also play a role in other forms of PH, as the increased presence of inflammatory macrophages was apparent in surgical pulmonary endarterectomy (PEA) material from CTEPH patients [21]. Moreover, in serum from eight CTEPH patients, increased expression of macrophage inflammatory protein-1α (CCL3) was detected, which can lead to the synthesis of inflammatory cytokines, vascular remodeling and recruitment of macrophages [42]. Recently, the accumulating evidence for the role for macrophages in PH has been extensively reviewed by Florentin et al. and Pullamsetti et al. [43,44]. Taken together, macrophages play a pivotal role in the pathogenesis of PH, through the production of inflammatory cytokines, initiation and proliferation of PASMCs and hypoxia factors.
Neutrophils
Neutrophils are early responders and are recruited to sites of acute inflammation, in response to chemokines produced by tissue-resident immune cells, such as macrophages. Neutrophils are known phagocytes, capable of ingesting microorganisms or particles. Besides their phagocytic role, neutrophils are able to degranulate and release antimicrobial contents. In PAH patients, an increased neutrophil-to-lymphocyte ratio in peripheral blood samples positively correlates with the NYHA FC and a negative ratio can even predict event-free survival [45,46]. In CTEPH patients, the neutrophil-to-lymphocyte ratio could predict postoperative mortality and might be used as a noninvasive measuring tool for operative risk stratification [47]. Currently, it is unclear if and how neutrophils contribute to PAH progression. In murine models, neutrophils accumulated at the site of inflammation/injury in the lungs of hypoxic PH and in monocrotaline (MCT)-induced PH rats [36,48]. Recent evidence suggests that the neutrophilic production of myeloperoxidase (MPO), a catalyst for reactive oxygen species (ROS) formation, can cause disease progression [49]. Plasma levels of MPO were found to be increased in PAH patients compared to healthy controls. Furthermore, hypoxia-exposed Mpo-/- mice showed a lower increase in RV pressure than wildtype mice [49], indicating a pathogenic role for neutrophils in PH through the production of MPO and adverse pulmonary vascular function.
Mast Cells
Mast cells (MCs) are long-living tissue-resident immune cells known for their important role in the immune system through their release of histamine and production of inflammatory cytokines. MCs are also known for their role in angiogenesis through their production of vascular endothelial growth factor (VEGF) and MC proteases, including chymase and tryptase [50,51,52]. Accumulating evidence indicates a role for MCs in the pathophysiology of PH. MCs are present in inflammatory lesions in IPAH patients [19], even in early perivascular cellular lesions in the lungs of IPAH patients [53]. In recent years, the role of MCs has been shown in proof-of-principle experiments. In Ws/Ws rats, in which MCs are absent due to a mutation in mast cell growth factor receptor c-kit [54], features of experimental PH, such as RVSP, PVR, RV hypertrophy and vascular remodeling were largely attenuated after pulmonary arterial banding or MCT treatment [55]. Furthermore, when degranulation of MCs was inhibited by ketotifen, the development of PH was reduced in several experimental rat PH models [55,56,57,58]. In a small clinical trial, nine PAH patients were treated with the MC inhibitors cromolyn and fexofenadine. In these patients, a decrease in VEGF levels and circulating proangiogenic myeloid cells was observed, together with an increase in exhaled nitric oxide (which is generally low in PAH), indicating this treatment might have a suppressive effect on MCs [59]. Mechanistically, MC proteases are believed to play an important role in the process of PH development and severity. MC proteases, such as chymase and tryptase, measured in the lung tissue, correlate with the severity of PH and pulmonary vascular remodeling [52,60,61,62,63]. Moreover, excessive MC infiltration and degranulation was detected in the lung tissue pf MCT-rats and not in the RV, indicating a release of proteases, such as tryptase, which contribute to pulmonary vascular remodeling [64]. Lastly, MCs are observed around distal pulmonary arteries, together with accumulated macrophages, in MCT-challenged rats, suggesting that MCs are involved in vascular remodeling in the lungs [64]. In summary, MCs are involved in PH pathobiology, most likely through the release of proangiogenic factors and MC proteases.
Natural Killer Cells
Natural killer (NK) cells comprise an important part of the innate immune system, as they provide rapid responses to virus-infected cells, but these cytotoxic cells are also known for regulating angiogenesis and vascular remodeling. Few studies have evaluated the possible contribution of NK cells in the pathogenesis of PAH and CTEPH. However, in both PAH patients and in rodent experimental PH models, impaired NK cell numbers and cytotoxicity were found [65]. Furthermore, in two independent genetic mouse models for NK cell dysfunction, involving deficiency of the NFIL3 transcription factor or the NK activating receptor NKp46, enhanced RV systolic pressures and RV hypertrophy were found. In both models, this experimental PH development was linked to increased interleukin-23 (IL-23) production, possibly due to NK cells or impairment, leading to the increased production of IL-23 by pulmonary macrophages and other myeloid cell types [66]. Interestingly, IL-23 is known for its production of inflammatory cytokines, such as IL-17A/F, IL-21 and IL-22, and for driving naïve T cells to a TH17 phenotype [67]. Taken together, NK cell defects may contribute to PAH pathogenesis by the aberrant regulation of pulmonary vascular remodeling, however further research is required to evaluate these findings.
2.2.2. Linking Innate and Adaptive Immunity
Dendritic Cells
DCs are key modulators between tolerance and immunity and are known to function as a bridge between innate and adaptive immunity. The main function of DCs is to capture, process and present antigens to T cells. DCs can be activated either by microbial stimuli through pattern recognition receptors, such as Toll-like receptors, or by inflammatory cytokines, which leads to the activation of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κβ) pathway. Next, DCs upregulate costimulatory molecules, produce various inflammatory cytokines, such as interleukin (IL)-6 and IL-12, and, together with antigen presentation, DCs promote T-cell activation, expansion and differentiation [68,69,70]. This process must be tightly controlled, as continuous DC activation could lead to severe side-effects, such as the presentation of self-antigens to T-cells, resulting in development of auto-immune diseases [69,71]. Rationally, DCs could also be involved in the pathophysiology of PAH [72].
In IPAH patients, DCs are increased in the lung, specifically accumulating around remodeled pulmonary arteries [73]. However, in the parenchyma, mostly immature DCs are observed, shown by an increased number of dendritic cell-specific intercellular adhesion molecule 3-grabbing non- integrin (DC-SIGN)+ DCs [73]. DCs can be divided into four main subtypes: type I conventional DCs (cDC1s), which are efficient at cross-presentation (i.e., presentation of exogenous antigens in the context of MHC Class I and elicitation of CD8+ T-cell responses); type II cDCs (cDC2s), which are capable of inducing CD4+ T-cell responses; plasmacytoid DCs (pDCs), which can produce large amounts of type I interferons to combat viral infections; and, finally, monocyte-derived DCs (mo-DCs) that arise during inflammation and produce large amounts of chemokines, attracting T-cells to the site of inflammation [74].
The numbers of cDCs and pDCs were increased both in total lung cell suspensions (either peripheral or perihilar samples) and in the larger pulmonary arteries of IPAH patients compared to controls [40]. Confocal microscopy analyses showed that pDCs were predominantly localized in the alveolar space, in proximity to blood vessels. In contrast, in peripheral blood, cDC numbers were decreased in IPAH patients [75] and, together with the increase in pulmonary cDCs, this suggests migration to the lungs. Currently, no data is available for different cDC subsets in the pathogenesis of PAH. Taken together, DCs are crucial in initiating adaptive immune responses and could be involved in the pathogenesis of PAH by antigen presentation and production of inflammatory cytokines.
2.2.3. Adaptive Immunity
T Cells
T cells are a vital part of the adaptive immune system. CD4+ T cells or T-helper (Th) cells, provide help by indirectly killing pathogens by supporting the activation of other cells in the immune system, such as B cells. CD8+ T cells are known as ‘cytotoxic T-cells’, which are able to directly kill pathogens through the release of granzymes, which induce apoptosis, and the pore-forming protein perforin, which creates holes in target-cell membranes. Gamma delta (γδ) T cells represent a small subset of T cells which are defined by the expression of heterodimeric T-cell receptors (TCRs) composed of γ and δ chains. An increased number of CD4+, CD8+ T-cells and γδ T cells was found in close proximity to the pulmonary arteries in IPAH lung biopsies using flow cytometry [40]. These CD4+ and CD8+ T cells are present in the adventitial space around the pulmonary vessels in IPAH patients [19]. In schistosomiasis-associated PAH and IPAH patients, increased peri-arterial CD4+ T cells were found as well [76].
Th cells are known to play an important role in many inflammatory and autoimmune diseases [77]. Th-cells can be roughly divided into Th1, Th2 and Th17 cells. Especially, Th17 cells are found in the pulmonary TLOs of IPAH patients [18]. Th17 cells are the main source of IL-17, IL-21, and IL-22. In remodeled PAs of IPAH patients, IL-21+ cells are present [78]. Th17 cells differentiate from naïve Th-cells in the presence of IL-1β, IL-6, and TGF-β [79]. In serum, both IL-1β and IL-6 are increased in IPAH patients compared to controls [80]. In CTD-PAH, Th17 cells and Th17-related cytokines were increased when compared to the healthy controls [81]. Work from our group has shown that the level of tumor necrosis factor alpha (TNF-α)-induced protein 3 (TNFAIP3) expression in DCs controls T-cell differentiation, because TNFAIP3-deficient DCs promote Th17-cell differentiation through increased expression of IL-1β, IL-6 and IL-23 [71,82]. DC-specific deletion of the TNFAIP3 gene also leads to increased NF-kB, creating a pro-inflammatory environment.
Follicular T-helper (Tfh) cells, expressing the CXCR5 chemokine receptor which contributes to their localization in B cell follicles, can support activated B cells under the influence of IL-21, IL-6, IL-12 and IL-27, leading to the induction of humoral immune responses. In IPAH patients’ TLOs, an increase was found in IL-21+ PD1+ Tfh cells [18]. In CTEPH patients, the histological evaluation of PEA material showed accumulation of CD3+ T cells in atherosclerotic and thrombotic lesions [21]. Little is known about T-cell function differences in CTEPH and more research is needed to provide evidence for a possible pathogenic role for T cells in CTEPH pathogenesis. Taken together, T cells are increased in IPAH lungs and CTEPH PEA material. It appears that a dysregulated Th17-immune response is present in PAH; however, more studies are needed to further elaborate this.
B Cells and Humoral Immune Responses
B cells are the effectors of the humoral immune response. Following antigen recognition by the B cell receptor they can present antigens, secrete cytokines and differentiate into memory B cells or plasma cells that produce large amounts of antibodies. Upon activation, B and T cells engage in a germinal center reaction, in which Tfh cells produce their canonical cytokine IL-21, which supports B cell survival, proliferation and differentiation. Moreover, activated T cells express CD40L, which interacts with its receptor CD40 in B cells to provide a co-stimulatory signal that is critical for B cell activation and germinal center formation. As mentioned above, the lungs of PAH patients contain TLOs containing B cells, T cells and DCs. These highly organized structures contain high endothelial venules, enabling circulating lymphocytes and stromal cells, including follicular dendritic cells that present antigens to B cells via Fc-receptors, to directly enter [83]. Importantly, the presence of IL-21+ Tfh cells, B cells that express activation-induced cytidine deaminase that is essential for immunoglobulin heavy chain class switch and antibody affinity maturation, and plasma cells, provide evidence for local and ongoing antibody production [18]. Next to the presence of TLOs in the lung, there is additional evidence supporting the notion that B-cell activation is dysregulated in IPAH and in connective tissue disease associated PAH (CTD-PAH) [84,85]. First, circulating plasma blasts are increased in IPAH patients [86]. Second, autoantibodies are present in approximately 40% of patients with IPAH [87]. These autoantibodies might be produced by the plasma cells located within TLOs in IPAH lungs [18,86], recognizing endothelial cell surface antigens [88]. Anti-endothelial autoantibodies promote apoptosis of endothelium, which contributes to vascular remodeling [86]. Furthermore, endothelial-specific IgA can promote cytokine production and the upregulation of adhesion molecules by endothelial cells [86,88,89,90]. Anti-endothelial IgG antibodies activate endothelial cells to a pro-adhesive and pro-inflammatory state [91]. In animal studies, the injection of autoantibodies from CTD-PAH patients into healthy mice leads to more abundant vascular and airway smooth muscle cell numbers and inflammatory pulmonary vasculopathy [92]. In MCT rats, high levels of plasma IgG were found that labeled lung vascular proteins. Moreover, the transfer of autoantibodies into rats caused pulmonary vascular remodeling and pulmonary hypertension [93].
In CTEPH, little is known about circulating and thrombus-resident B-cells. A well-known risk factor for CTEPH is a splenectomy and, considering that the spleen is important for B cell maturation, there might be a role for pathogenic B-cells in the CTEPH pathogenesis. Currently, studies are being performed using mass cytometry in PBMCs from CTEPH patients. Results from these studies are still in progress.
In summary, autoantibodies are found in IPAH patients, specifically targeting endothelial cell surface antigens. B-cells and plasma cell formation prior to this could play a major role in the pathogenesis of PAH.
3. Inflammatory Diagnostic and Prognostic Biomarkers in PAH and CTEPH
Inflammatory biomarkers might be useful as diagnostic and prognostic tools in PAH and CTEPH [21,32,42,80,94,95,96,97,98,99,100,101,102]; these are highlighted in Table 1. Inflammatory cytokines and chemokines can contribute directly to the recruitment of immune cells, the activation and proliferation of PASMCs, and endothelial dysfunction. Until now, the most prominent cytokine appears to be IL-6, which has many links to PAH pathogenesis. In animal models, PH development has been seen after the administration of recombinant IL-6 and also in IL-6 transgenic mice [103,104,105], whereas IL-6 knockout mice have shown resistance to hypoxia-induced PH development [106]. In clinical studies, IL-6 has shown to correlate to survival and quality of life in IPAH patients [80,107] and in predicting long-term responses to PEA in CTEPH patients [98]. In a recent study, cytokine clusters were made in PAH patients using machine learning. The analyses showed that the immune phenotypes were not dependent upon the subtypes within the WHO Group 1 PAH classification [108], indicating that immune phenotypes may vary within the WHO Group 1 subtypes. Furthermore, these findings might provide a framework to examine patient responses to emerging therapies targeting immunity in the future.
In tumor necrosis factor alpha (TNF-α)-overexpressing mice, the spontaneous development of PH was observed [109]. In a clinical study in CTEPH patients undergoing PEA, increased levels of TNF-α, IL-6 and IL-10 were found prior to surgery [97]. IL-6 and IL-10 were shown to peak immediately after surgery, while TNF-α decreased significantly within the first 24 h after PEA surgery [97].
In CTD-PAH, treatment-naïve patient baseline levels of placental growth factor (PlGF), sVEGFR-1, TNF-α, and VEGF-D were increased and could differentiate between healthy controls/IPAH and CTD-PAH. Moreover, after four months of PAH-targeted treatment, sVEGFR-1 levels were decreased, indicating that this growth factor is worthwhile to evaluate during therapies [99]. In a study of 206 PAH patients, angiopoietin 1 (Ang-1), VEGF and matrix metallopeptidase 9 (MMP-9) levels have been associated with increased risk of death and hospitalization at the 16-week follow-up point after baseline [101]. Many of the inflammatory biomarkers are still being investigated, because more pre-clinical, translational and clinical studies are needed to determine the clinical and prognostic value of these markers.
4. Immunomodulatory Therapy in PAH and CTEPH
Promising novel inflammatory therapeutic targets and ongoing clinical trials evaluating possible therapeutic drug compounds are highlighted in Figure 2. In addition, an overview of the attenuation of experimental PH by targeting immunomodulatory pathways is given in Supplementary Table S1.
Based on the evidence described above, targeting immune and inflammatory pathways may be sufficient to treat and prevent progression of the disease. Previous studies have shown that in the inflammatory MCT-rat PH model, anti-inflammatory therapies, such as dexamethasone, mycophenolate mofetil and the nuclear factor of activated T cell (NFAT) inhibition with cyclosporine can prevent and reverse the PH phenotype [110,111,112]. In SLE and MCTD-PAH patients, treatment with a combination of cyclophosphamide and glucocorticoids was possibly effective in lowering the PVR in patients with a less severe PH at baseline [113].
More targeted therapy, such as anti-IL-1 treatment was shown to prevent the PH phenotype in MCT-PH rats [114]. A current trial, conducted by the Virginia Commonwealth University, evaluating the efficacy of treatment of PAH patients with anakinra, an IL-1 receptor antagonist compound, will soon be finalized.
In recent studies, IL-6-specific antagonist treatment reversed experimental PH in MCT-PH and sugen/hypoxia (SU/Hx)-induced PH rat models [115]. In hypoxia-induced PH in mice, the attenuating effect of a blockade of IL-6 was also found [78]. Currently, the TRANSFORM-UK trial is running, in which PAH patients are being treated with anti-IL-6, and results of this study are expected soon [116].
In hypoxia-induced PH mice, increased expression levels were found for IL-17 and IL-21, signature genes for Th17 and Tfh cells, respectively [78]. Whereas the blockade of IL-17 showed no effects on the RVSP and the RV hypertrophy, IL-21-receptor knockout mice were resistant to hypoxia-induced PH [78]. Increased expression of M2 macrophage markers and IL-21, which can polarize macrophages towards an M2 phenotype, was detected in the lungs of IPAH patients who underwent lung transplantation. Together with the known prominent role of Il-21 in B-T cell interaction, these findings suggest that IL-21 is a potential target for treating PAH [78]. In autoimmune experimental arthritis, a combination of IL-6/IL-21 blockades have shown synergistic beneficial effects associated with strongly reduced Th17 differentiation [117].
Anti-TNF-α therapy (etanercept) has been demonstrated to attenuate the PH phenotype both in MCT-PH rats [118,119] as well as in SU/Hx-induced PH rats [120]. In endotoxemic pigs, anti-TNF-α therapy reversed the PAH phenotype [121]. Currently, no (pre-)clinical trials are available to determine the possible clinical effects of etanercept in patients.
Increasing knowledge on loss-of-function mutations in the BMPR2 signaling pathway have led to the initiation of studies evaluating possible novel therapeutic targets in this cascade. BMPR2, which is mainly expressed in vascular endothelial cells [122], is a member of the TGF-β receptor family and many studies have shown an important role for BMPR2 in the pathogenesis of PAH. Upon binding to bone morphogenetic proteins (BMPs), BMPR2 initiates intracellular signaling that ultimately leads to the inhibition of proliferation of vascular smooth muscle tissue. In smooth muscle cells, BMP signaling can be directly inhibited by TGF-β signaling and involved ligands are able to function as antagonists in competition for type II receptor binding [123]. BMPR2 loss-of-function mutations are a known cause for PAH development in patients and lead to more severe disease and increased risk of death when compared to PAH patients without a BMPR2 mutation [124]. In a recent study, BMP9 was shown to be a sensitive and specific biomarker of porto-pulmonary hypertension patients in order to predict transplant-free survival and the presence of PAH in liver disease [125]. In human IPAH lungs and in hypoxia-induced PH in mice, reduced BMPR2 expression-induced macrophage recruitment—involving enhanced production of the chemokine granulocyte macrophage colony-stimulating factor (GM-CSF)—led to the exacerbation of PAH features [126]. In rodent models, such as the MCT-rat, Su/Hx PH mouse model and in mice harboring a human BMPR2 mutation knock-in allele, BMPR2 activation can prevent vascular remodeling and can attenuate the PAH phenotype with endothelial growth and proliferation [127,128,129]. Recently, a therapeutic drug discovery company (MorphogenIX, Cambridge, UK) was founded for the development of BMPs as a novel treatment for PAH.
Targeting TGF-β signal pathways may also be an effective treatment for PAH, considering the upregulation of TGF-β downstream of the loss of function in BMPR2, which has shown a correlation with PH development [130,131]. In a recent study, treatment with immunoglobulin-Fc fusion protein TGF-β (TGFBRII-Fc), a selective TGF-β inhibitor targeting THF-b 1/3, has shown attenuation of the PH phenotype in MCT-PH rats and SU/Hx-induced PH rats and mice [132]. Currently, ligand traps, such as Sotatercept and Luspatercept, with high selectivity for members of the TGF-β superfamily are being investigated in phase II trials (PULSAR trial), as they were successful in previous phase I clinical trials. These ligand traps may rebalance BMPR2 signaling and restore vascular homeostasis.
Another potentially interesting compound in the BMPR2 pathway is calcineurin inhibitor FK506 (tacrolimus), which has been reported to increase the expression and activity of BMPR2. In MCT-PH and SU/Hx-induced PH rats and hypoxia-induced PH mice, FK506 was reported to reverse the severe PAH phenotype [133]. In IPAH patients, FK506 treatment reversed dysfunctional BMPR2 signaling in the pulmonary artery endothelial cells [133]. In a recent phase IIa trial, treatment with FK506 in 20 PAH patients showed increased expression of BMPR2, improvement of 6MWD and the serological and echocardiographic parameters of heart failure in some patients; however, these changes were not significant [134]. Nonetheless, FK506 was generally well tolerated and this study supports the initiation of a phase IIb efficacy trial. Significantly, tacrolimus is also well known for other immunosuppressive effects, such as T cell inhibition in the organ-transplant field, so the effects might not be exclusively limited to the BMPR2 pathway.
The nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κβ) is a ubiquitous transcription factor that is known for the regulation of many aspects of innate and adaptive immune functions. By inducing expression of various pro-inflammatory genes for cytokines and chemokines, NF-κβ is an important regulator for cell survival, proliferation and mobility [135]. Targeting NF-κβ may therefore be an interesting novel therapeutic pathway for the treatment of PAH. In MCT-PH rats, the NF-κβ was found to be activated and an NF-κβ-blocking treatment attenuated the PH phenotype [136,137,138]. This ameliorating effect was also found in SU-Hx rats, in which NF-κβ targeting severely reduced lung vascular lumen obliteration [139]. Furthermore, in MCT-treated transgenic mice overexpressing a cardiac-specific dominant-negative inhibitory binding partner of NF-κβ (IκBαP), the inhibition of NF-κβ prevented right ventricular hypertrophy (RVH) [140]. In PAH patients, NF-κβ has been shown to be highly activated in pulmonary lymphocytes, macrophages, endothelial cells and PASMCs [141].
Currently, several NF-κβ inhibitory compounds are available for the evaluation of treatment efficacy in PH patients. Bardoxolone methyl is a known inhibitor of NF-κβ and shows effects in suppressing the activation of pro-inflammatory mediators, enhancement of endothelial NO bioavailability, improvement of metabolic dysfunction, suppressing vascular proliferation and preventing maladaptive remodeling [142,143,144]. Currently, bardoxolone methyl is being evaluated in a phase II clinical trial in pulmonary hypertension patients (IPAH, CTD-PAH, WHO group III or group V PH).
Another promising compound is dimethyl fumarate (DMF), with potent anti-inflammatory effects through its inhibition of NF-κβ. DMF is an activating agent for the transcriptional regulator nuclear factor erythroid 2-related factor 2 (NRF2), which is known for its key regulation of antioxidant genes [145]. NRF2 function is linked to NFκB signaling with activation of NRF2, leading to inhibition of NFκB signaling and thereby inducing an anti-inflammatory response [146]. In the chronic hypoxia and SU/Hx-induced PH mouse model, DMF has been reported to attenuate the PAH phenotype [147]. Currently, no clinical trial for the evaluation of the clinical therapeutic value of DMF is being performed.
As previously described, accumulating evidence is beginning to show a pathological role for B cells and plasma cells in PH pathogenesis. Therefore, targeting CD20, which is a B-cell specific surface marker, could be a promising drug therapy to evaluate. In a case report, rituximab treatment significantly improved early onset PAH in a young patient suffering from SLE [148]. Currently, the National Institute of Allergy and Infectious Diseases (NIAD) is conducting a phase II prospective, double-blind, placebo-controlled, multi-center, randomized trial for evaluating the effect of treatment with rituximab in patients suffering from SSc-PAH.
Another interesting therapeutic target is leukotriene B4 (LTB4), a pro-inflammatory lipid mediator produced from arachidonic acid by the consecutive activities of 5-lipoxygenase, 5-lipoxygenase-activating protein, and leukotriene A4 hydrolase [149]. In MCT-PH rats, the LTB4 receptor antagonist (ONO4057) has been reported to reduce RVH after MCT treatment and prevent development of PH [150].
Nonspecific inhibition of Leukotriene A4 through bestatin (ubenimex) reversed the PH phenotype in MCT-PH and SU/Hx-induced PH rats [37]. Currently, a clinical phase II trial (LIBERTY trial) is evaluating the efficacy of bestatin treatment in 61 PAH patients. Preliminary results for the trial sponsor (Eiger Biopharmaceuticals, Palo Alto, CA, USA), however, show no significant treatment effect in comparison to placebo treatment.
5. Summary
Taken together, there is mounting evidence that the immune system plays a pivotal role in the pathogenesis of PAH and CTEPH. Both PAH and CTEPH histology demonstrated the extensive accumulation of immune cells. Further analyses in IPAH patient lungs and lung biopsy material from CTEPH patients provided compelling evidence for the activation of the innate immune system. The pathological involvement of macrophages, MCs and neutrophils by production of inflammatory cytokines, recruitment of other immune cells and local inflammation and damage was demonstrated. In the lungs of IPAH patients, increased numbers of DCs were observed, acting as a bridge between the innate and adaptive immune system by the presentation of antigens to T cells. DCs contribute to increased production of cytokines and chemokines, attracting other inflammatory cells to the site of inflammation. Dysregulated Th17 immunity was found in PAH patients, creating a pro-inflammatory auto-immune environment. Moreover, IPAH patients displayed an increase in circulating autoantibodies specifically targeting endothelial cell surface antigens. Extensive biomarker research revealed that many inflammatory and immune markers correlate with the hemodynamics and/or prognosis of PAH and CTEPH patients. However, further evaluation is required to investigate the applicability of these parameters in the clinical work-up of PAH and CTEPH patients. Currently, clinical trials are being performed to assess the value of promising inflammatory and immune targets defined in pre-clinical research in PAH. A combination of immunomodulatory therapies might be required besides current treatment based on vasodilatation alone, to establish the effective treatment and prevention of progression of this disease.
Supplementary Materials
The following are available online at https://www.mdpi.com/2077-0383/9/2/561/s1, Table S1: Pre-clinical therapeutic targets in experimental PH.
Author Contributions
Conceptualization, T.K. and M.K.; writing—original draft preparation, T.K.; writing—review and editing, M.K. and K.A.B.; visualization, T.K.; project administration, T.K. All authors have read and agreed to the published version of the manuscript.
Funding
T.K. was supported by an unrestricted grant by Actelion pharmaceuticals.
Acknowledgments
We would like to thank R.W. Hendriks and J.J.P. Collins for critically reading the manuscript and providing valuable feedback.
Conflicts of Interest
All authors declare that there is no conflict of interest.
Abbreviations
| Ang-1 | Angiopoietin 1 |
| APAH | associated PAH |
| BPA | Balloon pulmonary angioplasty |
| BMPR2 | Bone morphogenetic protein receptor type II |
| BMPs | Bone morphogenetic proteins |
| tacrolimus | Calcineurin inhibitor FK506 |
| CCL | Chemokine C-C motif ligand |
| CXCL | Chemokine C-X3-C motif ligand |
| CTEPH | Chronic thromboembolic pulmonary hypertension |
| CHD | Congenital heart disease |
| CTD-PAH | Connective tissue disease associated PAH |
| DC-SIGN | Dendritic cell- specific intercellular adhesion molecule-3 grabbing non- integrin |
| DCs | Dendritic cells |
| DMF | Dimethyl fumarate |
| Tfh | Follicular T-helper cells |
| FC | Functional class |
| γδ | Gamma delta |
| HPAH | Heritable PAH |
| TCRs | Heterodimeric T-cell receptors |
| HIF-1α | Hypoxia-inducible factor-1α |
| IPAH | Idiopathic PAH |
| IL | Interleukin |
| LTB4 | Leukotriene B4 |
| CCL3 | Macrophage inflammatory protein-1α |
| MHC | Major histocompatibility complex |
| MCs | Mast cells |
| MMP-9 | Matrix metallopeptidase 9 |
| MCTD | Mixed connective tissue diseases |
| MCT | Monocrotaline |
| mo-DCs | Monocyte-derived DCs |
| MPO | Myeloperoxidase |
| NIAD | National Institute of Allergy and Infectious Diseases |
| NK | Natural killer cells |
| NYHA | New York Heart Association |
| NF-κβ | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NFAT | Nuclear factor of activated T cell |
| Porto-pulmonary PAH | PAH due to liver disease |
| PPHN | Persistent pulmonary hypertension of the newborn |
| PlGF | Placental growth factor |
| pDCs | Plasmacytoid DCs |
| PAH | Pulmonary arterial hypertension |
| PAP | Pulmonary arterial pressures |
| PASMCs | Pulmonary arterial smooth muscle cells |
| PEA | Pulmonary endarterectomy |
| PH | Pulmonary hypertension |
| PVR | Pulmonary vascular resistance |
| PVOD | Pulmonary veno-occlusive disease |
| ROS | Reactive oxygen species |
| RELMα | Resistin-like molecule-α |
| RV | Right ventricular |
| RVH | Right ventricular hypertrophy |
| SU/Hx | Sugen/hypoxia |
| SLE | Systemic lupus erythematosus |
| SSc | Systemic sclerosis |
| TLOs | Tertiary lymphoid organs |
| Th | T-helper cell |
| TSP-1 | Thrombospondin-1 |
| TNFAIP3 | TNF Alpha Induced Protein 3 |
| NRF2 | Transcriptional regulator nuclear factor erythroid 2-related factor 2 |
| TGF-β | Transforming growth factor beta |
| TNF-α | Tumor necrosis factor alpha |
| cDC1s | Type 1 conventional DCs |
| cDC2s | Type 2 cDCs |
| VEGF | Vascular endothelial growth factor |
References
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Respir. J. 2015, 46, 903–975. [Google Scholar]
- Southgate, L.; Machado, R.D.; Graf, S.; Morrell, N.W. Molecular genetic framework underlying pulmonary arterial hypertension. Nat. Rev. Cardiol. 2019, 17, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Garg, L.; Akbar, G.; Agrawal, S.; Agarwal, M.; Khaddour, L.; Handa, R.; Garg, A.; Shah, M.; Patel, B.; Dalal, B.D. Drug-induced pulmonary arterial hypertension: A Review. Heart Fail. Rev. 2017, 22, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Sitbon, O.; Simonneau, G. Treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2004, 351, 1425–1436. [Google Scholar] [CrossRef]
- Benza, R.L.; Miller, D.P.; Barst, R.J.; Badesch, D.B.; Frost, A.E.; McGoon, A.D. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 2012, 142, 448–456. [Google Scholar] [CrossRef]
- Radegran, G.; Kjellstrom, B.; Ekmehag, B.; Larsen, F.; Rundqvist, B.; Blomquist, S.B.; Gustafsson, C.; Hesselstrand, R.; Karlsson, M.; Kornhall, B.; et al. Characteristics and survival of adult Swedish PAH and CTEPH patients 2000–2014. Scand. Cardiovasc. J. 2016, 50, 243–250. [Google Scholar] [CrossRef]
- Quadery, S.R.; Swift, A.J.; Billings, C.G.; Thompson, A.A.R.; Elliot, C.A.; Hurdman, J.; Charalampopoulos, A.; Sabroe, I.; Armstrong, I.J.; Hamilton, N.; et al. The impact of patient choice on survival in chronic thromboembolic pulmonary hypertension. Eur. Respir. J. 2018, 52. [Google Scholar] [CrossRef]
- Boucly, A.; Weatherald, J.; Savale, L.; Jais, X.; Cottin, V.; Prevot, G.; Picard, F.; de Groote, P.; Jevnikar, M.; Bergot, E.; et al. Risk assessment, prognosis and guideline implementation in pulmonary arterial hypertension. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [Green Version]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef]
- Matthews, D.T.; Hemnes, A.R. Current concepts in the pathogenesis of chronic thromboembolic pulmonary hypertension. Pulm. Circ. 2016, 6, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonderman, D.; Jakowitsch, J.; Adlbrecht, C.; Schemper, M.; Kyrle, P.A.; Schonauer, V.; Exner, M.; Klepetko, W.; Kneussl, M.P.; Maurer, G.; et al. Medical conditions increasing the risk of chronic thromboembolic pulmonary hypertension. Thromb. Haemost. 2005, 93, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res. 2017, 367, 643–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chazova, I.; Loyd, J.E.; Zhdanov, V.S.; Newman, J.H.; Belenkov, Y.; Meyrick, B. Pulmonary artery adventitial changes and venous involvement in primary pulmonary hypertension. Am. J. Pathol. 1995, 146, 389–397. [Google Scholar] [PubMed]
- Jonigk, D.; Golpon, H.; Bockmeyer, C.L.; Maegel, L.; Hoeper, M.M.; Gottlieb, J.; Nickel, N.; Hussein, K.; Maus, U.; Lehmann, U.; et al. Plexiform lesions in pulmonary arterial hypertension composition, architecture, and microenvironment. Am. J. Pathol. 2011, 179, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Wagenvoort, C.A. Pathology of pulmonary thromboembolism. Chest 1995, 107, 10S–17S. [Google Scholar] [CrossRef] [Green Version]
- Lang, I.M.; Dorfmuller, P.; Noordegraaf, A.V. The Pathobiology of Chronic Thromboembolic Pulmonary Hypertension. Ann. Am. Thorac. Soc. 2016, 13, S215–S221. [Google Scholar] [CrossRef]
- Perros, F.; Dorfmüller, P.; Montani, D.; Hammad, H.; Waelput, W.; Girerd, B.; Raymond, N.; Mercier, O.; Mussot, S.; Cohen-Kaminsky, S.; et al. Pulmonary lymphoid neogenesis in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 311–321. [Google Scholar] [CrossRef]
- Savai, R.; Pullamsetti, S.S.; Kolbe, J.; Bieniek, E.; Voswinckel, R.; Fink, L.; Scheed, A.; Ritter, C.; Dahal, B.K.; Vater, A.; et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 897–908. [Google Scholar] [CrossRef]
- Stacher, E.; Graham, B.B.; Hunt, J.M.; Gandjeva, A.; Groshong, S.D.; McLaughlin, V.V.; Jessup, M.; Grizzle, W.E.; Aldred, M.A.; Cool, C.D.; et al. Modern age pathology of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Quarck, R.; Wynants, M.; Verbeken, E.; Meyns, B.; Delcroix, M. Contribution of inflammation and impaired angiogenesis to the pathobiology of chronic thromboembolic pulmonary hypertension. Eur. Respir. J. 2015, 46, 431–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Kasmi, K.C.; Pugliese, S.C.; Riddle, S.R.; Poth, J.M.; Anderson, A.L.; Frid, M.G.; Li, M.; Pullamsetti, S.S.; Savai, R.; Nagel, M.A.; et al. Adventitial fibroblasts induce a distinct proinflammatory/profibrotic macrophage phenotype in pulmonary hypertension. J. Immunol. 2014, 193, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Groves, B.; Badesch, D.B.; Voelkel, N.F. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am. J. Pathol. 1994, 144, 275–285. [Google Scholar] [PubMed]
- Tan, S.Y.; Krasnow, M.A. Developmental origin of lung macrophage diversity. Development 2016, 143, 1318–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, M.; Giladi, A.; Gorki, A.D.; Solodkin, D.G.; Zada, M.; Hladik, A.; Miklosi, A.; Salame, T.M.; Halpern, K.B.; David, E.; et al. Lung Single-Cell Signaling Interaction Map Reveals Basophil Role in Macrophage Imprinting. Cell 2018, 175, 1031–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J.; et al. A molecular cell atlas of the human lung from single cell RNA sequencing. bioRxiv 2019. [Google Scholar] [CrossRef]
- Pugliese, S.C.; Kumar, S.; Janssen, W.J.; Graham, B.B.; Frid, M.G.; Riddle, S.R.; El Kasmi, K.C.; Stenmark, K.R. A Time- and Compartment-Specific Activation of Lung Macrophages in Hypoxic Pulmonary Hypertension. J. Immunol. 2017, 198, 4802–4812. [Google Scholar] [CrossRef]
- Franke-Ullmann, G.; Pförtner, C.; Walter, P.; Steinmüller, C.; Lohmann-Matthes, M.L.; Kobzik, L. Characterization of murine lung interstitial macrophages in comparison with alveolar macrophages in vitro. J. Immunol. 1996, 157, 3097–3104. [Google Scholar]
- Bedoret, D.; Wallemacq, H.; Marichal, T.; Desmet, C.; Quesada Calvo, F.; Henry, E.; Closset, R.; Dewals, B.; Thielen, C.; Gustin, P.; et al. Lung interstitial macrophages alter dendritic cell functions to prevent airway allergy in mice. J. Clin. Investig. 2009, 119, 3723–3738. [Google Scholar] [CrossRef]
- Laskin, D.L.; Weinberger, B.; Laskin, J.D. Functional heterogeneity in liver and lung macrophages. J. Leukoc. Biol. 2001, 70, 163–170. [Google Scholar]
- Florentin, J.; Coppin, E.; Vasamsetti, S.B.; Zhao, J.; Tai, Y.Y.; Tang, Y.; Zhang, Y.; Watson, A.; Sembrat, J.; Rojas, M.; et al. Inflammatory Macrophage Expansion in Pulmonary Hypertension Depends upon Mobilization of Blood-Borne Monocytes. J. Immunol. 2018, 200, 3612–3625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, O.; Marcos, E.; Perros, F.; Fadel, E.; Tu, L.; Humbert, M.; Dartevelle, P.; Simonneau, G.; Adnot, S.; Eddahibi, S. Role of endothelium-derived CC chemokine ligand 2 in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2007, 176, 1041–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abid, S.; Marcos, E.; Parpaleix, A.; Amsellem, V.; Breau, M.; Houssaini, A.; Vienney, N.; Lefevre, M.; Derumeaux, G.; Evans, S.; et al. CCR2/CCR5-mediated macrophage-smooth muscle cell crosstalk in pulmonary hypertension. Eur. Respir. J. 2019, 54, 1802308. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Mickael, C.; Kassa, B.; Sanders, L.; Hernandez-Saavedra, D.; E Koyanagi, D.; Kumar, S.; Pugliese, S.C.; Thomas, S.; McClendon, J.; et al. Interstitial Macrophage-Derived Thrombospondin-1 Contributes to Hypoxia-Induced Pulmonary Hypertension. Cardiovasc. Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Vergadi, E.; Chang, M.S.; Lee, C.; Liang, O.D.; Liu, X.; Fernandez-Gonzalez, A.; Mitsialis, A.; Kourembanas, S. Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation 2011, 123, 1986–1995. [Google Scholar] [CrossRef] [Green Version]
- Frid, M.G.; Brunetti, J.A.; Burke, D.L.; Carpenter, T.C.; Davie, N.J.; Reeves, J.T.; Roedersheimer, M.T.; Van Rooijen, N.; Stenmark, K.R. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am. J. Pathol. 2006, 168, 659–669. [Google Scholar] [CrossRef] [Green Version]
- Tian, W.; Jiang, X.; Tamosiuniene, R.; Sung, Y.K.; Qian, J.; Dhillon, G.; Gera, L.; Farkas, L.; Rabinovitch, M.; Zamanian, R.T.; et al. Bocking macrophage leukotriene b4 prevents endothelial injury and reverses pulmonary hypertension. Sci. Transl. Med. 2013, 5, 200ra117. [Google Scholar] [CrossRef] [Green Version]
- Tuder, R.M.; Chacon, M.; Alger, L.; Wang, J.; Taraseviciene-Stewart, L.; Kasahara, Y.; Cool, C.D.; Bishop, A.E.; Geraci, M.; Semenza, G.L.; et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: Evidence for a process of disordered angiogenesis. J. Pathol. 2001, 195, 367–374. [Google Scholar] [CrossRef]
- Kojima, H.; Tokunou, T.; Takahara, Y.; Sunagawa, K.; Hirooka, Y.; Ichiki, T.; Tsutsui, H. Hypoxia-inducible factor-1 alpha deletion in myeloid lineage attenuates hypoxia-induced pulmonary hypertension. Physiol. Rep. 2019, 7, e14025. [Google Scholar] [CrossRef] [Green Version]
- Marsh, L.M.; Jandl, K.; Grunig, G.; Foris, V.; Bashir, M.; Ghanim, B.; Klepetko, W.; Olschewski, H.; Olschewski, A.; Kwapiszewska, G. The inflammatory cell landscape in the lungs of patients with idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2018, 51, 1701214. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Fan, C.; Skinner, J.T.; Hunter, E.N.; Macdonald, A.A.; Illei, P.B.; Yamaji-Kegan, K.; Johns, R.A. RELMalpha Licenses Macrophages for Damage-Associated Molecular Pattern Activation to Instigate Pulmonary Vascular Remodeling. J. Immunol. 2019, 203, 2862–2871. [Google Scholar] [CrossRef] [PubMed]
- Zabini, D.; Heinemann, A.; Foris, V.; Nagaraj, C.; Nierlich, P.; Balint, Z.; Kwapiszewska, G.; Lang, I.M.; Klepetko, W.; Olschewski, H.; et al. Comprehensive analysis of inflammatory markers in chronic thromboembolic pulmonary hypertension patients. Eur. Respir. J. 2014, 44, 951–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florentin, J.; Dutta, P. Origin and production of inflammatory perivascular macrophages in pulmonary hypertension. Cytokine 2017, 100, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Pullamsetti, S.S.; Savai, R. Macrophage Regulation during Vascular Remodeling: Implications for Pulmonary Hypertension Therapy. Am. J. Respir. Cell Mol. Biol. 2017, 56, 556–558. [Google Scholar] [CrossRef] [PubMed]
- Ozpelit, E.; Akdeniz, B.; Özpelit, M.E.; Tas, S.; Bozkurt, S.; Tertemiz, K.C.; Sevinç, C.; Badak, Ö. Prognostic value of neutrophil-to-lymphocyte ratio in pulmonary arterial hypertension. J. Int. Med. Res. 2015, 43, 661–671. [Google Scholar] [CrossRef]
- Harbaum, L.; Baaske, K.M.; Simon, M.; Oqueka, T.; Sinning, C.; Glatzel, A.; Lüneburg, N.; Sydow, K.; Bokemeyer, C.; Klose, H. Exploratory analysis of the neutrophil to lymphocyte ratio in patients with pulmonary arterial hypertension. BMC Pulm. Med. 2017, 17, 72. [Google Scholar] [CrossRef] [Green Version]
- Yanartaş, M.; Kalkan, M.E.; Arslan, A.; Tas, S.G.; Koksal, C.; Bekiroğlu, N.; Yildizeli, B. Neutrophil/Lymphocyte Ratio Can Predict Postoperative Mortality in Patients with Chronic Thromboembolic Pulmonary Hypertension. Ann. Thorac. Cardiovasc. Surg. 2015, 21, 229–235. [Google Scholar] [CrossRef] [Green Version]
- Schultze, A.E.; Wagner, J.G.; White, S.M.; Roth, R.A. Early indications of monocrotaline pyrrole-induced lung injury in rats. Toxicol. Appl. Pharmacol. 1991, 109, 41–50. [Google Scholar] [CrossRef]
- Klinke, A.; Berghausen, E.; Friedrichs, K.; Molz, S.; Lau, D.; Remane, L.; Berlin, M.; Kaltwasser, C.; Adam, M.; Mehrkens, D.; et al. Myeloperoxidase aggravates pulmonary arterial hypertension by activation of vascular Rho-kinase. JCI Insight 2018, 3, e97530. [Google Scholar] [CrossRef]
- Theoharides, T.C. Mast cells and pancreatic cancer. N. Engl. J. Med. 2008, 358, 1860–1861. [Google Scholar] [CrossRef] [Green Version]
- Coussens, L.M.; Raymond, W.W.; Bergers, G.; Laig-Webster, M.; Behrendtsen, O.; Werb, Z.; Caughey, G.H.; Hanahan, D. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999, 13, 1382–1397. [Google Scholar] [CrossRef] [PubMed]
- Mitani, Y.; Ueda, M.; Maruyama, K.; Shimpo, H.; Kojima, A.; Matsumura, M.; Aoki, K.; Sakurai, M. Mast cell chymase in pulmonary hypertension. Thorax 1999, 54, 88–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heath, D.; Yacoub, M. Lung mast cells in plexogenic pulmonary arteriopathy. J. Clin. Pathol. 1991, 44, 1003–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilfillan, A.M.; Rivera, J. The tyrosine kinase network regulating mast cell activation. Immunol. Rev. 2009, 228, 149–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, J.; Yin, J.; Kukucka, M.; Yin, N.; Saarikko, I.; Sterner-Kock, A.; Fujii, H.; Leong-Poi, H.; Kuppe, H.; Schermuly, R.T.; et al. Mast cells promote lung vascular remodelling in pulmonary hypertension. Eur. Respir. J. 2011, 37, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Bartelds, B.; Van Loon, R.L.E.; Mohaupt, S.; Wijnberg, H.; Dickinson, M.G.; Boersma, B.; Takens, J.; Van Albada, M.; Berger, R.M. Mast cell inhibition improves pulmonary vascular remodeling in pulmonary hypertension. Chest 2012, 141, 651–660. [Google Scholar] [CrossRef]
- Dahal, B.K.; Kosanovic, D.; Kaulen, C.; Cornitescu, T.; Savai, R.; Hoffmann, J.; Reiss, I.K.M.; Ghofrani, A.; Weissmann, N.; Kuebler, W.M.; et al. Involvement of mast cells in monocrotaline-induced pulmonary hypertension in rats. Respir. Res. 2011, 12, 60. [Google Scholar] [CrossRef] [Green Version]
- Banasova, A.; Maxova, H.; Hampl, V.; Vizek, M.; Povysilova, V.; Novotna, J.; Vajnerova, O.; Hnilickova, O.; Herget, J. Prevention of mast cell degranulation by disodium cromoglycate attenuates the development of hypoxic pulmonary hypertension in rats exposed to chronic hypoxia. Respiration 2008, 76, 102–107. [Google Scholar]
- Farha, S.; Sharp, J.; Asosingh, K.; Park, M.; Comhair, S.A.; Tang, W.H.; Thomas, J.; Farver, C.; Hsieh, F.; Loyd, J.E.; et al. Mast cell number, phenotype, and function in human pulmonary arterial hypertension. Pulm. Circ. 2012, 2, 220–228. [Google Scholar] [CrossRef] [Green Version]
- Hamada, H.; Terai, M.; Kimura, H.; Hirano, K.; Oana, S.; Niimi, H. Increased expression of mast cell chymase in the lungs of patients with congenital heart disease associated with early pulmonary vascular disease. Am. J. Respir. Crit. Care Med. 1999, 160, 1303–1308. [Google Scholar] [CrossRef]
- Doggrell, S.A.; Wanstall, J.C. Vascular chymase: Pathophysiological role and therapeutic potential of inhibition. Cardiovasc. Res. 2004, 61, 653–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosanovic, D.; Luitel, H.; Dahal, B.K.; Cornitescu, T.; Janssen, W.; Danser, A.H.; Garrelds, I.M.; De Mey, J.G.; Fazzi, G.; Schiffers, P.; et al. Chymase: A multifunctional player in pulmonary hypertension associated with lung fibrosis. Eur. Respir. J. 2015, 46, 1084–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, D.J.; Thakker-Varia, S.; Wilson, F.J.; Poiani, G.J.; Tozzi, C.A. Role of proteolysis and apoptosis in regression of pulmonary vascular remodeling. Physiol. Res. 2000, 49, 577–585. [Google Scholar] [PubMed]
- Xu, J.; Wang, J.; Shao, C.; Zeng, X.; Sun, L.; Kong, H.; Xie, W.; Wang, H. New dynamic viewing of mast cells in pulmonary arterial hypertension (PAH): Contributors or outsiders to cardiovascular remodeling. J. Thorac. Dis. 2018, 10, 3016–3026. [Google Scholar] [CrossRef] [PubMed]
- Ormiston, M.L.; Chang, C.; Long, L.L.; Soon, E.; Jones, D.; Machado, R.; Treacy, C.; Toshner, M.R.; Campbell, K.; Riding, A.; et al. Impaired natural killer cell phenotype and function in idiopathic and heritable pulmonary arterial hypertension. Circulation 2012, 126, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Ratsep, M.T.; Moore, S.D.; Jafri, S.; Mitchell, M.; Brady, H.J.M.; Mandelboim, O.; Southwood, M.; Morrell, N.W.; Colucci, F.; Ormiston, M.L. Spontaneous pulmonary hypertension in genetic mouse models of natural killer cell deficiency. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, L977–L990. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, K.L.; Reizis, B. Dendritic cells: Arbiters of immunity and immunological tolerance. Cold Spring Harb. Perspect. Biol. 2012, 4, a007401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganguly, D.; Haak, S.; Sisirak, V.; Reizis, B. The role of dendritic cells in autoimmunity. Nat. Rev. Immunol. 2013, 13, 566–577. [Google Scholar] [CrossRef] [Green Version]
- Blanco, P.; Palucka, A.K.; Pascual, V.; Banchereau, J. Dendritic cells and cytokines in human inflammatory and autoimmune diseases. Cytokine Growth Factor Rev. 2008, 19, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Kool, M.; van Loo, G.; Waelput, W.; De Prijck, S.; Muskens, F.; Sze, M.; van Praet, J.; Branco-Madeira, F.; Janssens, S.; Reizis, B.; et al. The ubiquitin-editing protein A20 prevents dendritic cell activation, recognition of apoptotic cells, and systemic autoimmunity. Immunity 2011, 35, 82–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Uden, D.; Boomars, K.; Kool, M. Dendritic Cell Subsets and Effector Function in Idiopathic and Connective Tissue Disease-Associated Pulmonary Arterial Hypertension. Front. Immunol. 2019, 10, 11. [Google Scholar] [CrossRef] [PubMed]
- Perros, F.; Dorfmüller, P.; Souza, R.; Durand-Gasselin, I.; Mussot, S.; Mazmanian, M.; Hervé, P.; Emilie, D.; Simonneau, G.; Humbert, M. Dendritic cell recruitment in lesions of human and experimental pulmonary hypertension. Eur. Respir. J. 2007, 29, 462–468. [Google Scholar] [CrossRef] [Green Version]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yan, H.; Zhu, W.; Cui, Y.; Chen, J.; Wang, X.; Li, S.; Zhu, J. Impairment of monocyte-derived dendritic cells in idiopathic pulmonary arterial hypertension. J. Clin. Immunol. 2009, 29, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Mauad, T.; Pozzan, G.; Lancas, T.; Overbeek, M.J.; Souza, R.; Jardim, C.; Dolhnikoff, M.; Mello, G.; Pires-Neto, R.C.; Bernardi Fdel, C.; et al. Immunopathological aspects of schistosomiasis-associated pulmonary arterial hypertension. J. Infect. 2014, 68, 90–98. [Google Scholar] [CrossRef]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23-IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef]
- Hashimoto-Kataoka, T.; Hosen, N.; Sonobe, T.; Arita, Y.; Yasui, T.; Masaki, T.; Minami, M.; Inagaki, T.; Miyagawa, S.; Sawa, Y.; et al. Interleukin-6/interleukin-21 signaling axis is critical in the pathogenesis of pulmonary arterial hypertension. Proc. Natl. Acad. Sci. USA 2015, 112, E2677–E2686. [Google Scholar] [CrossRef] [Green Version]
- Van Hamburg, J.P.; Tas, S.W. Molecular mechanisms underpinning T helper 17 cell heterogeneity and functions in rheumatoid arthritis. J. Autoimmun. 2018, 87, 69–81. [Google Scholar] [CrossRef]
- Soon, E.; Holmes, A.M.; Treacy, C.M.; Doughty, N.J.; Southgate, L.; Machado, R.D.; Trembath, R.C.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010, 122, 920–927. [Google Scholar] [CrossRef] [Green Version]
- Gaowa, S.; Zhou, W.; Yu, L.; Zhou, X.; Liao, K.; Yang, K.; Lu, Z.; Jiang, H.; Chen, X. Effect of Th17 and Treg axis disorder on outcomes of pulmonary arterial hypertension in connective tissue diseases. Mediat. Inflamm. 2014, 2014, 247372. [Google Scholar] [CrossRef] [PubMed]
- Vroman, H.; Bergen, I.M.; Van Hulst, J.A.; Van Nimwegen, M.; Van Uden, D.; Schuijs, M.J.; Pillai, S.Y.; Van Loo, G.; Hammad, H.; Lambrecht, B.N.; et al. TNF-alpha-induced protein 3 levels in lung dendritic cells instruct TH2 or TH17 cell differentiation in eosinophilic or neutrophilic asthma. J. Allergy Clin. Immunol. 2018, 141, 1620–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitzalis, C.; Jones, G.W.; Bombardieri, M.; Jones, S.A. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat. Rev. Immunol. 2014, 14, 447–462. [Google Scholar] [CrossRef] [PubMed]
- De Bourcy, C.F.A.; Dekker, C.L.; Davis, M.M.; Nicolls, M.R.; Quake, S.R. Dynamics of the human antibody repertoire after B cell depletion in systemic sclerosis. Sci. Immunol. 2017, 2, eaan8289. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, S.; Taraseviciene-Stewart, L.; Huber, L.C.; Speich, R.; Voelkel, N.F. Peripheral blood B lymphocytes derived from patients with idiopathic pulmonary arterial hypertension express a different RNA pattern compared with healthy controls: A cross sectional study. Respir. Res. 2008, 9, 20. [Google Scholar] [CrossRef] [Green Version]
- Blum, L.K.; Cao, R.R.L.; Sweatt, A.J.; Bill, M.; Lahey, L.J.; Hsi, A.C.; Lee, C.S.; Kongpachith, S.; Ju, C.H.; Mao, R.; et al. Circulating plasmablasts are elevated and produce pathogenic anti-endothelial cell autoantibodies in idiopathic pulmonary arterial hypertension. Eur. J. Immunol. 2018, 48, 874–884. [Google Scholar] [CrossRef]
- Rich, S.; Kieras, K.; Hart, K.; Groves, B.M.; Stobo, J.D.; Brundage, B.H. Antinuclear antibodies in primary pulmonary hypertension. J. Am. Coll. Cardiol. 1986, 8, 1307–1311. [Google Scholar] [CrossRef] [Green Version]
- Arends, S.J.; Damoiseaux, J.; Duijvestijn, A.; Debrus-Palmans, L.; Boomars, K.; Broers, B.; Tervaert, J.W.; van Paassen, P. Prevalence of anti-endothelial cell antibodies in idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2010, 35, 923–925. [Google Scholar] [CrossRef] [Green Version]
- Dib, H.; Tamby, M.C.; Bussone, G.; Regent, A.; Berezne, A.; Lafine, C.; Broussard, C.; Simonneau, G.; Guillevin, L.; Witko-Sarsat, V.; et al. Targets of anti-endothelial cell antibodies in pulmonary hypertension and scleroderma. Eur. Respir. J. 2012, 39, 1405–1414. [Google Scholar] [CrossRef] [Green Version]
- Tamby, M.C.; Chanseaud, Y.; Humbert, M.; Fermanian, J.; Guilpain, P.; Garcia-de-la-Pena-Lefebvre, P.; Brunet, S.; Servettaz, A.; Weill, B.; Simonneau, G.; et al. Anti-endothelial cell antibodies in idiopathic and systemic sclerosis associated pulmonary arterial hypertension. Thorax 2005, 60, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Arends, S.J.; Damoiseaux, J.G.M.C.; Duijvestijn, A.M.; Debrus-Palmans, L.; Vroomen, M.; Boomars, K.A.; Rocca, H.-P.B.-L.; Reutelingsperger, C.P.M.; Tervaert, J.W.C.; Van Paassen, P. Immunoglobulin G anti-endothelial cell antibodies: Inducers of endothelial cell apoptosis in pulmonary arterial hypertension? Clin. Exp. Immunol. 2013, 174, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.O.; Kill, A.; Kutsche, M.; Guenther, J.; Rose, A.; Tabeling, C.; Witzenrath, M.; Kühl, A.A.; Heidecke, H.; Ghofrani, A.; et al. Vascular receptor autoantibodies in pulmonary arterial hypertension associated with systemic sclerosis. Am. J. Respir. Crit. Care Med. 2014, 190, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Colvin, K.L.; Cripe, P.J.; Ivy, D.D.; Stenmark, K.R.; Yeager, M.E. Bronchus-associated lymphoid tissue in pulmonary hypertension produces pathologic autoantibodies. Am. J. Respir. Crit. Care Med. 2013, 188, 1126–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cracowski, J.L.; Chabot, F.; Labarere, J.; Faure, P.; Degano, B.; Schwebel, C.; Chaouat, A.; Reynaud-Gaubert, M.; Cracowski, C.; Sitbon, O.; et al. Proinflammatory cytokine levels are linked to death in pulmonary arterial hypertension. Eur. Respir. J. 2014, 43, 915–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Säleby, J.; Bouzina, H.; Lundgren, J.; Rådegran, G. Angiogenic and inflammatory biomarkers in the differentiation of pulmonary hypertension. Scand. Cardiovasc. J. 2017, 51, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Okada, O.; Tanabe, N.; Tanaka, Y.; Terai, M.; Takiguchi, Y.; Masuda, M.; Nakajima, N.; Hiroshima, K.; Inadera, H.; et al. Plasma monocyte chemoattractant protein-1 and pulmonary vascular resistance in chronic thromboembolic pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2001, 164, 319–324. [Google Scholar] [CrossRef]
- Langer, F.; Schramm, R.; Bauer, M.; Tscholl, D.; Kunihara, T.; Schäfers, H.-J. Cytokine response to pulmonary thromboendarterectomy. Chest 2004, 126, 135–141. [Google Scholar] [CrossRef]
- Soon, E.; Holmes, A.; Treacy, C.; Barker, L.; Suntharalingam, J.; Southwood, M.; Nicklin, P.; Walker, C.; Budd, D.; Jenkins, D.; et al. Inflammatory cytokines are elevated in patients with operable chronic thromboembolic pulmonary hypertension and predict outcome post-endarterectomy. Thorax 2010, 65, A45. [Google Scholar] [CrossRef] [Green Version]
- Kylhammar, D.; Hesselstrand, R.; Nielsen, S.; Scheele, C.; Rådegran, G. Angiogenic and inflammatory biomarkers for screening and follow-up in patients with pulmonary arterial hypertension. Scand. J. Rheumatol. 2018, 47, 319–324. [Google Scholar] [CrossRef]
- Low, A.; George, S.; Howard, L.; Bell, N.; Millar, A.; Tulloh, R. Lung Function, Inflammation, and Endothelin-1 in Congenital Heart Disease-Associated Pulmonary Arterial Hypertension. J. Am. Heart Assoc. 2018, 7, e007249. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.A.; Davey, R.; Rao, Y.; Shen, K.; Benza, R.L.; Raina, A. Association between cytokines and functional, hemodynamic parameters, and clinical outcomes in pulmonary arterial hypertension. Pulm. Circ. 2018, 8, 2045894018794051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorfmüller, P.; Zarka, V.; Durand-Gasselin, I.; Monti, G.; Balabanian, K.; Garcia, G.; Capron, F.; Coulomb-Lhermine, A.; Marfaing-Koka, A.; Simonneau, G.; et al. Chemokine RANTES in severe pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2002, 165, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Steiner, M.K.; Syrkina, O.L.; Kolliputi, N.; Mark, E.J.; Hales, C.A.; Waxman, A.B. Interleukin-6 overexpression induces pulmonary hypertension. Circ. Res. 2009, 104, 236–244. [Google Scholar] [CrossRef]
- Golembeski, S.M.; West, J.; Tada, Y.; Fagan, K.A. Interleukin-6 causes mild pulmonary hypertension and augments hypoxia-induced pulmonary hypertension in mice. Chest 2005, 128, 572S–573S. [Google Scholar] [CrossRef] [PubMed]
- Miyata, M.; Sakuma, F.; Yoshimura, A.; Ishikawa, H.; Nishimaki, T.; Kasukawa, R. Pulmonary hypertension in rats. 2. Role of interleukin-6. Int. Arch. Allergy Immunol. 1995, 108, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Savale, L.; Tu, L.; Rideau, D.; Izziki, M.; Maitre, B.; Adnot, S.; Eddahibi, S. Impact of interleukin-6 on hypoxia-induced pulmonary hypertension and lung inflammation in mice. Respir. Res. 2009, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- Matura, L.A.; Ventetuolo, C.E.; Palevsky, H.I.; Lederer, D.J.; Horn, E.M.; Mathai, S.C.; Pinder, D.; Archer-Chicko, C.; Bagiella, E.; Roberts, K.E.; et al. Interleukin-6 and tumor necrosis factor-alpha are associated with quality of life-related symptoms in pulmonary arterial hypertension. Ann. Am. Thorac. Soc. 2015, 12, 370–375. [Google Scholar] [CrossRef] [Green Version]
- Sweatt, A.J.; Hedlin, H.K.; Balasubramanian, V.; Hsi, A.; Blum, L.K.; Robinson, W.H.; Haddad, F.; Hickey, P.M.; Condliffe, R.; Lawrie, A.; et al. Discovery of Distinct Immune Phenotypes Using Machine Learning in Pulmonary Arterial Hypertension. Circ. Res. 2019, 124, 904–919. [Google Scholar] [CrossRef] [Green Version]
- Fujita, M.; Mason, R.J.; Cool, C.; Shannon, J.M.; Hara, N.; Fagan, K.A. Pulmonary hypertension in TNF-alpha-overexpressing mice is associated with decreased VEGF gene expression. J. Appl. Physiol. 2002, 93, 2162–2170. [Google Scholar] [CrossRef] [Green Version]
- Price, L.C.; Montani, D.; Tcherakian, C.; Dorfmuller, P.; Souza, R.; Gambaryan, N.; Chaumais, M.C.; Shao, D.M.; Simonneau, G.; Howard, L.S.; et al. Dexamethasone reverses monocrotaline-induced pulmonary arterial hypertension in rats. Eur. Respir. J. 2011, 37, 813–822. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, C.; Takahashi, M.; Morimoto, H.; Izawa, A.; Ise, H.; Hongo, M.; Hoshikawa, Y.; Ito, T.; Miyashita, H.; Kobayashi, E.; et al. Mycophenolate mofetil attenuates pulmonary arterial hypertension in rats. Biochem. Biophys. Res. Commun. 2006, 349, 781–788. [Google Scholar] [CrossRef]
- Bonnet, S.; Rochefort, G.Y.; Sutendra, G.; Archer, S.L.; Haromy, A.; Webster, L.; Hashimoto, K.; Bonnet, S.N.; Michelakis, E.D. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc. Natl. Acad. Sci. USA 2007, 104, 11418–11423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jais, X.; Launay, D.; Yaici, A.; Le Pavec, J.; Tcherakian, C.; Sitbon, O.; Simonneau, G.; Humbert, M. Immunosuppressive therapy in lupus- and mixed connective tissue disease-associated pulmonary arterial hypertension: A retrospective analysis of twenty-three cases. Arthritis Rheum. 2008, 58, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Voelkel, N.F.; Tuder, R.M.; Bridges, J.; Arend, W.P. Interleukin-1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline. Am. J. Respir. Cell Mol. Biol. 1994, 11, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Phan, C.; Tu, L.; Le Hiress, M.; Thuillet, R.; Jutant, E.M.; Fadel, E.; Savale, L.; Huertas, A.; Humbert, M.; et al. Ectopic upregulation of membrane-bound IL6R drives vascular remodeling in pulmonary arterial hypertension. J. Clin. Investig. 2018, 128, 1956–1970. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Sánchez, J.; Harlow, L.; Church, C.; Gaine, S.; Knightbridge, E.; Bunclark, K.; Gor, D.; Bedding, A.; Morrell, N.; A Corris, P.; et al. Clinical trial protocol for TRANSFORM-UK: A therapeutic open-label study of tocilizumab in the treatment of pulmonary arterial hypertension. Pulm. Circ. 2018, 8, 2045893217735820. [Google Scholar] [CrossRef] [Green Version]
- Roeleveld, D.M.; Marijnissen, R.J.; Walgreen, B.; Helsen, M.M.; van den Bersselaar, L.; van de Loo, F.A.; van Lent, P.L.; van der Kraan, P.M.; van den Berg, W.B.; Koenders, M.I. Higher efficacy of anti-IL-6/IL-21 combination therapy compared to monotherapy in the induction phase of Th17-driven experimental arthritis. PLoS ONE 2017, 12, e0171757. [Google Scholar] [CrossRef]
- Wang, Q.; Zuo, X.R.; Wang, Y.Y.; Xie, W.P.; Wang, H.; Zhang, M. Monocrotaline-induced pulmonary arterial hypertension is attenuated by TNF-alpha antagonists via the suppression of TNF-alpha expression and NF-kappaB pathway in rats. Vascul. Pharmacol. 2013, 58, 71–77. [Google Scholar] [CrossRef]
- Zhang, L.L.; Lu, J.; Li, M.T.; Wang, Q.; Zeng, X.F. Preventive and remedial application of etanercept attenuate monocrotaline-induced pulmonary arterial hypertension. Int. J. Rheum. Dis. 2016, 19, 192–198. [Google Scholar] [CrossRef]
- Hurst, L.A.; Dunmore, B.J.; Long, L.; Crosby, A.; Al-Lamki, R.; Deighton, J.; Southwood, M.; Yang, X.; Nikolic, M.Z.; Herrera, B.; et al. TNFalpha drives pulmonary arterial hypertension by suppressing the BMP type-II receptor and altering NOTCH signalling. Nat. Commun. 2017, 8, 14079. [Google Scholar] [CrossRef]
- Mutschler, D.; Wikström, G.; Lind, L.; Larsson, A.; Lagrange, A.; Eriksson, M. Etanercept reduces late endotoxin-induced pulmonary hypertension in the pig. J. Interferon Cytokine Res. 2006, 26, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Pousada, G.; Lupo, V.; Castro-Sánchez, S.; Álvarez-Satta, M.; Sánchez-Monteagudo, A.; Baloira, A.; Espinós, C.; Valverde, D. Molecular and functional characterization of the BMPR2 gene in Pulmonary Arterial Hypertension. Sci. Rep. 2017, 7, 1923. [Google Scholar] [CrossRef] [PubMed]
- Rol, N.; Kurakula, K.B.; Happe, C.; Bogaard, H.J.; Goumans, M.J. TGF-beta and BMPR2 Signaling in PAH: Two Black Sheep in One Family. Int. J. Mol. Sci. 2018, 19, 2585. [Google Scholar] [CrossRef] [Green Version]
- Evans, J.D.; Girerd, B.; Montani, D.; Wang, X.J.; Galie, N.; Austin, E.D.; Elliott, G.; Asano, K.; Grunig, E.; Yan, Y.; et al. BMPR2 mutations and survival in pulmonary arterial hypertension: An individual participant data meta-analysis. Lancet Respir. Med. 2016, 4, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Nikolic, I.; Yung, L.M.; Yang, P.; Malhotra, R.; Paskin-Flerlage, S.D.; Dinter, T.; Bocobo, G.A.; Tumelty, K.E.; Faugno, A.J.; Troncone, L.; et al. Bone Morphogenetic Protein 9 Is a Mechanistic Biomarker of Portopulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2019, 199, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Sawada, H.; Saito, T.; Nickel, N.P.; Alastalo, T.P.; Glotzbach, J.P.; Chan, R.; Haghighat, L.; Fuchs, G.; Januszyk, M.; Cao, A.; et al. Reduced BMPR2 expression induces GM-CSF translation and macrophage recruitment in humans and mice to exacerbate pulmonary hypertension. J. Exp. Med. 2014, 211, 263–280. [Google Scholar] [CrossRef] [Green Version]
- Hensley, M.K.; Levine, A.; Gladwin, M.T.; Lai, Y.C. Emerging therapeutics in pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L769–L781. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, A.M.; Xia, W.; Holmes, M.D.; Hodge, S.; Danilov, S.; Curiel, D.T.; Morrell, N.W.; Reynolds, P.N. Bone morphogenetic protein type 2 receptor gene therapy attenuates hypoxic pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L1182–L1192. [Google Scholar] [CrossRef] [Green Version]
- Long, L.; Ormiston, M.; Yang, X.; Southwood, M.; Gräf, S.; Machado, R.D.; Mueller, M.; Kinzel, B.; Yung, L.M.; Wilkinson, J.M.; et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat. Med. 2015, 21, 777–785. [Google Scholar] [CrossRef] [Green Version]
- Morrell, N.W.; Yang, X.; Upton, P.D.; Jourdan, K.B.; Morgan, N.; Sheares, K.K.; Trembath, R.C. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-beta(1) and bone morphogenetic proteins. Circulation 2001, 104, 790–795. [Google Scholar] [CrossRef] [Green Version]
- Long, L.; Crosby, A.; Yang, X.; Southwood, M.; Upton, P.D.; Kim, D.K.; Morrell, N.W. Altered bone morphogenetic protein and transforming growth factor-beta signaling in rat models of pulmonary hypertension: Potential for activin receptor-like kinase-5 inhibition in prevention and progression of disease. Circulation 2009, 119, 566–576. [Google Scholar] [CrossRef] [Green Version]
- Yung, L.M.; Nikolic, I.; Paskin-Flerlage, S.D.; Pearsall, R.S.; Kumar, R.; Yu, P.B. A Selective Transforming Growth Factor-beta Ligand Trap Attenuates Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2016, 194, 1140–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiekerkoetter, E.; Tian, X.; Cai, J.; Hopper, R.K.; Sudheendra, D.; Li, C.G.; El-Bizri, N.; Sawada, H.; Haghighat, R.; Chan, R.; et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J. Clin. Investig. 2013, 123, 3600–3613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiekerkoetter, E.; Sung, Y.K.; Sudheendra, D.; Scott, V.; Del Rosario, P.; Bill, M.; Haddad, F.; Long-Boyle, J.; Hedlin, H.; Zamanian, R.T. Randomised placebo-controlled safety and tolerability trial of FK506 (tacrolimus) for pulmonary arterial hypertension. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-kappaB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, S.; Egashira, K.; Chen, L.; Nakano, K.; Iwata, E.; Miyagawa, M.; Tsujimoto, H.; Hara, K.; Morishita, R.; Sueishi, K.; et al. Nanoparticle-mediated delivery of nuclear factor kappaB decoy into lungs ameliorates monocrotaline-induced pulmonary arterial hypertension. Hypertension 2009, 53, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Kaminski, P.M.; Edwards, J.G.; Yeh, A.; Wolin, M.S.; Frishman, W.H.; Gewitz, M.H.; Mathew, R. Pyrrolidine dithiocarbamate restores endothelial cell membrane integrity and attenuates monocrotaline-induced pulmonary artery hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L1250–L1259. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wei, C.; Kim, I.K.; Janssen-Heininger, Y.; Gupta, S. Inhibition of nuclear factor-kappaB in the lungs prevents monocrotaline-induced pulmonary hypertension in mice. Hypertension 2014, 63, 1260–1269. [Google Scholar] [CrossRef] [Green Version]
- Farkas, D.; Alhussaini, A.A.; Kraskauskas, D.; Kraskauskiene, V.; Cool, C.D.; Nicolls, M.R.; Natarajan, R.; Farkas, L. Nuclear factor kappaB inhibition reduces lung vascular lumen obliteration in severe pulmonary hypertension in rats. Am. J. Respir. Cell Mol. Biol. 2014, 51, 413–425. [Google Scholar] [CrossRef]
- Kumar, S.; Wei, C.; Thomas, C.M.; Kim, I.K.; Seqqat, R.; Kumar, R.; Baker, K.M.; Jones, W.K.; Gupta, S. Cardiac-specific genetic inhibition of nuclear factor-kappaB prevents right ventricular hypertrophy induced by monocrotaline. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1655–H1666. [Google Scholar] [CrossRef] [Green Version]
- Price, L.C.; Caramori, G.; Perros, F.; Meng, C.; Gambaryan, N.; Dorfmuller, P.; Montani, D.; Casolari, P.; Zhu, J.; Dimopoulos, K.; et al. Nuclear factor kappa-B is activated in the pulmonary vessels of patients with end-stage idiopathic pulmonary arterial hypertension. PLoS ONE 2013, 8, e75415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.Y.; Yang, Y.X.; Zhe, H.; He, Z.X.; Zhou, S.F. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: An update on its pharmacokinetic and pharmacodynamic properties. Drug Des. Devel. Ther. 2014, 8, 2075–2088. [Google Scholar] [PubMed] [Green Version]
- Heiss, E.H.; Schachner, D.; Werner, E.R.; Dirsch, V.M. Active NF-E2-related factor (Nrf2) contributes to keep endothelial NO synthase (eNOS) in the coupled state: Role of reactive oxygen species (ROS), eNOS, and heme oxygenase (HO-1) levels. J. Biol. Chem. 2009, 284, 31579–31586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.Y.; Rubin, L.J. Metabolic dysfunction in pulmonary hypertension: From basic science to clinical practice. Eur. Respir. Rev. 2017, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.; Hayes, J. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Khor, T.O.; Xu, C.; Shen, G.; Jeong, W.-S.; Yu, S.; Kong, A.-N. Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem. Pharmacol. 2008, 76, 1485–1489. [Google Scholar] [CrossRef] [Green Version]
- Grzegorzewska, A.P.; Seta, F.; Han, R.; Czajka, C.A.; Makino, K.; Stawski, L.; Isenberg, J.S.; Browning, J.L.; Trojanowska, M. Dimethyl Fumarate ameliorates pulmonary arterial hypertension and lung fibrosis by targeting multiple pathways. Sci. Rep. 2017, 7, 41605. [Google Scholar] [CrossRef] [Green Version]
- Hennigan, S.; Channick, R.N.; Silverman, G.J. Rituximab treatment of pulmonary arterial hypertension associated with systemic lupus erythematosus: A case report. Lupus 2008, 17, 754–756. [Google Scholar] [CrossRef]
- Samuelsson, B.; Dahlen, S.E.; Lindgren, J.A.; Rouzer, C.A.; Serhan, C.N. Leukotrienes and lipoxins: Structures, biosynthesis, and biological effects. Science 1987, 237, 1171–1176. [Google Scholar] [CrossRef]
- Tian, W.; Jiang, X.; Sung, Y.K.; Qian, J.; Yuan, K.; Nicolls, M.R. Leukotrienes in pulmonary arterial hypertension. Immunol. Res. 2014, 58, 387–393. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Immunohistopathology in IPAH and CTEPH. Left: schematic overview of cells involved in tertiary lymphoid organs (TLOs) in idiopathic pulmonary arterial hypertension (IPAH) patients. In the pulmonary hypertension (PH) situation, endothelial hyperproliferation is visible in the tunica intima with plexiform lesion formation in the lumen of the artery. Furthermore, smooth muscle cell (SMC) hyperplasia is visible in the tunica media of the pulmonary artery. Surrounding the tunica adventitia is a combination of B cells, T cells, mast cells, dendritic cells, neutrophils and macrophages. Right: schematic overview of vascular remodeling and inflammation in the thrombotic material of chronic thromboembolic pulmonary hypertension (CTEPH) patients. Between the (neo)intimal vascular wall and the tunica media, an influx of inflammatory cells such as B cells, T cells, neutrophils and macrophages is visible. Moreover, the enhanced presence of pro-inflammatory mediators, such as interleukin (IL)-6, IL-8, chemokine (C-C motif) ligands 2 and 3 (CCL2 and CCL3), C-X-C motif chemokines 10 and 12 (CXCL10 and CXCL12) is present.
Figure 1.
Immunohistopathology in IPAH and CTEPH. Left: schematic overview of cells involved in tertiary lymphoid organs (TLOs) in idiopathic pulmonary arterial hypertension (IPAH) patients. In the pulmonary hypertension (PH) situation, endothelial hyperproliferation is visible in the tunica intima with plexiform lesion formation in the lumen of the artery. Furthermore, smooth muscle cell (SMC) hyperplasia is visible in the tunica media of the pulmonary artery. Surrounding the tunica adventitia is a combination of B cells, T cells, mast cells, dendritic cells, neutrophils and macrophages. Right: schematic overview of vascular remodeling and inflammation in the thrombotic material of chronic thromboembolic pulmonary hypertension (CTEPH) patients. Between the (neo)intimal vascular wall and the tunica media, an influx of inflammatory cells such as B cells, T cells, neutrophils and macrophages is visible. Moreover, the enhanced presence of pro-inflammatory mediators, such as interleukin (IL)-6, IL-8, chemokine (C-C motif) ligands 2 and 3 (CCL2 and CCL3), C-X-C motif chemokines 10 and 12 (CXCL10 and CXCL12) is present.

Figure 2.
Immunomodulatory therapeutic targets in PAH. Schematic overview of (pre-)clinical targets for immunomodulatory therapy in pulmonary arterial hypertension (PAH) patients. Pre-clinical animal PH model signs are mentioned in the legend in the bottom right corner. Interleukin (IL); transforming growth factor beta (TGF-β); bone morphogenetic protein (BMP); bone morphogenetic protein receptor type II (BMPR2); tumor necrosis factor-a (TNF-α); nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κβ,); Leukotriene A4/B4 (LTA4/B4). Janus kinases (JAK); signal transducer and activator of transcription proteins (STAT); small mothers against decapentaplegic (SMAD); TNF receptor-associated factor (TRAF); tumor necrosis factor receptor type 1-associated death domain (TRADD); receptor-interacting protein (RIP); inhibitor of κβ (Iκ-β); 5-lipoxygenase activating protein (FLAP); arachidonate 5-lipoxygenase (5-LO).
Figure 2.
Immunomodulatory therapeutic targets in PAH. Schematic overview of (pre-)clinical targets for immunomodulatory therapy in pulmonary arterial hypertension (PAH) patients. Pre-clinical animal PH model signs are mentioned in the legend in the bottom right corner. Interleukin (IL); transforming growth factor beta (TGF-β); bone morphogenetic protein (BMP); bone morphogenetic protein receptor type II (BMPR2); tumor necrosis factor-a (TNF-α); nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κβ,); Leukotriene A4/B4 (LTA4/B4). Janus kinases (JAK); signal transducer and activator of transcription proteins (STAT); small mothers against decapentaplegic (SMAD); TNF receptor-associated factor (TRAF); tumor necrosis factor receptor type 1-associated death domain (TRADD); receptor-interacting protein (RIP); inhibitor of κβ (Iκ-β); 5-lipoxygenase activating protein (FLAP); arachidonate 5-lipoxygenase (5-LO).


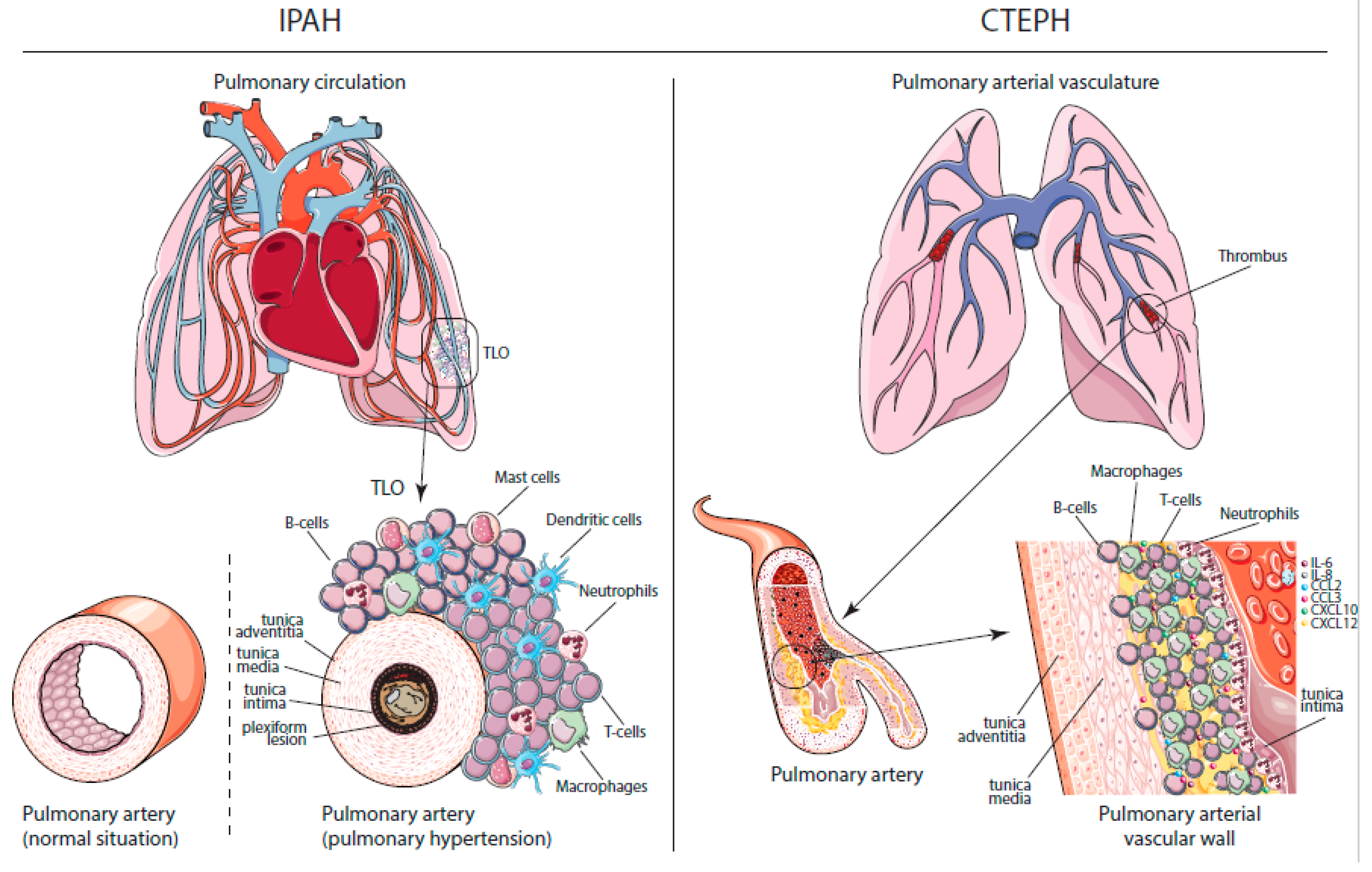{kind=link}
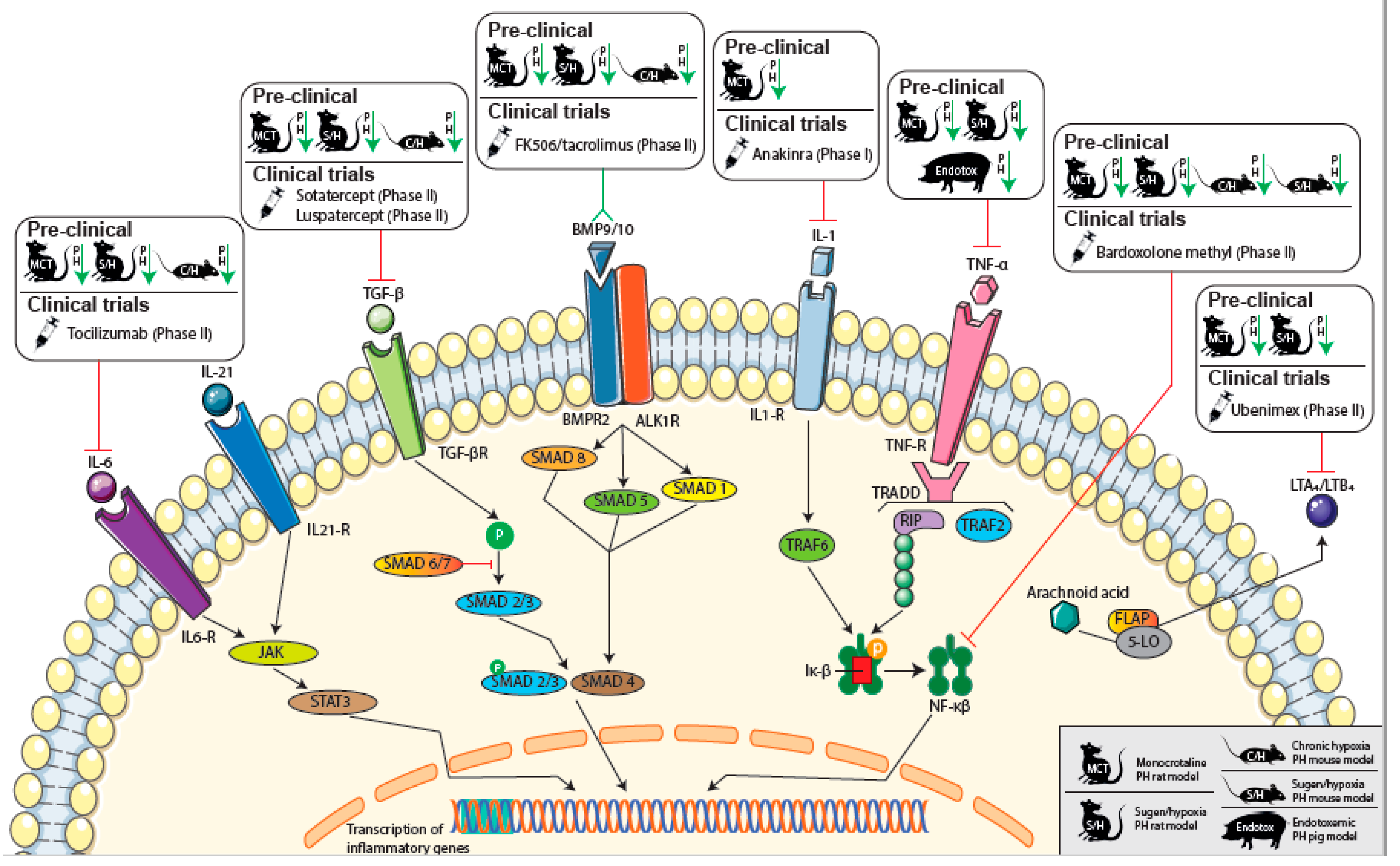{kind=link}
Table 1.
Circulating cytokine/chemokine Levels in patients with pulmonary arterial hypertension or chronic thromboembolic pulmonary hypertension.
Table 1.
Circulating cytokine/chemokine Levels in patients with pulmonary arterial hypertension or chronic thromboembolic pulmonary hypertension.
| PAH | CTEPH | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Biomarker | Incident Patients | Prevalent Patients | Hemodynamic Correlation | Prognosis | Etiology | Ref. | Prevalent Patients | Hemodynamic Correlation | Prognosis | Ref. |
| IL-1α | ↑[94] | ↑ | N/A | + | IPAH, HPAH | [94] | ||||
| IL-1β | ↑[94] | ↑ | − | + [94] | IPAH, HPAH, CHD-PAH | [80,94] | ↑[98] | − | − | [21,96,98] |
| IL-2 | = | ↑ | − | − | PAH, HPAH | [80,94] | ↑ | − | − | [98] |
| IL-4 | = | ↑ | − | − | PAH, HPAH | [80,94] | ↑ | − | − | [98] |
| IL-5 | N/A | = | − | − | PAH, HPAH | [80] | = | − | − | [98] |
| IL-6 | ↑[94,95,99] | ↑ | + | + | IPAH, HPAH, CTD-PAH, CHD-PAH | [80,94,95,99,100] | ↑[42,97] | + | + [97,98] | [21,42,95,97,98] |
| IL-8 | = | ↑ | − | + [80] | IPAH, HPAH, CTD-PAH, CHD-PAH | [80,94,95,99,100] | ↑[42,97,98] | − | + [97,98] | [42,95,97,98] |
| IL-10 | = | ↑ | − | + [80] | IPAH, HPAH | [80,94] | ↑ | − | + [97,98] | [21,97,98] |
| IL-12 | = | ↑ | − | + [80] | IPAH, HPAH | [80,94] | = | − | − | [98] |
| IL-13 | = | = | − | + [94] | IPAH, HPAH | [80,94] | = | − | − | [98] |
| IFN-γ | = | = | − | − | IPAH, HPAH | [79,93] | = | − | − | [98] |
| TNF-α | ↑[94,95,99] | ↑ | N/A | + [94] | IPAH, HPAH, CTD-PAH, CHD-PAH | [80,94,95,99,100] | = | − | + [96,97] | [95,96,97] |
| MMP-9 | ↑ | ↑ | + | + | PAH | [101] | ↑ | − | N/A | [21] |
| VEGF | = | ↑ | N/A | + [101] | IPAH, HPAH, CHD-PAH | [80,94,100,101] | = | − | N/A | [21] |
| CCL-2 | = | ↑[33] | N/A | − | IPAH | [33,94] | ↑ | − | N/A | [21,42] |
| MIG | N/A | ↑ | − | N/A | IPAH | [42] | ↑ | − | N/A | [42] |
| CCL-3 | = | = | − | N/A | IPAH | [42] | ↑ | + | N/A | [21,42] |
| CXCL-10 | N/A | ↑ | − | N/A | IPAH | [42] | ↑ | + | N/A | [42] |
| CCL-5 | N/A | ↑ | − | N/A | IPAH, PAH | [42,102] | ↓ | − | N/A | [42] |
| CX3CL-1 | = | = | − | N/A | IPAH | [42] | = | − | N/A | [42] |
| CXCL-12 | = | = | − | N/A | IPAH | [42] | = | − | N/A | [42] |
Pulmonary arterial hypertension (PAH); idiopathic PAH (IPAH); hereditary PAH (HPAH); congenital heart disease-associated PAH (CHD-PAH); connective tissue disease-associated PAH (CTD-PAH); chronic thromboembolic pulmonary hypertension (CTEPH); no significant differences (=); positive correlation (+); no significant correlation (−); not assessed (N/A); interleukin (IL); interferon gamma (IFN-γ); tumor necrosis factor-a (TNF-α); matrix metallopeptidase 9 (MMP-9); vascular endothelial growth factor (VEGF); chemokine (CCL) (C-C motif) ligand; monokine-induced by interferon-γ (MIG); C-X-C motif chemokine (CXCL); C-X3-C motif ligand (CX3CL).
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Koudstaal, T.; Boomars, K.A.; Kool, M. Pulmonary Arterial Hypertension and Chronic Thromboembolic Pulmonary Hypertension: An Immunological Perspective. J. Clin. Med. 2020, 9, 561. https://doi.org/10.3390/jcm9020561
AMA Style
Koudstaal T, Boomars KA, Kool M. Pulmonary Arterial Hypertension and Chronic Thromboembolic Pulmonary Hypertension: An Immunological Perspective. Journal of Clinical Medicine. 2020; 9(2):561. https://doi.org/10.3390/jcm9020561
Chicago/Turabian StyleKoudstaal, Thomas, Karin A. Boomars, and Mirjam Kool. 2020. "Pulmonary Arterial Hypertension and Chronic Thromboembolic Pulmonary Hypertension: An Immunological Perspective" Journal of Clinical Medicine 9, no. 2: 561. https://doi.org/10.3390/jcm9020561
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.