Short and Long Term Clinical and Immunologic Follow up after Bone Marrow Mesenchymal Stromal Cell Therapy in Progressive Multiple Sclerosis—A Phase I Study

Abstract
:1. Introduction
2. Material and Methods
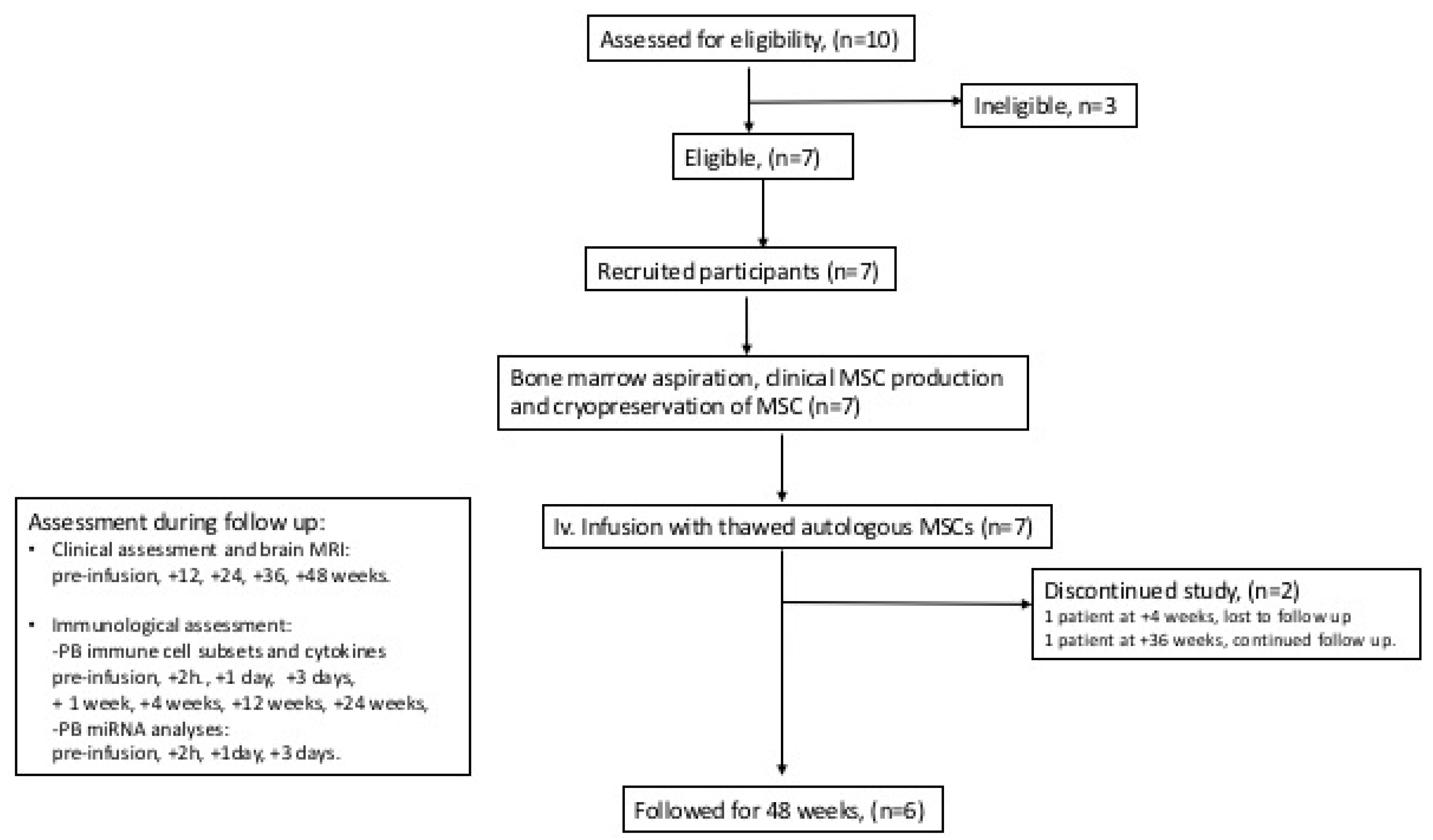
2.1. Clinical Study Design
2.2. MS Subjects, Healthy MSC Donors, and Ethics
2.3. MSC Therapy
2.4. Safety
2.5. Clinical Evaluation
2.6. Preparation of Peripheral Blood Mononuclear Cells and Plasma
2.7. Flow Cytometric Analysis of Peripheral Immune Cells
2.8. Cytokine Analyses and microRNA Analyses
2.9. In Vitro Phenotypic and Immunosuppressive Analyses of MSC
2.10. Statistical Analyses
3. Results
3.1. Characteristics of Clinical Study Subjects
3.2. MSC Treatment and Safety
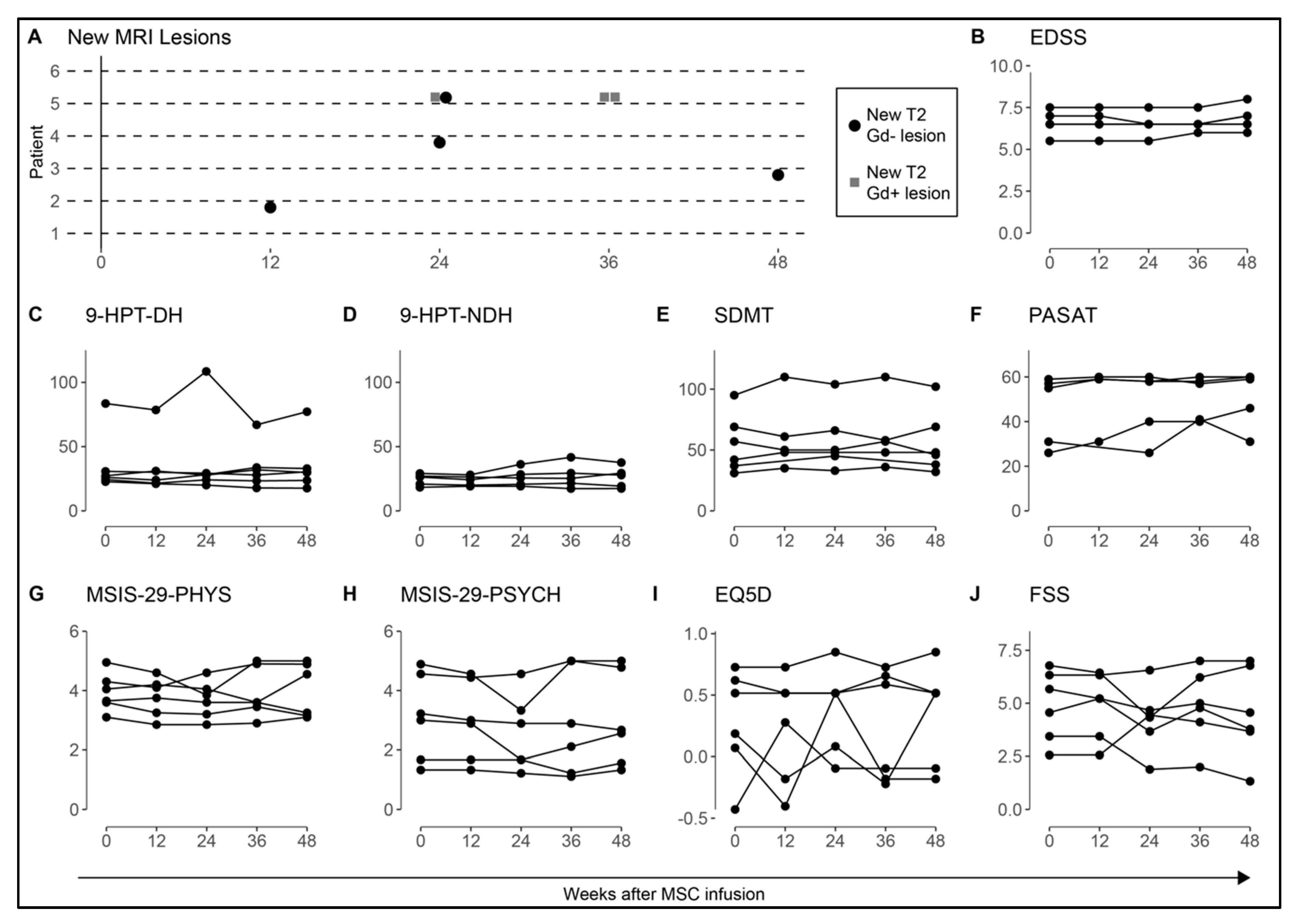
3.3. MRI and Clinical Assessment
3.4. Plasma Cytokine and miR Levels after MSC Infusion
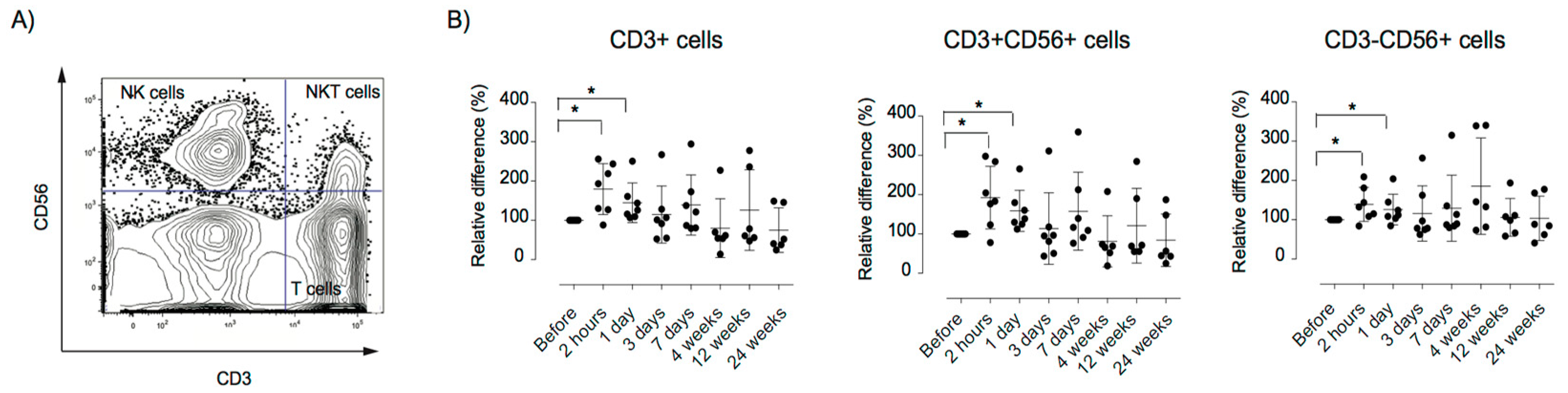
3.5. Alterations in the Peripheral Immune Repertoire after MSC Therapy
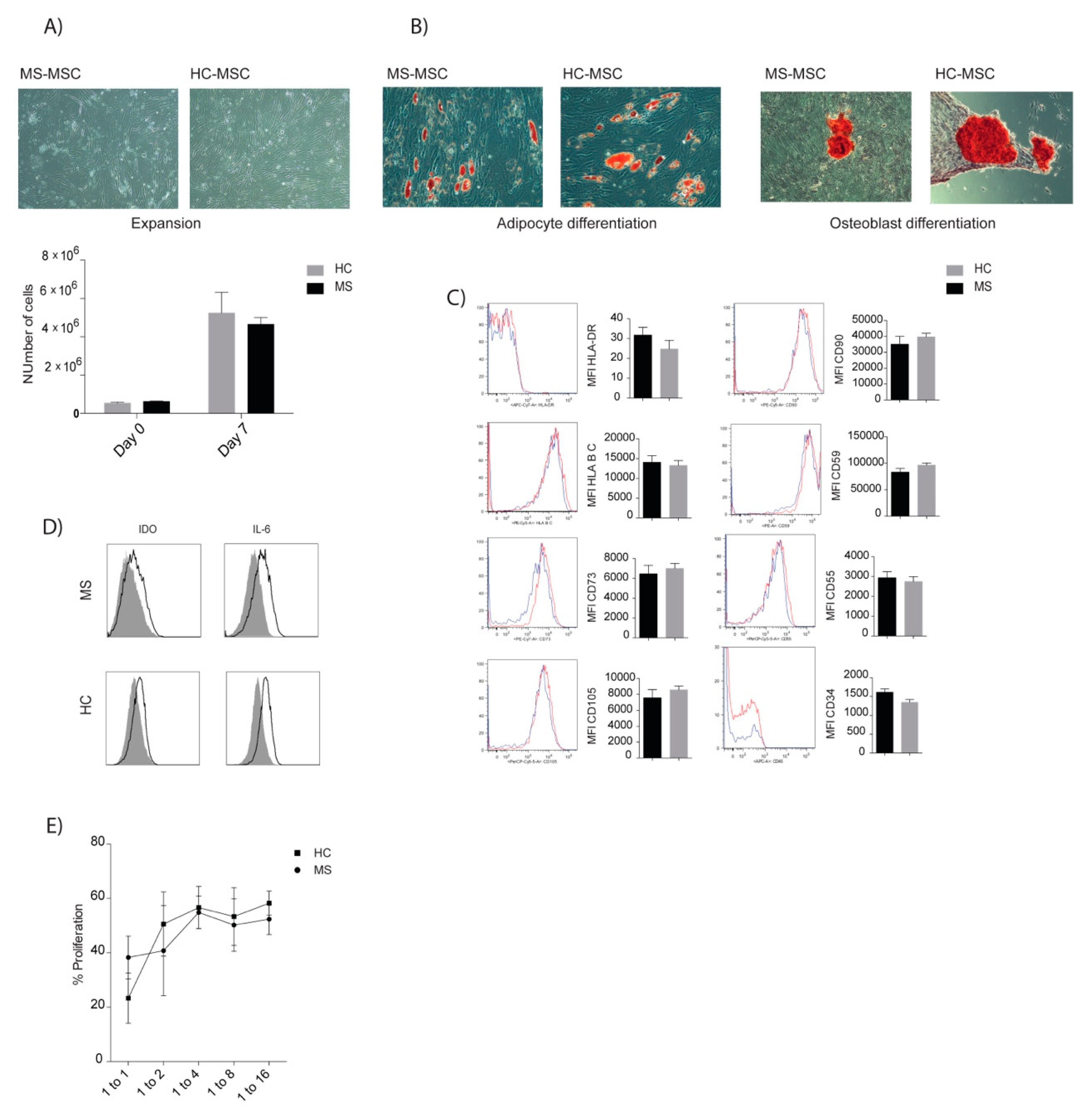
3.6. Comparable Characteristics of MSCs of MS Patients and Healthy Donors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Ebers, G.C. Natural history of primary progressive multiple sclerosis. Mult. Scler. J. 2004, 10 (Suppl. 1), S8–S15, discussion S13–S15. [Google Scholar] [CrossRef] [PubMed]
- Dendrou, C.A.; Fugger, L. Immunomodulation in multiple sclerosis: Promises and pitfalls. Curr. Opin. Immunol. 2017, 49, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Gaitan, M.I.; Ysrraelit, M.C.; Fiol, M.P. Progressive multiple sclerosis: From pathogenic mechanisms to treatment. Brain 2017, 140, 527–546. [Google Scholar] [CrossRef]
- Tur, C.; Montalban, X. Progressive MS trials: Lessons learned. Mult. Scler. J. 2017, 23, 1583–1592. [Google Scholar] [CrossRef]
- Förster, M.; Küry, P.; Aktas, O.; Warnke, C.; Havla, J.; Hohlfeld, R.; Mares, J.; Hartung, H.P.; Kremer, D. Managing Risks with Immune Therapies in Multiple Sclerosis. Drug Saf. 2019, 42, 633–647. [Google Scholar] [CrossRef]
- Galipeau, J.; Sensebé, L. Mesenchymal Stromal Cells: Clinical Challenges and Therapeutic Opportunities. Cell Stem Cell 2018, 22, 824–833. [Google Scholar] [CrossRef]
- Le Blanc, K.; Davies, L.C. Mscs-cells with many sides. Cytotherapy 2018, 20, 273–278. [Google Scholar] [CrossRef]
- Najar, M.; Bouhtit, F.; Melki, R.; Afif, H.; Hamal, A.; Fahmi, H.; Merimi, M.; Lagneaux, L. Mesenchymal Stromal Cell-Based Therapy: New Perspectives and Challenges. J. Clin. Med. 2019, 8, 626. [Google Scholar] [CrossRef]
- Galleu, A.; Riffo-Vasquez, Y.; Trento, C.; Lomas, C.; Dolcetti, L.; Cheung, T.S.; von Bonin, M.; Barbieri, L.; Halai, K.; Ward, S.; et al. Apoptosis in mesenchymal stromal cells induces in vivo recipient-mediated immunomodulation. Sci. Transl. Med. 2017, 9, eaam7828. [Google Scholar] [CrossRef]
- Moll, G.; Rasmusson-Duprez, I.; von Bahr, L.; Connolly-Andersen, A.M.; Elgue, G.; Funke, L.; Hamad, O.A.; Lonnies, H.; Magnusson, P.U.; Sanchez, J.; et al. Are therapeutic human mesenchymal stromal cells compatible with human blood? Stem Cells 2012, 30, 1565–1574. [Google Scholar] [CrossRef] [PubMed]
- Jitschin, R.; Mougiakakos, D.; Von Bahr, L.; Völkl, S.; Moll, G.; Ringdén, O.; Kiessling, R.; Linder, S.; Le Blanc, K. Alterations in the Cellular Immune Compartment of Patients Treated with Third-Party Mesenchymal Stromal Cells Following Allogeneic Hematopoietic Stem Cell Transplantation. Stem Cells 2013, 31, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, A.; Laroni, A.; Freedman, M.S. Mesenchymal stem cells for the treatment of multiple sclerosis and other neurological diseases. Lancet Neurol. 2011, 10, 649–656. [Google Scholar] [CrossRef]
- Meirelles Lda, S.; Fontes, A.M.; Covas, D.T.; Caplan, A.I. Mechanisms involved in the therapeutic properties of mesenchymal stem cells. Cytokine Growth Factor Rev. 2009, 20, 419–427. [Google Scholar] [CrossRef]
- Teixeira, F.G.; Carvalho, M.M.; Sousa, N.; Salgado, A.J. Mesenchymal stem cells secretome: A new paradigm for central nervous system regeneration? Cell. Mol. Life Sci. 2013, 70, 3871–3882. [Google Scholar] [CrossRef]
- Keto, J.; Kaartinen, T.; Salmenniemi, U.; Castrén, J.; Partanen, J.; Hänninen, A.; Korhonen, M.; Lähteenmäki, K.; Itälä-Remes, M.; Nystedt, J. Immunomonitoring of MSC-Treated GvHD Patients Reveals Only Moderate Potential for Response Prediction but Indicates Treatment Safety. Mol. Ther. Methods Clin. Dev. 2018, 9, 109–118. [Google Scholar] [CrossRef]
- Karussis, D.; Karageorgiou, C.; Vaknin-Dembinsky, A.; Gowda-Kurkalli, B.; Gomori, J.M.; Kassis, I.; Bulte, J.W.M.; Petrou, P.; Ben-Hur, T.; Abramsky, O.; et al. Safety and immunological effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Arch. Neurol. 2010, 67, 1187–1194. [Google Scholar] [CrossRef]
- Carlsson, P.O.; Schwarcz, E.; Korsgren, O.; Le Blanc, K. Preserved beta-cell function in type 1 diabetes by mesenchymal stromal cells. Diabetes 2015, 64, 587–592. [Google Scholar] [CrossRef]
- Liang, J.; Zhang, H.; Hua, B.; Wang, H.; Lu, L.; Shi, S.; Hou, Y.; Zeng, X.; Gilkeson, G.S.; Sun, L. Allogenic mesenchymal stem cells transplantation in refractory systemic lupus erythematosus: A pilot clinical study. Ann. Rheum. Dis. 2010, 69, 1423–1429. [Google Scholar] [CrossRef]
- Xu, J.; Wang, D.; Liu, D.; Fan, Z.; Zhang, H.; Liu, O.; Ding, G.; Gao, R.; Zhang, C.; Ding, Y.; et al. Allogeneic mesenchymal stem cell treatment alleviates experimental and clinical Sjögren syndrome. Blood 2012, 120, 3142–3151. [Google Scholar] [CrossRef]
- Ghoryani, M.; Shariati-Sarabi, Z.; Tavakkol-Afshari, J.; Ghasemi, A.; Poursamimi, J.; Mohammadi, M. Amelioration of clinical symptoms of patients with refractory rheumatoid arthritis following treatment with autologous bone marrow-derived mesenchymal stem cells: A successful clinical trial in Iran. Biomed. Pharmacother. 2018, 109, 1834–1840. [Google Scholar] [CrossRef] [PubMed]
- Zappia, E.; Casazza, S.; Pedemonte, E.; Benvenuto, F.; Bonanni, I.; Gerdoni, E.; Giunti, D.; Ceravolo, A.; Cazzanti, F.; Frassoni, F.; et al. Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing t-cell anergy. Blood 2005, 106, 1755–1761. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, Y.; Chen, J.; Cui, Y.; Lü, M.; Elias, S.B.; Mitchell, J.B.; Hammill, L.; Vanguri, P.; Chopp, M. Human bone marrow stromal cell treatment improves neurological functional recovery in EAE mice. Exp. Neurol. 2005, 195, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Gerdoni, E.; Gallo, B.; Casazza, S.; Musio, S.; Bonanni, I.; Pedemonte, E.; Mantegazza, R.; Frassoni, F.; Mancardi, G.; Pedotti, R.; et al. Mesenchymal stem cells effectively modulate pathogenic immune response in experimental autoimmune encephalomyelitis. Ann. Neurol. 2007, 61, 219–227. [Google Scholar] [CrossRef]
- Kassis, I.; Grigoriadis, N.; Gowda-Kurkalli, B.; Mizrachi-Kol, R.; Ben-Hur, T.; Slavin, S.; Abramsky, O.; Karussis, D. Neuroprotection and Immunomodulation with Mesenchymal Stem Cells in Chronic Experimental Autoimmune Encephalomyelitis. Arch. Neurol. 2008, 65, 753–761. [Google Scholar] [CrossRef]
- Lanza, C.; Morando, S.; Voci, A.; Canesi, L.; Principato, M.C.; Serpero, L.D.; Mancardi, G.; Uccelli, A.; Vergani, L. Neuroprotective mesenchymal stem cells are endowed with a potent antioxidant effect in vivo. J. Neurochem. 2009, 110, 1674–1684. [Google Scholar] [CrossRef]
- Kemp, K.; Hares, K.; Mallam, E.; Heesom, K.J.; Scolding, N.; Wilkins, A. Mesenchymal stem cell-secreted superoxide dismutase promotes cerebellar neuronal survival. J. Neurochem. 2010, 114, 1569–1580. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Y.; Lü, M.; Cui, Y.; Chen, J.; Noffsinger, L.; Elias, S.B.; Chopp, M. Bone marrow stromal cells reduce axonal loss in experimental autoimmune encephalomyelitis mice. J. Neurosci. Res. 2006, 84, 587–595. [Google Scholar] [CrossRef]
- Gordon, D.; Pavlovska, G.; Uney, J.B.; Wraith, D.C.; Scolding, N.J. Human Mesenchymal Stem Cells Infiltrate the Spinal Cord, Reduce Demyelination, and Localize to White Matter Lesions in Experimental Autoimmune Encephalomyelitis. J. Neuropathol. Exp. Neurol. 2010, 69, 1087–1095. [Google Scholar] [CrossRef]
- Rafei, M.; Campeau, P.M.; Aguilar-Mahecha, A.; Buchanan, M.; Williams, P.; Birman, E.; Yuan, S.; Young, Y.K.; Boivin, M.N.; Forner, K.; et al. Mesenchymal Stromal Cells Ameliorate Experimental Autoimmune Encephalomyelitis by Inhibiting CD4 Th17 T Cells in a CC Chemokine Ligand 2-Dependent Manner. J. Immunol. 2009, 182, 5994–6002. [Google Scholar] [CrossRef]
- Mohyeddin Bonab, M.; Yazdanbakhsh, S.; Lotfi, J.; Alimoghaddom, K.; Talebian, F.; Hooshmand, F.; Ghavamzadeh, A.; Nikbin, B. Does mesenchymal stem cell therapy help multiple sclerosis patients? Report of a pilot study. Iran. J. Immunol. 2007, 4, 50–57. [Google Scholar] [PubMed]
- Yamout, B.; Hourani, R.; Salti, H.; Barada, W.; El-Hajj, T.; Al-Kutoubi, A.; Herlopian, A.; Baz, E.K.; Mahfouz, R.; Khalil-Hamdan, R.; et al. Bone marrow mesenchymal stem cell transplantation in patients with multiple sclerosis: A pilot study. J. Neuroimmunol. 2010, 227, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Connick, P.; Kolappan, M.; Crawley, C.; Webber, D.J.; Patani, R.; Michell, A.W.; Du, M.Q.; Luan, S.L.; Altmann, D.R.; Thompson, A.J.; et al. Autologous mesenchymal stem cells for the treatment of secondary progressive multiple sclerosis: An open-label phase 2a proof-of-concept study. Lancet Neurol. 2012, 11, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Bonab, M.M.; Sahraian, M.A.; Aghsaie, A.; Karvigh, S.A.; Hosseinian, S.M.; Nikbin, B.; Lotfi, J.; Khorramnia, S.; Motamed, M.R.; Togha, M.; et al. Autologous mesenchymal stem cell therapy in progressive multiple sclerosis: An open label study. Curr. Stem Cell Res. Ther. 2012, 7, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Llufriu, S.; Sepulveda, M.; Blanco, Y.; Marin, P.; Moreno, B.; Berenguer, J.; Gabilondo, I.; Martinez-Heras, E.; Sola-Valls, N.; Arnaiz, J.A.; et al. Randomized placebo-controlled phase ii trial of autologous mesenchymal stem cells in multiple sclerosis. PLoS ONE 2014, 9, e113936. [Google Scholar] [CrossRef]
- Cohen, J.A.; Imrey, P.B.; Planchon, S.M.; Bermel, R.A.; Fisher, E.; Fox, R.J.; Bar-Or, A.; Sharp, S.L.; Skaramagas, T.T.; Jagodnik, P.; et al. Pilot trial of intravenous autologous culture-expanded mesenchymal stem cell transplantation in multiple sclerosis. Mult. Scler. J. 2018, 24, 501–511. [Google Scholar] [CrossRef] [Green Version]
- Kurtzke, J.F. Rating neurologic impairment in multiple sclerosis: An expanded disability status scale (EDSS). Neurology 1983, 33, 1444. [Google Scholar] [CrossRef] [Green Version]
- Prockop, D.J.; Brenner, M.; Fibbe, W.E.; Horwitz, E.; Le Blanc, K.; Phinney, D.G.; Simmons, P.J.; Sensebé, L.; Keating, A. Defining the risks of mesenchymal stromal cell therapy. Cytotherapy 2010, 12, 576–578. [Google Scholar] [CrossRef]
- Suna, L.; Akiyamab, K.; Zhanga, H.; Yamazab, T.; Houa, Y.; Zhaoa, S.; Xua, T.; Leb, A.; Shib, S. Mesenchymal stem cell transplantation reverses multi-organ dysfunction in systemic lupus erythematosus mice and humans. Stem Cells 2009, 27, 1421–1432. [Google Scholar]
- de Oliveira, G.L.; de Lima, K.W.; Colombini, A.M.; Pinheiro, D.G.; Panepucci, R.A.; Palma, P.V.; Brum, D.G.; Covas, D.T.; Simoes, B.P.; de Oliveira, M.C.; et al. Bone marrow mesenchymal stromal cells isolated from multiple sclerosis patients have distinct gene expression profile and decreased suppressive function compared with healthy counterparts. Cell Transplant. 2015, 24, 151–165. [Google Scholar] [CrossRef] [Green Version]
- Redondo, J.; Sarkar, P.; Kemp, K.; Heesom, K.J.; Wilkins, A.; Scolding, N.J.; Rice, C.M. Dysregulation of Mesenchymal Stromal Cell Antioxidant Responses in Progressive Multiple Sclerosis. Stem Cells Transl. Med. 2018, 7, 748–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polman, C.H.; Reingold, S.C.; Banwell, B.; Clanet, M.; Cohen, J.A.; Filippi, M.; Fujihara, K.; Havrdova, E.; Hutchinson, M.; Kappos, L.; et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the mcdonald criteria. Ann. Neurol. 2011, 69, 292–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Blanc, K.; Frassoni, F.; Ball, L.; Locatelli, F.; Roelofs, H.; Lewis, I.; Lanino, E.; Sundberg, B.; Bernardo, M.E.; Remberger, M.; et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: A phase ii study. Lancet 2008, 371, 1579–1586. [Google Scholar] [CrossRef]
- Rudick, R.; Antel, J.; Confavreux, C.; Cutter, G.; Ellison, G.; Fischer, J.; Lublin, F.; Miller, A.; Petkau, J.; Rao, S.; et al. Clinical outcomes assessment in multiple sclerosis. Ann. Neurol. 1996, 40, 469–479. [Google Scholar] [CrossRef]
- Benedict, R.H.; Fischer, J.S.; Archibald, C.J.; Arnett, P.A.; Beatty, W.W.; Bobholz, J.; Chelune, G.J.; Fisk, J.D.; Langdon, D.W.; Caruso, L.; et al. Minimal neuropsychological assessment of ms patients: A consensus approach. Clin. Neuropsychol. 2002, 16, 381–397. [Google Scholar] [CrossRef]
- Hobart, J.; Lamping, D.; Fitzpatrick, R.; Riazi, A.; Thompson, A. The Multiple Sclerosis Impact Scale (MSIS-29): A new patient-based outcome measure. Brain 2001, 124, 962–973. [Google Scholar] [CrossRef]
- Krupp, L.B.; LaRocca, N.G.; Muir-Nash, J.; Steinberg, A.D. The fatigue severity scale. Application to patients with multiple sclerosis and systemic lupus erythematosus. Arch. Neurol. 1989, 46, 1121–1123. [Google Scholar] [CrossRef]
- Rabin, R.; De Charro, F. EQ-5D: A measure of health status from the EuroQol Group. Ann. Med. 2001, 33, 337–343. [Google Scholar] [CrossRef]
- Iacobaeus, E.; Douagi, I.; Jitschin, R.; Marcusson-Ståhl, M.; Andrén, A.T.; Gavin, C.; Lefsihane, K.; Davies, L.C.; Mougiakakos, D.; Kadri, N.; et al. Phenotypic and functional alterations of myeloid-derived suppressor cells during the disease course of multiple sclerosis. Immunol. Cell Boil. 2018, 96, 820–830. [Google Scholar] [CrossRef]
- Blondal, T.; Nielsen, S.J.; Baker, A.; Andreasen, D.; Mouritzen, P.; Teilum, M.W.; Dahlsveen, I.K. Assessing sample and miRNA profile quality in serum and plasma or other biofluids. Methods 2013, 59, S1–S6. [Google Scholar] [CrossRef]
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T.; Dalamagas, T.; Hatzigeorgiou, A.G. Diana-mirpath v3.0: Deciphering microrna function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Won, K.J.; Park, S.W.; Lee, H.W.; Kim, B.; Kim, J.H.; Kim, D.K. Mesenchymal stem cells regulate the proliferation of T cells via the growth-related oncogene/CXC chemokine receptor, CXCR2. Cell. Immunol. 2012, 279, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gavin, C.; Meinke, S.; Heldring, N.; Heck, K.A.; Achour, A.; Iacobaeus, E.; Höglund, P.; Le Blanc, K.; Kadri, N. The Complement System Is Essential for the Phagocytosis of Mesenchymal Stromal Cells by Monocytes. Front. Immunol. 2019, 10, 2249. [Google Scholar] [CrossRef] [PubMed]
- Kishk, N.A.; Abokrysha, N.T.; Gabr, H. Possible induction of acute disseminated encephalomyelitis (adem)-like demyelinating illness by intrathecal mesenchymal stem cell injection. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2013, 20, 310–312. [Google Scholar] [CrossRef] [PubMed]
- Mallam, E.; Kemp, K.; Wilkins, A.; Rice, C.; Scolding, N. Characterization of in vitro expanded bone marrow-derived mesenchymal stem cells from patients with multiple sclerosis. Mult. Scler. J. 2010, 16, 909–918. [Google Scholar] [CrossRef]
- Mazzanti, B.; Aldinucci, A.; Biagioli, T.; Barilaro, A.; Urbani, S.; Pozzo, S.D.; Amato, M.P.; Siracusa, G.; Crescioli, C.; Manuelli, C.; et al. Differences in mesenchymal stem cell cytokine profiles between MS patients and healthy donors: Implication for assessment of disease activity and treatment. J. Neuroimmunol. 2008, 199, 142–150. [Google Scholar] [CrossRef]
- Riordan, N.H.; Morales, I.; Fernández, G.; Allen, N.; Fearnot, N.E.; Leckrone, M.E.; Markovich, D.J.; Mansfield, D.; Avila, D.; Patel, A.N.; et al. Clinical feasibility of umbilical cord tissue-derived mesenchymal stem cells in the treatment of multiple sclerosis. J. Transl. Med. 2018, 16, 57. [Google Scholar] [CrossRef] [Green Version]
- Francois, M.; Copland, I.B.; Yuan, S.; Romieu-Mourez, R.; Waller, E.K.; Galipeau, J. Cryopreserved mesenchymal stromal cells display impaired immunosuppressive properties as a result of heat-shock response and impaired interferon-gamma licensing. Cytotherapy 2012, 14, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Xiao, B.; Ma, X.; Qu, M.; Li, Y.; Nagarkatti, P.; Nagarkatti, M.; Zhou, J. MicroRNAs associated with the pathogenesis of multiple sclerosis. J. Neuroimmunol. 2016, 295, 148–161. [Google Scholar] [CrossRef]
- Heldring, N.; Mäger, I.; Wood, M.J.; Le Blanc, K.; Andaloussi, S.E. Therapeutic Potential of Multipotent Mesenchymal Stromal Cells and Their Extracellular Vesicles. Hum. Gene Ther. 2015, 26, 506–517. [Google Scholar] [CrossRef]
- Pers, Y.M.; Maumus, M.; Bony, C.; Jorgensen, C.; Noël, D. Contribution of microRNAs to the immunosuppressive function of mesenchymal stem cells. Biochimie 2018, 155, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.P.; Wang, L.H. Disease implication of hyper-hippo signalling. Open Biol. 2016, 6, 160119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karp, J.M.; Teo, G.S.L. Mesenchymal Stem Cell Homing: The Devil Is in the Details. Cell Stem Cell 2009, 4, 206–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mäkelä, T.; Takalo, R.; Arvola, O.; Haapanen, H.; Yannopoulos, F.; Blanco, R.; Ahvenjarvi, L.; Kiviluoma, K.; Kerkelä, E.; Nystedt, J.; et al. Safety and biodistribution study of bone marrow–derived mesenchymal stromal cells and mononuclear cells and the impact of the administration route in an intact porcine model. Cytotherapy 2015, 17, 392–402. [Google Scholar] [CrossRef]
- von Bahr, L.; Batsis, I.; Moll, G.; Hagg, M.; Szakos, A.; Sundberg, B.; Uzunel, M.; Ringden, O.; Le Blanc, K. Analysis of tissues following mesenchymal stromal cell therapy in humans indicates limited long-term engraftment and no ectopic tissue formation. Stem Cells 2012, 30, 1575–1578. [Google Scholar] [CrossRef]
- de Witte, S.F.H.; Luk, F.; Sierra Parraga, J.M.; Gargesha, M.; Merino, A.; Korevaar, S.S.; Shankar, A.S.; O’Flynn, L.; Elliman, S.J.; Roy, D.; et al. Immunomodulation by therapeutic mesenchymal stromal cells (msc) is triggered through phagocytosis of msc by monocytic cells. Stem Cells 2018, 36, 602–615. [Google Scholar] [CrossRef] [Green Version]
- Leibacher, J.; Dauber, K.; Ehser, S.; Brixner, V.; Kollar, K.; Vogel, A.; Spohn, G.; Schäfer, R.; Seifried, E.; Henschler, R. Human mesenchymal stromal cells undergo apoptosis and fragmentation after intravenous application in immune-competent mice. Cytotherapy 2017, 19, 61–74. [Google Scholar] [CrossRef]
- Budkowska, M.; Ostrycharz, E.; Wojtowicz, A.; Marcinowska, Z.; Wozniak, J.; Ratajczak, M.Z.; Dolegowska, B. A circadian rhythm in both complement cascade (comc) activation and sphingosine-1-phosphate (s1p) levels in human peripheral blood supports a role for the comc-s1p axis in circadian changes in the number of stem cells circulating in peripheral blood. Stem Cell Rev. Rep. 2018, 14, 677–685. [Google Scholar] [CrossRef] [Green Version]
- McCoy, C.E. Mir-155 dysregulation and therapeutic intervention in multiple sclerosis. Adv. Exp. Med. Biol. 2017, 1024, 111–131. [Google Scholar]
- Arruda, L.C.; Lorenzi, J.C.; Sousa, A.P.; Zanette, D.L.; Palma, P.V.; Panepucci, R.A.; Brum, D.S.; Barreira, A.A.; Covas, D.T.; Simoes, B.P.; et al. Autologous hematopoietic sct normalizes mir-16, -155 and -142-3p expression in multiple sclerosis patients. Bone Marrow Transplant. 2015, 50, 380–389. [Google Scholar] [CrossRef]
- Nauta, A.J.; Kruisselbrink, A.B.; Lurvink, E.; Willemze, R.; Fibbe, W.E. Mesenchymal stem cells inhibit generation and function of both cd34+-derived and monocyte-derived dendritic cells. J. Immunol. 2006, 177, 2080–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Ge, W.; Li, C.; You, S.; Liao, L.; Han, Q.; Deng, W.; Zhao, R.C. Effects of Mesenchymal Stem Cells on Differentiation, Maturation, and Function of Human Monocyte-Derived Dendritic Cells. Stem Cells Dev. 2004, 13, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Groh, M.; Maitra, B.; Szekely, E.; Koç, O. Human mesenchymal stem cells require monocyte-mediated activation to suppress alloreactive T cells. Exp. Hematol. 2005, 33, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Rozenberg, A.; Rezk, A.; Boivin, M.N.; Darlington, P.J.; Nyirenda, M.; Li, R.; Jalili, F.; Winer, R.; Artsy, E.A.; Uccelli, A.; et al. Human mesenchymal stem cells impact th17 and th1 responses through a prostaglandin e2 and myeloid-dependent mechanism. Stem Cells Transl. Med. 2016, 5, 1506–1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasandan, A.B.; Jahnavi, S.; Shashank, C.; Prasad, P.; Kumar, A.; Prasanna, S.J. Human Mesenchymal stem cells program macrophage plasticity by altering their metabolic status via a PGE2-dependent mechanism. Sci. Rep. 2016, 6, 38308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laranjeira, P.; Gomes, J.; Pedreiro, S.; Pedrosa, M.; Martinho, A.; Antunes, B.; Ribeiro, T.; Santos, F.; Domingues, R.; Abecasis, M.; et al. Human Bone Marrow-Derived Mesenchymal Stromal Cells Differentially Inhibit Cytokine Production by Peripheral Blood Monocytes Subpopulations and Myeloid Dendritic Cells. Stem Cells Int. 2015, 2015, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elyaman, W.; Kivisakk, P.; Reddy, J.; Chitnis, T.; Raddassi, K.; Imitola, J.; Bradshaw, E.; Kuchroo, V.K.; Yagita, H.; Sayegh, M.H.; et al. Distinct functions of autoreactive memory and effector cd4+ t cells in experimental autoimmune encephalomyelitis. Am. J. Pathol. 2008, 173, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Muraro, P.A.; Douek, D.C.; Packer, A.; Chung, K.; Guenaga, F.J.; Cassiani-Ingoni, R.; Campbell, C.; Memon, S.; Nagle, J.W.; Hakim, F.T.; et al. Thymic output generates a new and diverse TCR repertoire after autologous stem cell transplantation in multiple sclerosis patients. J. Exp. Med. 2005, 201, 805–816. [Google Scholar] [CrossRef]
- Nielsen, B.R.; Ratzer, R.; Bornsen, L.; von Essen, M.R.; Christensen, J.R.; Sellebjerg, F. Characterization of naive, memory and effector t cells in progressive multiple sclerosis. J. Neuroimmunol. 2017, 310, 17–25. [Google Scholar] [CrossRef]
- Sahraian, M.A.; Bonab, M.M.; Baghbanian, S.M.; Owji, M.; Moghadasi, A.N. Therapeutic Use of Intrathecal Mesenchymal Stem Cells in patients with Multiple Sclerosis: A Pilot Study with Booster Injection. Immunol. Investig. 2018, 48, 160–168. [Google Scholar] [CrossRef]
- Uccelli, A.; Laroni, A.; Brundin, L.; Clanet, M.; Fernandez, O.; Nabavi, S.M.; Muraro, P.A.; Oliveri, R.S.; Radue, E.W.; Sellner, J.; et al. MEsenchymal StEm cells for Multiple Sclerosis (MESEMS): A randomized, double blind, cross-over phase I/II clinical trial with autologous mesenchymal stem cells for the therapy of multiple sclerosis. Trials 2019, 20, 263. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Patients | 7 |
|---|---|
| MS subdiagnosis, SPMS/PPMS | 5/2 |
| Sex, female/male | 6/1 |
| Age, median (range) | 40 (23–49) |
| Disease duration, years, median (range) | 12 (5–19) |
| MRI T2 lesion number, median (range) | 20 (20–50) |
| Number of Gd T2, median * | 0 |
| Relapses within 2 years, median (range) | 1.5 (0–3) |
| EDSS, median (range) | 6.5 (5.5–7.0) |
| 9-HPT dominant hand, median (range) | 26.1 (17.7–83.5) |
| 9-HPT non-dominant hand, median (range) | 19.6 (18.1–27) |
| SDMT, median (range) | 50 (31–95) |
| PASAT, median (range) | 54 (26–59) |
| MSIS-29 PHYS, median (range) | 3.65 (2.6–4.95) |
| MSIS-29 PSYCH, median (range) | 3 (1.3–4.9) |
| FSS, median (range) | 5.8 (2.6–6.8) |
| EQ5D, median (range) | 0.52 (0.1–0.73) |
| Prior disease modifying treatment | interferon beta (n = 4) |
| glatiramer acetate (n = 3) | |
| fingolimod (n = 2) | |
| natalizumab (n = 2) | |
| mitoxantrone (n = 1) | |
| rituximab (n = 2) |
| +2 h after MSC Infusion | |||
|---|---|---|---|
| miRNA | SD | Fold Change | p-Value |
| hsa-miR-193a-5p | 1.2 | 8.1 | 0.00060 |
| hsa-miR-365a-3p | 1.4 | 4.8 | 0.0050 |
| hsa-miR-34a-5p | 1.1 | 4.6 | 0.0020 |
| hsa-miR-100-5p | 1.3 | 4.2 | 0.0061 |
| hsa-miR-375 | 1.4 | 2.5 | 0.048 |
| hsa-miR-215-5p | 0.60 | 1.8 | 0.011 |
| hsa-miR-192-5p | 0.50 | 1.6 | 0.010 |
| hsa-miR-16-5p | 0.61 | 1.5 | 0.044 |
| hsa-miR-140-3p | 0.40 | 1.3 | 0.034 |
| hsa-miR-335-5p | 1.1 | −2.8 | 0.012 |
| hsa-miR-15b-5p | 0.76 | −1.8 | 0.024 |
| hsa-miR-543 | 0.64 | −1.7 | 0.020 |
| hsa-miR-374b-5p | 0.65 | −1.6 | 0.040 |
| hsa-miR-155-5p | 0.43 | −1.5 | 0.014 |
| hsa-miR-584-5p | 0.35 | −1.5 | 0.0053 |
| hsa-miR-126-3p | 0.41 | −1.3 | 0.031 |
| hsa-let-7d-3p | 0.28 | −1.2 | 0.031 |
| hsa-miR-30d-5p | 0.31 | −1.2 | 0.048 |
| hsa-miR-362-3p | 0.33 | −1.2 | 0.049 |
| +24 h after MSC infusion | |||
| hsa-miR-332-3p | 0.5 | −1.4 | 0.044 |
| +3 days after MSC infusion | |||
| hsa-miR-375 | 2.4 | 4.9 | 0.046 |
| hsa-miR-143-3p | 0.65 | −1.6 | 0.049 |
| hsa-miR-29a-3p | 0.53 | −1.5 | 0.049 |
| hsa-miR-133a-3p | 0.21 | −1.2 | 0.021 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iacobaeus, E.; Kadri, N.; Lefsihane, K.; Boberg, E.; Gavin, C.; Törnqvist Andrén, A.; Lilja, A.; Brundin, L.; Blanc, K.L. Short and Long Term Clinical and Immunologic Follow up after Bone Marrow Mesenchymal Stromal Cell Therapy in Progressive Multiple Sclerosis—A Phase I Study. J. Clin. Med. 2019, 8, 2102. https://doi.org/10.3390/jcm8122102
Iacobaeus E, Kadri N, Lefsihane K, Boberg E, Gavin C, Törnqvist Andrén A, Lilja A, Brundin L, Blanc KL. Short and Long Term Clinical and Immunologic Follow up after Bone Marrow Mesenchymal Stromal Cell Therapy in Progressive Multiple Sclerosis—A Phase I Study. Journal of Clinical Medicine. 2019; 8(12):2102. https://doi.org/10.3390/jcm8122102
Chicago/Turabian StyleIacobaeus, Ellen, Nadir Kadri, Katia Lefsihane, Erik Boberg, Caroline Gavin, Anton Törnqvist Andrén, Anders Lilja, Lou Brundin, and Katarina Le Blanc. 2019. "Short and Long Term Clinical and Immunologic Follow up after Bone Marrow Mesenchymal Stromal Cell Therapy in Progressive Multiple Sclerosis—A Phase I Study" Journal of Clinical Medicine 8, no. 12: 2102. https://doi.org/10.3390/jcm8122102