
Dysregulation of TGFβ1 Activity in Cancer and Its Influence on the Quality of Anti-Tumor Immunity

Abstract
:
1. Introduction
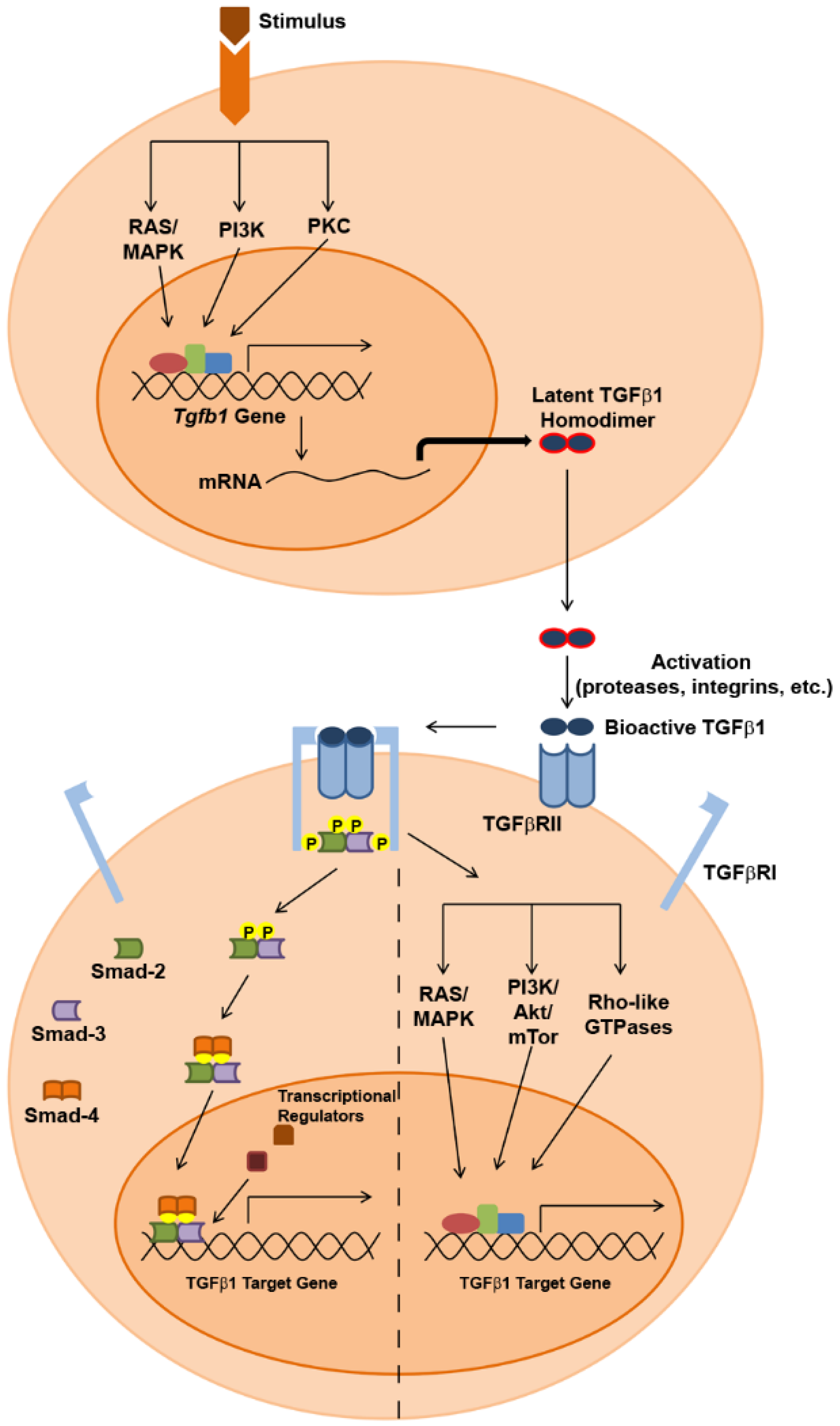
2. TGFβ1 Expression and Signaling
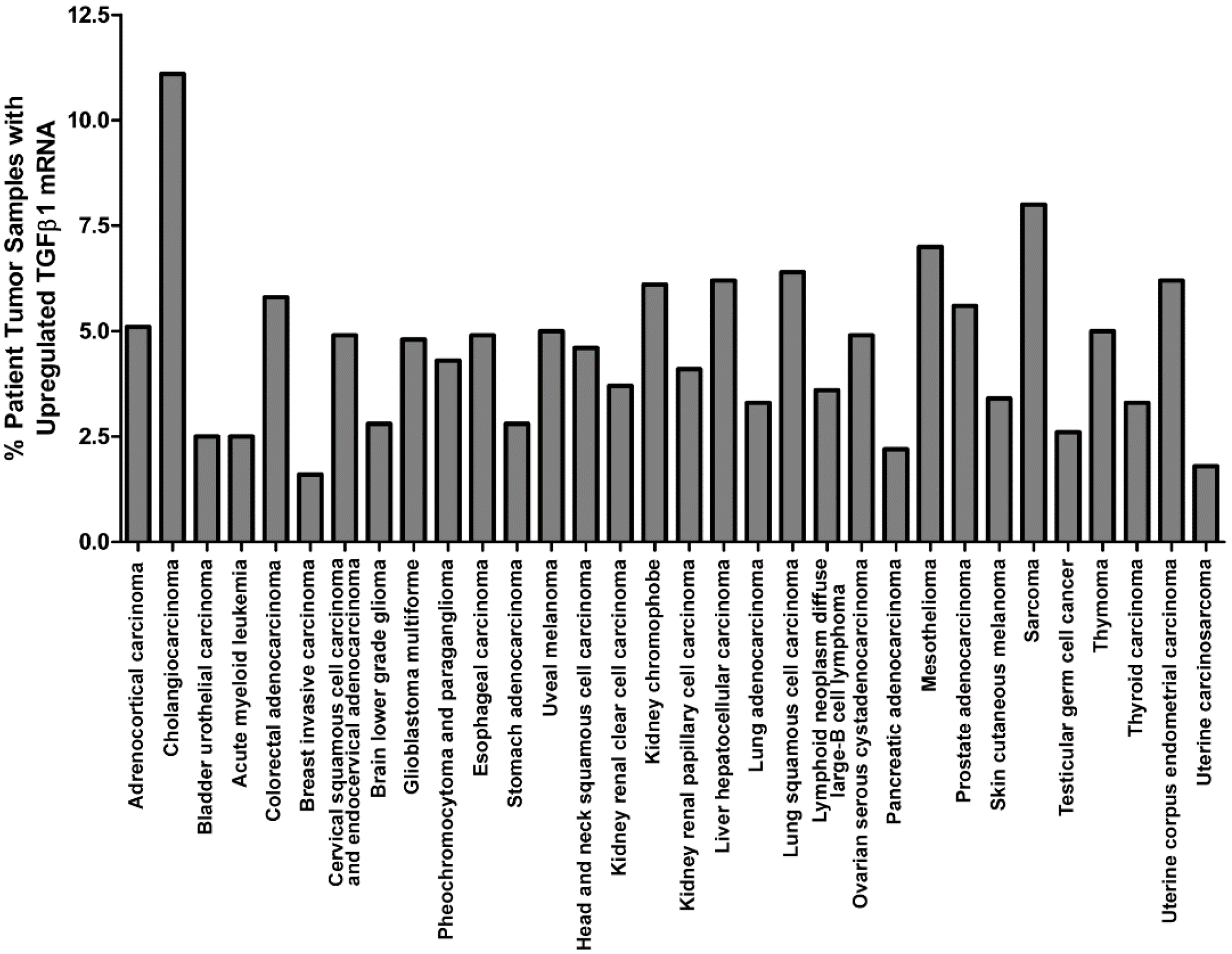
3. Overexpression of TGFβ1 in Cancer
4. TGFβ1 in Tumor Immune Suppression and Escape
4.1. TGFβ1 Influence on the Function of Dendritic Cells and Their Hematopoietic Precursors
4.2. TGFβ1 Influence on Tumor-Associated Macrophages and Neutrophils
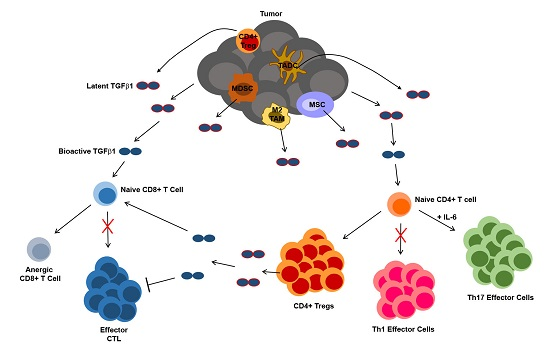
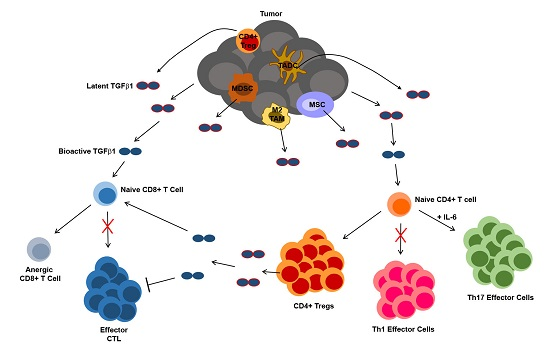
4.3. TGFβ1 Influence on T Cells
4.4. TGFβ1 Influence on Natural Killer Cells
5. Strategies for Interfering with TGFβ1-Mediated Suppression of Anti-Tumor Immunity
6. Clinical Trials Targeting TGFβ1 in the Context of Cancer Immunotherapy
7. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Massagué, J. How cells read TGF-beta signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.J.; Blobe, G.C. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim. Biophys. Acta 2008, 1782, 197–228. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Gao, S.; Wang, Z. Transcriptional regulation of the TGF-beta1 promoter by androgen receptor. Biochem. J. 2008, 416, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, D.E.; Ferris, M.; Nguyen, H.; Abboud, E.; Brody, A.R. TNF-alpha induces TGF-beta1 expression in lung fibroblasts at the transcriptional level via AP-1 activation. J. Cell. Mol. Med. 2009, 13, 1866–1876. [Google Scholar] [CrossRef] [PubMed]
- Fraser, D.; Brunskill, N.; Ito, T.; Phillips, A. Long-term exposure of proximal tubular epithelial cells to glucose induces transforming growth factor-beta 1 synthesis via an autocrine PDGF loop. Am. J. Pathol. 2003, 163, 2565–2574. [Google Scholar] [CrossRef] [PubMed]
- Bascom, C.C.; Wolfshohl, J.R.; Coffey, R.J.; Madisen, L.; Webb, N.R.; Purchio, A.R.; Derynck, R.; Moses, H.L. Complex regulation of transforming growth factor beta 1, beta 2, and beta 3 mRNA expression in mouse fibroblasts and keratinocytes by transforming growth factors beta 1 and beta 2. Mol. Cell. Biol. 1989, 9, 5508–5515. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.Q.; Freire-de-Lima, C.G.; Schiemann, W.P.; Bratton, D.L.; Vandivier, R.W.; Henson, P.M. Transcriptional and translational regulation of TGF-beta production in response to apoptotic cells. J. Immunol. 2008, 181, 3575–3585. [Google Scholar] [CrossRef] [PubMed]
- Chegini, N.; Tang, X.M.; Ma, C. Regulation of transforming growth factor-beta1 expression by granulocyte macrophage-colony-stimulating factor in leiomyoma and myometrial smooth muscle cells. J. Clin. Endocrinol. Metab. 1999, 84, 4138–4143. [Google Scholar] [PubMed]
- Grewal, J.S.; Mukhin, Y.V.; Garnovskaya, M.N.; Raymond, J.R.; Greene, E.L. Serotonin 5-HT2A receptor induces TGF-beta1 expression in mesangial cells via ERK: Proliferative and fibrotic signals. Am. J. Physiol. 1999, 276, F922–F930. [Google Scholar] [PubMed]
- Li, Z.-D.; Bork, J.P.; Krueger, B.; Patsenker, E.; Schulze-Krebs, A.; Hahn, E.G.; Schuppan, D. VEGF induces proliferation, migration, and TGF-beta1 expression in mouse glomerular endothelial cells via mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Biochem. Biophys. Res. Commun. 2005, 334, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; Mulder, K.M. Requirement of Ras/MAPK pathway activation by transforming growth factor beta for transforming growth factor beta 1 production in a Smad-dependent pathway. J. Biol. Chem. 2000, 275, 30765–30773. [Google Scholar] [CrossRef] [PubMed]
- Birchenall-Roberts, M.C.; Ruscetti, F.W.; Kasper, J.; Lee, H.D.; Friedman, R.; Geiser, A.; Sporn, M.B.; Roberts, A.B.; Kim, S.J. Transcriptional regulation of the transforming growth factor beta 1 promoter by v-src gene products is mediated through the AP-1 complex. Mol. Cell. Biol. 1990, 10, 4978–4983. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-Y.; Ito, K.; Hayashi, R.; Jazrawi, E.P.I.; Barnes, P.J.; Adcock, I.M. NF-kappaB and activator protein 1 response elements and the role of histone modifications in IL-1beta-induced TGF-beta1 gene transcription. J. Immunol. 2006, 176, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Geiser, A.G.; Busam, K.J.; Kim, S.J.; Lafyatis, R.; O’Reilly, M.A.; Webbink, R.; Roberts, A.B.; Sporn, M.B. Regulation of the transforming growth factor-beta 1 and -beta 3 promoters by transcription factor Sp1. Gene 1993, 129, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Glick, A.; Sporn, M.; Roberts, A. Characterization of the promoter region of the human transforming growth factor-beta 1 gene. J. Biol. Chem. 1989, 264, 402–408. [Google Scholar] [PubMed]
- Kinjyo, I.; Inoue, H.; Hamano, S.; Fukuyama, S.; Yoshimura, T.; Koga, K.; Takaki, H.; Himeno, K.; Takaesu, G.; Kobayashi, T.; et al. Loss of SOCS3 in T helper cells resulted in reduced immune responses and hyperproduction of interleukin 10 and transforming growth factor-beta 1. J. Exp. Med. 2006, 203, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Kubiczkova, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. TGF-β—An excellent servant but a bad master. J. Transl. Med. 2012, 10, 183. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [PubMed]
- Papageorgis, P.; Lambert, A.W.; Ozturk, S.; Gao, F.; Pan, H.; Manne, U.; Alekseyev, Y.O.; Thiagalingam, A.; Abdolmaleky, H.M.; Lenburg, M.; et al. Smad signaling is required to maintain epigenetic silencing during breast cancer progression. Cancer Res. 2010, 70, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.; Cheung, E.; Petrakis, T.G.; Howell, M.; Kraus, W.L.; Hill, C.S. Smads orchestrate specific histone modifications and chromatin remodeling to activate transcription. EMBO J. 2006, 25, 4490–4502. [Google Scholar] [CrossRef] [PubMed]
- Xi, Q.; Wang, Z.; Zaromytidou, A.-I.; Zhang, X.H.-F.; Chow-Tsang, L.-F.; Liu, J.X.; Kim, H.; Barlas, A.; Manova-Todorova, K.; Kaartinen, V.; et al. A poised chromatin platform for TGF-β access to master regulators. Cell 2011, 147, 1511–1524. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, A.; Howe, P.H. The tale of transforming growth factor-beta (TGFbeta) signaling: A soigné enigma. IUBMB Life 2009, 61, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Papageorgis, P. TGFβ signaling in tumor initiation, epithelial-to-mesenchymal transition, and metastasis. J. Oncol. 2015, 2015, 587193. [Google Scholar] [CrossRef] [PubMed]
- Tucker, R.F.; Shipley, G.D.; Moses, H.L.; Holley, R.W. Growth inhibitor from BSC-1 cells closely related to platelet type beta transforming growth factor. Science 1984, 226, 705–707. [Google Scholar] [CrossRef] [PubMed]
- Ristow, H.J. BSC-1 growth inhibitor/type beta transforming growth factor is a strong inhibitor of thymocyte proliferation. Proc. Natl. Acad. Sci. USA 1986, 83, 5531–5533. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Pol, J.A.; Talkad, V.D.; Klos, D.J.; Hamilton, P.D. Suppression of the EGF-dependent induction of c-myc proto-oncogene expression by transforming growth factor beta in a human breast carcinoma cell line. Biochem. Biophys. Res. Commun. 1987, 144, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Pietenpol, J.A.; Holt, J.T.; Stein, R.W.; Moses, H.L. Transforming growth factor beta 1 suppression of c-myc gene transcription: Role in inhibition of keratinocyte proliferation. Proc. Natl. Acad. Sci. USA 1990, 87, 3758–3762. [Google Scholar] [CrossRef] [PubMed]
- Hannon, G.J.; Beach, D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature 1994, 371, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Datto, M.B.; Li, Y.; Panus, J.F.; Howe, D.J.; Xiong, Y.; Wang, X.F. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc. Natl. Acad. Sci. USA 1995, 92, 5545–5549. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Kato, J.Y.; Solomon, M.J.; Sherr, C.J.; Massague, J.; Roberts, J.M.; Koff, A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994, 8, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Pillaire, M.J.; Casagrande, F.; Malecaze, F.; Manenti, S.; Darbon, J.M. Regulation by transforming growth factor-beta 1 of G1 cyclin-dependent kinases in human retinal epithelial cells. Exp. Eye Res. 1999, 68, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Djaborkhel, R.; Tvrdík, D.; Eckschlager, T.; Raska, I.; Müller, J. Cyclin A down-regulation in TGFbeta1-arrested follicular lymphoma cells. Exp. Cell Res. 2000, 261, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasagakis, K.; Thölke, D.; Farthmann, B.; Eberle, J.; Mansmann, U.; Orfanos, C.E. Elevated plasma levels of transforming growth factor (TGF)-beta1 and TGF-beta2 in patients with disseminated malignant melanoma. Br. J. Cancer 1998, 77, 1492–1494. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.M.; Anscher, M.S.; Murase, T.; Abbott, B.D.; Iglehart, J.D.; Jirtle, R.L. Elevated plasma transforming growth factor-beta 1 levels in breast cancer patients decrease after surgical removal of the tumor. Ann. Surg. 1995, 222, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Ivanović, V.; Demajo, M.; Krtolica, K.; Krajnović, M.; Konstantinović, M.; Baltić, V.; Prtenjak, G.; Stojiljković, B.; Breberina, M.; Nesković-Konstantinović, Z.; et al. Elevated plasma TGF-beta1 levels correlate with decreased survival of metastatic breast cancer patients. Clin. Chim. Acta. 2006, 371, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Friess, H.; Yamanaka, Y.; Büchler, M.; Ebert, M.; Beger, H.G.; Gold, L.I.; Korc, M. Enhanced expression of transforming growth factor beta isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology 1993, 105, 1846–1856. [Google Scholar] [CrossRef] [PubMed]
- Mitropoulos, D.; Kiroudi, A.; Christelli, E.; Serafetinidis, E.; Zervas, A.; Anastasiou, I.; Dimopoulos, C. Expression of transforming growth factor beta in renal cell carcinoma and matched non-involved renal tissue. Urol. Res. 2004, 32, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Xiong, B.; Gong, L.-L.; Zhang, F.; Hu, M.-B.; Yuan, H.-Y. TGF beta1 expression and angiogenesis in colorectal cancer tissue. World J. Gastroenterol. 2002, 8, 496–498. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ma, L.; He, Q.; Zhang, S.; Zhang, C.; Jia, W. TGF-β1 expression is associated with invasion and metastasis of intrahepatic cholangiocarcinoma. Biol. Res. 2015, 48, 26. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-C.; Tang, S.-J.; Sun, G.-H.; Sun, K.-H. CXCR7 mediates TGFβ1-promoted EMT and tumor-initiating features in lung cancer. Oncogene 2015, 35, 2123–2132. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-W.; Koh, Y.; Kim, D.-W.; Ahn, Y.-O.; Kim, T.M.; Han, S.-W.; Oh, D.-Y.; Lee, S.-H.; Im, S.-A.; Kim, T.-Y.; et al. Clinical Implications of VEGF, TGF-β1, and IL-1β in Patients with Advanced Non-small Cell Lung Cancer. Cancer Res. Treat. 2013, 45, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Lu, Q.; Shen, B.; Huang, X.; Shen, L.; Zheng, X.; Huang, R.; Yan, J.; Guo, H. TGFβ1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci. Rep. 2015, 5, 11924. [Google Scholar] [CrossRef] [PubMed]
- Terabe, M.; Matsui, S.; Park, J.-M.; Mamura, M.; Noben-Trauth, N.; Donaldson, D.D.; Chen, W.; Wahl, S.M.; Ledbetter, S.; Pratt, B.; et al. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: Abrogation prevents tumor recurrence. J. Exp. Med. 2003, 198, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Puig, P.E.; Roux, S.; Parcellier, A.; Schmitt, E.; Solary, E.; Kroemer, G.; Martin, F.; Chauffert, B.; Zitvogel, L. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J. Exp. Med. 2005, 202, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Dumitriu, I.E.; Dunbar, D.R.; Howie, S.E.; Sethi, T.; Gregory, C.D. Human dendritic cells produce TGF-beta 1 under the influence of lung carcinoma cells and prime the differentiation of CD4+CD25+Foxp3+ regulatory T cells. J. Immunol. 2009, 182, 2795–2807. [Google Scholar] [CrossRef] [PubMed]
- Kadin, M.E.; Cavaille-Coll, M.W.; Gertz, R.; Massagué, J.; Cheifetz, S.; George, D. Loss of receptors for transforming growth factor beta in human T-cell malignancies. Proc. Natl. Acad. Sci. USA 1994, 91, 6002–6006. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, S.; Wang, J.; Myeroff, L.; Parsons, R.; Sun, L.; Lutterbaugh, J.; Fan, R.S.; Zborowska, E.; Kinzler, K.W.; Vogelstein, B. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 1995, 268, 1336–1338. [Google Scholar] [CrossRef] [PubMed]
- Garrigue-Antar, L.; Muñoz-Antonia, T.; Antonia, S.J.; Gesmonde, J.; Vellucci, V.F.; Reiss, M. Missense mutations of the transforming growth factor beta type II receptor in human head and neck squamous carcinoma cells. Cancer Res. 1995, 55, 3982–3987. [Google Scholar] [PubMed]
- Guo, Y.; Jacobs, S.C.; Kyprianou, N. Down-regulation of protein and mRNA expression for transforming growth factor-beta (TGF-beta1) type I and type II receptors in human prostate cancer. Int. J. Cancer 1997, 71, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.Y.; Ahn, H.J.; Zelner, D.J.; Shaw, J.W.; Lang, S.; Kato, M.; Oefelein, M.G.; Miyazono, K.; Nemeth, J.A.; Kozlowski, J.M.; et al. Loss of expression of transforming growth factor beta type I and type II receptors correlates with tumor grade in human prostate cancer tissues. Clin. Cancer Res. 1996, 2, 1255–1261. [Google Scholar] [PubMed]
- Lu, S.-L.; Herrington, H.; Reh, D.; Weber, S.; Bornstein, S.; Wang, D.; Li, A.G.; Tang, C.-F.; Siddiqui, Y.; Nord, J.; et al. Loss of transforming growth factor-beta type II receptor promotes metastatic head-and-neck squamous cell carcinoma. Genes Dev. 2006, 20, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Schutte, M.; Hruban, R.H.; Hedrick, L.; Cho, K.R.; Nadasdy, G.M.; Weinstein, C.L.; Bova, G.S.; Isaacs, W.B.; Cairns, P.; Nawroz, H.; et al. DPC4 gene in various tumor types. Cancer Res. 1996, 56, 2527–2530. [Google Scholar] [PubMed]
- Hahn, S.A.; Hoque, A.T.; Moskaluk, C.A.; da Costa, L.T.; Schutte, M.; Rozenblum, E.; Seymour, A.B.; Weinstein, C.L.; Yeo, C.J.; Hruban, R.H.; et al. Homozygous deletion map at 18q21.1 in pancreatic cancer. Cancer Res. 1996, 56, 490–494. [Google Scholar] [PubMed]
- Eppert, K.; Scherer, S.W.; Ozcelik, H.; Pirone, R.; Hoodless, P.; Kim, H.; Tsui, L.C.; Bapat, B.; Gallinger, S.; Andrulis, I.L.; et al. MADR2 maps to 18q21 and encodes a TGFbeta-regulated MAD-related protein that is functionally mutated in colorectal carcinoma. Cell 1996, 86, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Sjöblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, M.; Doody, J.; Timokhina, I.; Massagué, J. A mechanism of repression of TGFbeta/Smad signaling by oncogenic Ras. Genes Dev. 1999, 13, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, M.; Doody, J.; Massagué, J. Opposing BMP and EGF signalling pathways converge on the TGF-beta family mediator Smad1. Nature 1997, 389, 618–622. [Google Scholar] [PubMed]
- Kleeff, J.; Ishiwata, T.; Maruyama, H.; Friess, H.; Truong, P.; Büchler, M.W.; Falb, D.; Korc, M. The TGF-beta signaling inhibitor Smad 7 enhances tumorigenicity in pancreatic cancer. Oncogene 1999, 18, 5363–5372. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, S.C.; Mariani, A.; Reinholz, M.M.; Keeney, G.L.; Spelsberg, T.C.; Podratz, K.C.; Janknecht, R. Overexpression of the TGF-beta antagonist Smad 7 in endometrial cancer. Gynecol. Oncol. 2005, 96, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, P.; Agrawal, A.; Bhushan, R.; Chavalmane, A.K.; Kalathur, R.; Takahashi, T.; Kondaiah, P. Expression profiling of genes regulated by TGF-beta: Differential regulation in normal and tumour cells. BMC Genom. 2007, 8, 98. [Google Scholar] [CrossRef] [PubMed]
- Lebrun, J.-J.; Lebrun, J.-J. The dual role of TGFβ in human cancer: From tumor suppression to cancer metastasis. ISRN Mol. Biol. 2012, 2012, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.L.; Jegathesan, M.; Darnell, R.B. Dendritic cell maturation is required for the cross-tolerization of CD8+ T cells. Nat. Immunol. 2001, 2, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Hawiger, D.; Inaba, K.; Dorsett, Y.; Guo, M.; Mahnke, K.; Rivera, M.; Ravetch, J.V.; Steinman, R.M.; Nussenzweig, M.C. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 2001, 194, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Dhodapkar, M.V.; Steinman, R.M.; Krasovsky, J.; Munz, C.; Bhardwaj, N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J. Exp. Med. 2001, 193, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef] [PubMed]
- Joffre, O.; Nolte, M.A.; Spörri, R.; Reis e Sousa, C. Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunol. Rev. 2009, 227, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Tsumura, H.; Miwa, M.; Inaba, K. Contrasting effects of TGF-beta 1 and TNF-alpha on the development of dendritic cells from progenitors in mouse bone marrow. Stem Cells 1997, 15, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yang, J.; Qian, J.; Wezeman, M.; Kwak, L.W.; Yi, Q. Tumor evasion of the immune system: Inhibiting p38 MAPK signaling restores the function of dendritic cells in multiple myeloma. Blood 2006, 107, 2432–2439. [Google Scholar] [CrossRef] [PubMed]
- Oosterhoff, D.; Lougheed, S.; van de Ven, R.; Lindenberg, J.; van Cruijsen, H.; Hiddingh, L.; Kroon, J.; van den Eertwegh, A.J.M.; Hangalapura, B.; Scheper, R.J.; et al. Tumor-mediated inhibition of human dendritic cell differentiation and function is consistently counteracted by combined p38 MAPK and STAT3 inhibition. Oncoimmunology 2012, 1, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Poliakov, A.; Liu, C.; Liu, Y.; Deng, Z.; Wang, J.; Cheng, Z.; Shah, S.V.; Wang, G.-J.; Zhang, L.; et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int. J. Cancer 2009, 124, 2621–2633. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.G.; Megiel, C.; Russell, S.M.; Bingham, B.; Arger, N.; Woo, T.; Epstein, A.L. Functional characterization of human Cd33+ and Cd11b+ myeloid-derived suppressor cell subsets induced from peripheral blood mononuclear cells co-cultured with a diverse set of human tumor cell lines. J. Transl. Med. 2011, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Marvel, D.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J. Clin. Investig. 2015, 125, 3356–3364. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Revy, P.; Regnault, A.; Lepelletier, Y.; Dy, M.; Brousse, N.; Amigorena, S.; Hermine, O.; Durandy, A. TGF-beta 1 prevents the noncognate maturation of human dendritic Langerhans cells. J. Immunol. 1999, 162, 4567–4575. [Google Scholar] [PubMed]
- Laouar, Y.; Town, T.; Jeng, D.; Tran, E.; Wan, Y.; Kuchroo, V.K.; Flavell, R.A. TGF-beta signaling in dendritic cells is a prerequisite for the control of autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2008, 105, 10865–10870. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, R.; Larmonier, C.B.; Thurston, R.D.; Midura-Kiela, M.T.; Zheng, S.G.; Ghishan, F.K.; Kiela, P.R. Dendritic cell-specific disruption of TGF-β receptor II leads to altered regulatory T cell phenotype and spontaneous multiorgan autoimmunity. J. Immunol. 2012, 189, 3878–3893. [Google Scholar] [CrossRef] [PubMed]
- Hargadon, K.M.; Bishop, J.D.; Brandt, J.P.; Hand, Z.C.; Ararso, Y.T.; Forrest, O.A. Melanoma-derived factors alter the maturation and activation of differentiated tissue-resident dendritic cells. Immunol. Cell Biol. 2016, 94, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Belladonna, M.L.; Volpi, C.; Bianchi, R.; Vacca, C.; Orabona, C.; Pallotta, M.T.; Boon, L.; Gizzi, S.; Fioretti, M.C.; Grohmann, U.; et al. Cutting edge: Autocrine TGF-beta sustains default tolerogenesis by IDO-competent dendritic cells. J. Immunol. 2008, 181, 5194–5198. [Google Scholar] [CrossRef] [PubMed]
- Hargadon, K.M. Tumor-altered dendritic cell function: Implications for anti-tumor immunity. Front. Immunol. 2013, 4, 192. [Google Scholar] [CrossRef] [PubMed]
- Scarlett, U.K.; Rutkowski, M.R.; Rauwerdink, A.M.; Fields, J.; Escovar-Fadul, X.; Baird, J.; Cubillos-Ruiz, J.R.; Jacobs, A.C.; Gonzalez, J.L.; Weaver, J.; et al. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J. Exp. Med. 2012, 209, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Sisirak, V.; Faget, J.; Gobert, M.; Goutagny, N.; Vey, N.; Treilleux, I.; Renaudineau, S.; Poyet, G.; Labidi-Galy, S.I.; Goddard-Leon, S.; et al. Impaired IFN-α production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progression. Cancer Res. 2012, 72, 5188–5197. [Google Scholar] [CrossRef] [PubMed]
- Sisirak, V.; Vey, N.; Goutagny, N.; Renaudineau, S.; Malfroy, M.; Thys, S.; Treilleux, I.; Labidi-Galy, S.I.; Bachelot, T.; Dezutter-Dambuyant, C.; et al. Breast cancer-derived transforming growth factor-β and tumor necrosis factor-α compromise interferon-α production by tumor-associated plasmacytoid dendritic cells. Int. J. Cancer 2013, 133, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Standiford, T.J.; Kuick, R.; Bhan, U.; Chen, J.; Newstead, M.; Keshamouni, V.G. TGF-β-induced IRAK-M expression in tumor-associated macrophages regulates lung tumor growth. Oncogene 2011, 30, 2475–2484. [Google Scholar] [CrossRef] [PubMed]
- Machado, C.M.L.; Andrade, L.N.S.; Teixeira, V.R.; Costa, F.F.; Melo, C.M.; dos Santos, S.N.; Nonogaki, S.; Liu, F.-T.; Bernardes, E.S.; Camargo, A.A.; et al. Galectin-3 disruption impaired tumoral angiogenesis by reducing VEGF secretion from TGFβ1-induced macrophages. Cancer Med. 2014, 3, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Alleva, D.G.; Walker, T.M.; Elgert, K.D. Induction of macrophage suppressor activity by fibrosarcoma-derived transforming growth factor-beta 1: Contrasting effects on resting and activated macrophages. J. Leukoc. Biol. 1995, 57, 919–928. [Google Scholar] [PubMed]
- Cao, Q.; Wang, Y.; Zheng, D.; Sun, Y.; Wang, Y.; Lee, V.W.S.; Zheng, G.; Tan, T.K.; Ince, J.; Alexander, S.I.; et al. IL-10/TGF-beta-modified macrophages induce regulatory T cells and protect against adriamycin nephrosis. J. Am. Soc. Nephrol. 2010, 21, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Steidl, C.; Lee, T.; Shah, S.P.; Farinha, P.; Han, G.; Nayar, T.; Delaney, A.; Jones, S.J.; Iqbal, J.; Weisenburger, D.D.; et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N. Engl. J. Med. 2010, 362, 875–885. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Kurahara, H.; Shinchi, H.; Mataki, Y.; Maemura, K.; Noma, H.; Kubo, F.; Sakoda, M.; Ueno, S.; Natsugoe, S.; Takao, S. Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J. Surg. Res. 2011, 167, e211–e219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.C.; Gao, J.; Wang, J.; Rao, Z.G.; Wang, B.C.; Gao, J.F. Tumor-associated macrophages infiltration is associated with peritumoral lymphangiogenesis and poor prognosis in lung adenocarcinoma. Med. Oncol. 2011, 28, 1447–1452. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Sadot, E.; Basturk, O.; Klimstra, D.S.; Gönen, M.; Lokshin, A.; Do, R.K.G.; D’Angelica, M.I.; DeMatteo, R.P.; Kingham, T.P.; Jarnagin, W.R.; et al. Tumor-associated neutrophils and malignant progression in intraductal papillary mucinous neoplasms. Ann. Surg. 2015, 262, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Hu, P.; Donskov, F.; Wang, G.; Liu, Q.; Du, J. Tumor-associated neutrophils as a new prognostic factor in cancer: A systematic review and meta-analysis. PLoS ONE 2014, 9, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.-M.; Jing, Y.-Y.; Yu, G.-F.; Kou, X.-R.; Ye, F.; Gao, L.; Li, R.; Zhao, Q.-D.; Yang, Y.; Lu, Z.-H.; et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014, 352, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Tham, M.; Tan, K.W.; Keeble, J.; Wang, X.; Hubert, S.; Barron, L.; Tan, N.S.; Kato, M.; Prevost-Blondel, A.; Angeli, V.; et al. Melanoma-initiating cells exploit M2 macrophage TGFβ and arginase pathway for survival and proliferation. Oncotarget 2014, 5, 12027–12042. [Google Scholar] [CrossRef] [PubMed]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Van den Broek, M.E.; Kägi, D.; Ossendorp, F.; Toes, R.; Vamvakas, S.; Lutz, W.K.; Melief, C.J.; Zinkernagel, R.M.; Hengartner, H. Decreased tumor surveillance in perforin-deficient mice. J. Exp. Med. 1996, 184, 1781–1790. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Thia, K.Y.; Street, S.E.; MacGregor, D.; Godfrey, D.I.; Trapani, J.A. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J. Exp. Med. 2000, 192, 755–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penn, I. Tumors of the immunocompromised patient. Annu. Rev. Med. 1988, 39, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Dantal, J.; Soulillou, J.-P. Immunosuppressive drugs and the risk of cancer after organ transplantation. N. Engl. J. Med. 2009, 353, 1371–1373. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.M.; Patel, A.; Hunt, M.J.; Tefany, F.J.; Barnetson, R.S.C. Spontaneous regression of human melanoma/nonmelanoma skin cancer: Association with infiltrating CD4+ T cells. World J. Surg. 1995, 19, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Yamshchikov, G.; Thompson, L.; Ross, W.G.; Galavotti, H.; Aquila, W.; Deacon, D.; Caldwell, J.; Patterson, J.W.; Hunt, D.F.; Slingluff, C.L. Analysis of a natural immune response against tumor antigens in a melanoma survivor: Lessons applicable to clinical trial evaluations. Clin. Cancer Res. 2001, 7, 909s–916s. [Google Scholar] [PubMed]
- Anichini, A.; Scarito, A.; Molla, A.; Parmiani, G.; Mortarini, R. Differentiation of CD8+ T cells from tumor-invaded and tumor-free lymph nodes of melanoma patients: Role of common gamma-chain cytokines. J. Immunol. 2003, 171, 2134–2141. [Google Scholar] [CrossRef] [PubMed]
- Zippelius, A.; Batard, P.; Rubio-godoy, V.; Bioley, G.; Lie, D.; Lejeune, F.; Rimoldi, D.; Guillaume, P.; Meidenbauer, N.; Mackensen, A.; et al. Effector function of human tumor-specific CD8 T cells in melanoma lesions: A state of local functional tolerance effector function of human tumor-specific CD8 T cells in melanoma lesions: A state of local functional tolerance. Cancer Res. 2004, 64, 2865–2873. [Google Scholar] [CrossRef] [PubMed]
- Mortarini, R.; Piris, A.; Maurichi, A.; Molla, A.; Bersani, I.; Bono, A.; Bartoli, C.; Santinami, M.; Lombardo, C.; Ravagnani, F.; et al. Lack of terminally differentiated tumor-specific CD8+ T cells at tumor site in spite of antitumor immunity to self-antigens in human metastatic melanoma. Cancer Res. 2003, 63, 2535–2545. [Google Scholar] [PubMed]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Xu, D.; Liu, Z.; Shi, M.; Zhao, P.; Fu, B.; Zhang, Z.; Yang, H.; Zhang, H.; Zhou, C.; et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology 2007, 132, 2328–2339. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Sanjabi, S.; Flavell, R.A. Transforming growth factor-β controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 2006, 25, 455–471. [Google Scholar] [CrossRef] [PubMed]
- Marie, J.C.; Liggitt, D.; Rudensky, A.Y. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-β receptor. Immunity 2006, 25, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.K.; Schietinger, A.; Liggitt, H.D.; Tan, X.; Funk, S.; Freeman, G.J.; Ratliff, T.L.; Greenberg, N.M.; Greenberg, P.D. Cell-intrinsic abrogation of TGF-β signaling delays but does not prevent dysfunction of self/tumor-specific CD8 T cells in a murine model of autochthonous prostate cancer. J. Immunol. 2012, 189, 3936–3946. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Bai, O.; Yuan, J.; Chibbar, R.; Slattery, K.; Wei, Y.; Deng, Y.; Xiang, J. Tumor apoptotic bodies inhibit CTL responses and antitumor immunity via membrane-bound transforming growth factor-beta1 inducing CD8+ T-cell anergy and CD4+ Tr1 cell responses. Cancer Res. 2009, 69, 7756–7766. [Google Scholar] [CrossRef] [PubMed]
- Shvedova, A.A.; Kisin, E.R.; Yanamala, N.; Tkach, A.V.; Gutkin, D.W.; Star, A.; Shurin, G.V.; Kagan, V.E.; Shurin, M.R. MDSC and TGFβ are required for facilitation of tumor growth in the lungs of mice exposed to carbon nanotubes. Cancer Res. 2015, 75, 1615–1623. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.-J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.A.; Meyer, J.R.; Greco, S.J.; Corcoran, K.E.; Bryan, M.; Rameshwar, P. Mesenchymal stem cells protect breast cancer cells through regulatory T cells: Role of mesenchymal stem cell-derived TGF-beta. J. Immunol. 2010, 184, 5885–5894. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Pan, P.-Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.-H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Shurin, G.V.; Ma, Y.; Shurin, M.R. Immunosuppressive mechanisms of regulatory dendritic cells in cancer. Cancer Microenviron. 2013, 6, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kitani, A.; Strober, W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J. Exp. Med. 2001, 194, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-L.; Pittet, M.J.; Gorelik, L.; Flavell, R.A.; Weissleder, R.; von Boehmer, H.; Khazaie, K. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 419–424. [Google Scholar] [CrossRef] [PubMed]
- De Jong, E.; Suddason, T.; Lord, G.M. Translational mini-review series on Th17 cells: Development of mouse and human T helper 17 cells. Clin. Exp. Immunol. 2010, 159, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Restifo, N.P. T(H)17 cells in tumour immunity and immunotherapy. Nat. Rev. Immunol. 2010, 10, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yi, T.; Kortylewski, M.; Pardoll, D.M.; Zeng, D.; Yu, H. IL-17 can promote tumor growth through an IL-6–Stat3 signaling pathway. J. Exp. Med. 2009, 206, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Li, H.; Yusuf, N.; Elmets, C.A.; Li, J.; Mountz, J.D.; Xu, H. IL-17 promotes tumor development through the induction of tumor promoting microenvironments at tumor sites and myeloid-derived suppressor cells. J. Immunol. 2010, 184, 2281–2288. [Google Scholar] [CrossRef] [PubMed]
- De Simone, V.; Franzè, E.; Ronchetti, G.; Colantoni, A.; Fantini, M.C.; di Fusco, D.; Sica, G.S.; Sileri, P.; MacDonald, T.T.; Pallone, F.; et al. Th17-type cytokines, IL-6 and TNF-α synergistically activate STAT3 and NF-κB to promote colorectal cancer cell growth. Oncogene 2015, 34, 3493–3503. [Google Scholar] [CrossRef] [PubMed]
- Prabhala, R.H.; Pelluru, D.; Fulciniti, M.; Prabhala, H.K.; Nanjappa, P.; Song, W.; Pai, C.; Amin, S.; Tai, Y.-T.; Richardson, P.G.; et al. Elevated IL-17 produced by TH17 cells promotes myeloma cell growth and inhibits immune function in multiple myeloma. Blood 2010, 115, 5385–5392. [Google Scholar] [CrossRef] [PubMed]
- Iida, T.; Iwahashi, M.; Katsuda, M.; Ishida, K.; Nakamori, M.; Nakamura, M.; Naka, T.; Ojima, T.; Ueda, K.; Hayata, K.; et al. Tumor-infiltrating CD4+ Th17 cells produce IL-17 in tumor microenvironment and promote tumor progression in human gastric cancer. Oncol. Rep. 2011, 25, 1271–1277. [Google Scholar] [PubMed]
- Zhang, J.-P.; Yan, J.; Xu, J.; Pang, X.-H.; Chen, M.-S.; Li, L.; Wu, C.; Li, S.-P.; Zheng, L. Increased intratumoral IL-17-producing cells correlate with poor survival in hepatocellular carcinoma patients. J. Hepatol. 2009, 50, 980–989. [Google Scholar] [CrossRef] [PubMed]
- Waldhauer, I.; Steinle, A. NK cells and cancer immunosurveillance. Oncogene 2008, 27, 5932–5943. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Sui, Q.; Zhang, J.; Sun, X.; Zhang, C.; Han, Q.; Tian, Z. NK cells are the crucial antitumor mediators when STAT3-mediated immunosuppression is blocked in hepatocellular carcinoma. J. Immunol. 2014, 193, 2016–2023. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.; Bhattacharya-Chatterjee, M.; O’Malley, B.W.; Chatterjee, S.K. Inhibition of NK cell activity through TGF-1 by down-regulation of NKG2D in a murine model of head and neck cancer. J. Immunol. 2005, 175, 5541–5550. [Google Scholar] [CrossRef] [PubMed]
- Donatelli, S.S.; Zhou, J.-M.; Gilvary, D.L.; Eksioglu, E.A.; Chen, X.; Cress, W.D.; Haura, E.B.; Schabath, M.B.; Coppola, D.; Wei, S.; et al. TGF-inducible microRNA-183 silences tumor-associated natural killer cells. Proc. Natl. Acad. Sci. USA 2014, 111, 4203–4208. [Google Scholar] [CrossRef] [PubMed]
- Viel, S.; Marçais, A.; Guimaraes, F.S.-F.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal. 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-C.; Lee, K.-M.; Kim, D.-W.; Heo, D.S. Elevated TGF-1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J. Immunol. 2004, 172, 7335–7340. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.J.; Heo, D.S.; Kang, S.H.; Lee, K.H.; Kim, W.S.; Kim, G.P.; Lee, J.A.; Lee, K.S.; Bang, Y.J.; Kim, N.K. Natural killer cell activity depression in peripheral blood and ascites from gastric cancer patients with high TGF-beta 1 expression. Anticancer Res. 1997, 18, 1591–1596. [Google Scholar]
- Kim, S.; Buchlis, G.; Fridlender, Z.G.; Sun, J.; Kapoor, V.; Cheng, G.; Haas, A.; Cheung, H.K.; Zhang, X.; Corbley, M.; et al. Systemic blockade of transforming growth factor-beta signaling augments the efficacy of immunogene therapy. Cancer Res. 2008, 68, 10247–10256. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, E.; Kapoor, V.; Cheung, H.K.; Ling, L.E.; DeLong, P.A.; Kaiser, L.R.; Albelda, S.M. Soluble type II transforming growth factor-beta receptor inhibits established murine malignant mesothelioma tumor growth by augmenting host antitumor immunity. Clin. Cancer Res. 2004, 10, 5907–5918. [Google Scholar] [CrossRef] [PubMed]
- Takaku, S.; Terabe, M.; Ambrosino, E.; Peng, J.; Lonning, S.; McPherson, J.M.; Berzofsky, J.A. Blockade of TGF-beta enhances tumor vaccine efficacy mediated by CD8(+) T cells. Int. J. Cancer 2010, 126, 1666–1674. [Google Scholar] [PubMed]
- Penafuerte, C.; Bautista-Lopez, N.; Bouchentouf, M.; Birman, E.; Forner, K.; Galipeau, J. Novel TGF-beta antagonist inhibits tumor growth and angiogenesis by inducing IL-2 receptor-driven STAT1 activation. J. Immunol. 2011, 186, 6933–6944. [Google Scholar] [CrossRef] [PubMed]
- Conroy, H.; Galvin, K.C.; Higgins, S.C.; Mills, K.H.G. Gene silencing of TGF-β1 enhances antitumor immunity induced with a dendritic cell vaccine by reducing tumor-associated regulatory T cells. Cancer Immunol. Immunother. 2012, 61, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Sakuraba, H.; Ishiguro, Y.; Yamagata, K.; Munakata, A.; Nakane, A. Blockade of TGF-β accelerates mucosal destruction through epithelial cell apoptosis. Biochem. Biophys. Res. Commun. 2007, 359, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues-Díez, R.; Rayego-Mateos, S.; Orejudo, M.; Aroeira, L.S.; Selgas, R.; Ortiz, A.; Egido, J.; Ruiz-Ortega, M. TGF-beta blockade increases renal inflammation caused by the C-terminal module of the CCN2. Mediat. Inflamm. 2015, 2015, 506041. [Google Scholar] [CrossRef] [PubMed]
- Lacouture, M.E.; Morris, J.C.; Lawrence, D.P.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Berzofsky, J.A.; Hsu, F.J.; Guitart, J. Cutaneous keratoacanthomas/squamous cell carcinomas associated with neutralization of transforming growth factor β by the monoclonal antibody fresolimumab (GC1008). Cancer Immunol. Immunother. 2015, 64, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yang, X.J.; Kundu, S.D.; Pins, M.; Javonovic, B.; Meyer, R.; Kim, S.-J.; Greenberg, N.M.; Kuzel, T.; Meagher, R.; et al. Blockade of transforming growth factor-{beta} signaling in tumor-reactive CD8(+) T cells activates the antitumor immune response cycle. Mol. Cancer Ther. 2006, 5, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yang, X.; Pins, M.; Javonovic, B.; Kuzel, T.; Kim, S.-J.; van Parijs, L.; Greenberg, N.M.; Liu, V.; Guo, Y.; et al. Adoptive transfer of tumor-reactive transforming growth factor-beta-insensitive CD8+ T cells: Eradication of autologous mouse prostate cancer. Cancer Res. 2005, 65, 1761–1769. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Jang, T.L.; Yang, X.; Park, I.; Meyer, R.E.; Kundu, S.; Pins, M.; Javonovic, B.; Kuzel, T.; Kim, S.-J.; et al. Infiltration of tumor-reactive transforming growth factor-beta insensitive CD8+ T cells into the tumor parenchyma is associated with apoptosis and rejection of tumor cells. Prostate 2006, 66, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Gate, D.; Danielpour, M.; Rodriguez, J.; Kim, G.-B.; Levy, R.; Bannykh, S.; Breunig, J.J.; Kaech, S.M.; Flavell, R.A.; Town, T. T-cell TGF-β signaling abrogation restricts medulloblastoma progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3458–E3466. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, Z.; Muranski, P.; Palmer, D.C.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. Inhibition of TGF-β signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy. Gene Ther. 2013, 20, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.E.; Dotti, G.; Lu, A.; Khalil, M.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M.; Bollard, C.M. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-beta receptor. J. Immunother. 2008, 31, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Lacuesta, K.; Buza, E.; Hauser, H.; Granville, L.; Pule, M.; Corboy, G.; Finegold, M.; Weiss, H.; Chen, S.Y.; Brenner, M.K.; et al. Assessing the safety of cytotoxic T lymphocytes transduced with a dominant negative transforming growth factor-beta receptor. J. Immunother. 2006, 29, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.-L.; Qin, W.-J.; Wen, W.-H.; Tian, F.; Song, B.; Zhang, Q.; Lee, C.; Zhong, W.; Guo, Y.-L.; Wang, H. TGF-beta insensitive dendritic cells: An efficient vaccine for murine prostate cancer. Cancer Immunol. Immunother. 2007, 56, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Wang, L.; Qin, W.; Wang, F.; Song, B.; Li, Y.; Wen, W.; Zhang, Z.; Zhu, K.; Zhang, Q.; et al. Vaccination with transforming growth factor-beta insensitive dendritic cells suppresses pulmonary metastases of renal carcinoma in mice. Cancer Lett. 2008, 271, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.-H.; Hong, S.-O.; Kim, J.H.; Noh, K.H.; Song, K.-H.; Lee, Y.-H.; Jeon, J.-H.; Kim, D.-W.; Seo, J.H.; Kim, T.W. The siRNA cocktail targeting interleukin 10 receptor and transforming growth factor-β receptor on dendritic cells potentiates tumour antigen-specific CD8(+) T cell immunity. Clin. Exp. Immunol. 2015, 181, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Baird, J.R.; Tesone, A.J.; Rutkowski, M.R.; Scarlett, U.K.; Camposeco-Jacobs, A.L.; Anadon-Arnillas, J.; Harwood, N.M.; Korc, M.; Fiering, S.N.; et al. Reprogramming tumor-associated dendritic cells in vivo using miRNA mimetics triggers protective immunity against ovarian cancer. Cancer Res. 2012, 72, 1683–1693. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the TGFβ pathway for cancer therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, J.P.; Kindler, H.L.; Papasavvas, E.; Sun, J.; Jacobs-Small, M.; Hull, J.; Schwed, D.; Ranganathan, A.; Newick, K.; Heitjan, D.F.; et al. Immunological effects of the TGFβ-blocking antibody GC1008 in malignant pleural mesothelioma patients. Oncoimmunology 2013, 2, e26218. [Google Scholar] [CrossRef] [PubMed]
- Senzer, N.; Barve, M.; Kuhn, J.; Melnyk, A.; Beitsch, P.; Lazar, M.; Lifshitz, S.; Magee, M.; Oh, J.; Mill, S.W.; et al. Phase I trial of & quot;bi-shRNAi(furin)/GMCSF DNA/autologous tumor cell & quot; vaccine (FANG) in advanced cancer. Mol. Ther. 2012, 20, 679–686. [Google Scholar] [PubMed]
- Senzer, N.; Barve, M.; Nemunaitis, J.; Kuhn, J.; Melnyk, A.; Beltsch, P.; Magee, M.; Oh, J.; Bedell, C.; Kumar, P.; et al. Long term follow up: Phase I trial of “bi-shRNA furin/GMCSF DNA/Autologous tumor cell” immunotherapy (FANG™) in advanced cancer. J. Vaccines Vaccin. 2013, 4, 8. [Google Scholar]
- Nemunaitis, J.; Barve, M.; Orr, D.; Kuhn, J.; Magee, M.; Lamont, J.; Bedell, C.; Wallraven, G.; Pappen, B.O.; Roth, A.; et al. Summary of bi-shRNA/GM-CSF augmented autologous tumor cell immunotherapy (FANG™) in advanced cancer of the liver. Oncology 2014, 87, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Ghisoli, M.; Barve, M.; Schneider, R.; Mennel, R.; Lenarsky, C.; Wallraven, G.; Pappen, B.O.; LaNoue, J.; Kumar, P.; Nemunaitis, D.; et al. Pilot trial of FANG immunotherapy in ewing’s sarcoma. Mol. Ther. 2015, 23, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- Ghisoli, M.; Barve, M.; Mennel, R.; Lenarsky, C.; Horvath, S.; Wallraven, G.; Pappen, B.O.; Whiting, S.; Rao, D.; Senzer, N.; et al. Three year follow up of GMCSF/bi-shRNA(furin) DNA transfected autologous tumor immunotherapy (Vigil(™)) in metastatic advanced ewing’s sarcoma. Mol. Ther. 2016. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial Identifier | Description of Therapy | Cancer | Status |
|---|---|---|---|
| Adoptive Cell Transfer Therapies | |||
| NCT00368082 | LMP-specific DNR-CTL | EBV+ lymphoma | Phase I; ongoing, not recruiting |
| NCT02065362 | LMP/BARF1/EBNA1-specific DNR-CTL ± lymphodepletion | EBV+ nasopharyngeal carcinoma | Phase I; currently recruiting |
| NCT00889954 | HER2 CAR/EBV-specific DNR-CTL | Advanced stage HER2+ malignancies | Phase I; ongoing, not recruiting |
| NCT02379520 | E6/E7-specific DNR-CTL | HPV-related/HPV+ cancers | Phase I; recruiting |
| NCT01955460 | Lymphodepletion + DNRII TIL + high-dose IL-2 | Melanoma | Phase I; recruiting |
| Autologous Tumor Cell Vaccines | |||
| NCT01061840 | Vigil™ (FANG™) bi-shRNAfurin + GM-CSF vaccine | Ewing sarcoma, non-small cell lung cancer, liver cancer, thyroid cancer | Phase I; ongoing, not recruiting |
| NCT01453361 | Vigil™ (FANG™) bi-shRNAfurin + GM-CSF vaccine | Advanced melanoma (Stage IIIc/IV) | Phase II; ongoing, not recruiting |
| NCT01505166 | Vigil™ (FANG™) bi-shRNAfurin + GM-CSF vaccine | Colorectal carcinoma with liver metastases | Phase II; ongoing, not recruiting |
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hargadon, K.M. Dysregulation of TGFβ1 Activity in Cancer and Its Influence on the Quality of Anti-Tumor Immunity. J. Clin. Med. 2016, 5, 76. https://doi.org/10.3390/jcm5090076
Hargadon KM. Dysregulation of TGFβ1 Activity in Cancer and Its Influence on the Quality of Anti-Tumor Immunity. Journal of Clinical Medicine. 2016; 5(9):76. https://doi.org/10.3390/jcm5090076
Chicago/Turabian StyleHargadon, Kristian M. 2016. "Dysregulation of TGFβ1 Activity in Cancer and Its Influence on the Quality of Anti-Tumor Immunity" Journal of Clinical Medicine 5, no. 9: 76. https://doi.org/10.3390/jcm5090076