
Current Treatment of Juvenile Myelomonocytic Leukemia


1
Division of Pediatric Hematology and Oncology, Department of Pediatrics and Adolescent Medicine, Medical Center, Faculty of Medicine, University of Freiburg, 79106 Freiburg, Germany
2
German Cancer Consortium (DKTK), 79106 Freiburg, Germany
*
Author to whom correspondence should be addressed.
J. Clin. Med. 2021, 10(14), 3084; https://doi.org/10.3390/jcm10143084
Submission received: 22 June 2021
/
Revised: 9 July 2021
/
Accepted: 10 July 2021
/
Published: 13 July 2021
(This article belongs to the Special Issue Advance in the Treatment of Pediatric Leukemia)
Abstract
:Juvenile myelomonocytic leukemia (JMML) is a rare pediatric leukemia characterized by mutations in five canonical RAS pathway genes. The diagnosis is made by typical clinical and hematological findings associated with a compatible mutation. Although this is sufficient for clinical decision-making in most JMML cases, more in-depth analysis can include DNA methylation class and panel sequencing analysis for secondary mutations. NRAS-initiated JMML is heterogeneous and adequate management ranges from watchful waiting to allogeneic hematopoietic stem cell transplantation (HSCT). Upfront azacitidine in KRAS patients can achieve long-term remissions without HSCT; if HSCT is required, a less toxic preparative regimen is recommended. Germline CBL patients often experience spontaneous resolution of the leukemia or exhibit stable mixed chimerism after HSCT. JMML driven by PTPN11 or NF1 is often rapidly progressive, requires swift HSCT and may benefit from pretransplant therapy with azacitidine. Because graft-versus-leukemia alloimmunity is central to cure high risk patients, the immunosuppressive regimen should be discontinued early after HSCT.
1. Introduction
JMML is a pediatric leukemia with shared features of myelodysplastic and myeloproliferative neoplasms, usually manifesting during early childhood with leukocytosis, thrombocytopenia, pronounced monocytosis, splenomegaly, immature precursors on peripheral blood (PB) smear, and bone marrow (BM) blast count below 20% [1,2,3]. Its clinical and hematological picture, as well as natural history and outcome, are remarkably diverse [4]. The common molecular denominator of JMML is the deregulation of the intracellular Ras signal transduction pathway, caused in >90% of cases by mutations in one (or, rarely, more than one) of five primordial genes (PTPN11, NRAS, KRAS, NF1, or CBL) [5]. For most patients, allogeneic hematopoietic stem cell transplantation (HSCT) is the only curative treatment option, in contrast to a smaller percentage of children who survive long-term without HSCT and eventually experience spontaneous clinical remissions [6,7]. Clinical and molecular risk factors were established to help predict the disease course and guide therapeutic decisions, including age at diagnosis, percentage of fetal hemoglobin (HbF), platelet count, and aberrant DNA methylation patterns [8,9]. In this article, we review the current knowledge of genetic and epigenetic properties of JMML and provide detailed recommendations for the clinical management of children diagnosed with this challenging disorder.
2. The Origin of JMML: The Ras Pathway
The Ras pathway is a sequence of kinases in the cell that serves as a chain of communication between extracellular mitogens and the cell nucleus [10]. External cytokine signals, relayed through receptor tyrosine kinases and intracellular adapter proteins, lead to guanosine exchange factor-mediated transformation of Ras proteins into their active guanosine triphosphate (GTP)-bound state (reviewed in more detail in [11,12]). The Ras signal is terminated by intrinsic Ras phosphatase activity, which converts Ras back to an inactive guanosine diphosphate (GDP)-bound configuration. An additional layer of regulation is provided by GTPase activating proteins (GAPs). Effects of Ras activation include the subsequent phosphorylation of Raf, Mek, and Erk kinases [13,14,15,16,17], activation of the mammalian target of rapamycin (mTOR) axis via phosphoinositide 3-kinase (PI3K) [18], and others [19]. Among nuclear targets are the transcription factors Jun and Fos [20].
Genetic mutations in specific Ras pathway components (PTPN11, NRAS, KRAS, NF1, or CBL), resulting in net hyperactivation of the Ras-GTP-GDP loop, are present in hematopoietic cells of >90% of children diagnosed with JMML [4,21,22,23,24,25,26]. These can be traced back to early myeloid stem/progenitor cell compartments [27,28,29], and they are found in patient cord blood samples [24], substantiating their role as initiating events and suggesting the inception of the leukemogenic sequence before birth [30].
Somatic mutations in exons 3 or 13 of the PTPN11 gene are present in ~35% of JMML cases [22,31], resulting in a gain-of-function of the nonreceptor tyrosine phosphatase Shp2 [32]. Somatic mutations in NRAS or KRAS codons 12, 13, or 61, accounting for ~25% of JMML cases [4,25,33], freeze Ras in its active GTP-bound form by inhibition of GTPase activity or resistance to GAPs [4]. Somatic PTPN11, NRAS, and KRAS mutations occur in heterozygous form in JMML, indicating strong cell-transforming capacity already in monoallelic fashion.
Two congenital developmental disorders predispose to JMML: NF-1 and CBL syndrome [26,34,35,36]. Here, the germline of the patient carries a monoallelic loss-of-function mutation of the NF1 or CBL gene, which may have been inherited or arisen de novo. JMML develops after somatic biallelic inactivation of the respective gene in hematopoietic progenitor cells, predominantly by mitotic gene recombination resulting in uniparental isodisomy [21,37]. NF1 functions as a Ras-GAP and thus negatively regulates the Ras pathway [38,39]. Indicative features in children with JMML/NF-1 are the presence of ≥6 cutaneous café au lait spots and/or the family history; other characteristics of NF-1, such as neurofibromas, optic pathway gliomas, bone lesions and neurological abnormalities, usually manifest only later. Overall, 10–15% of JMML cases are driven by NF1 [33,40]. CBL is a E3 ubiquitin ligase mediating the decay of receptor tyrosine kinases in the Ras pathway. Mutations targeting exons 8 or 9account for ~15% of JMML cases [26,33]. CBL syndrome, a Noonan-like rasopathy, has a wide phenotypic spectrum. Features include impaired growth, facial anomalies, developmental delay, cryptorchidism, autoimmune phenomena, and notably, neurovasculitis [26,37]. However, it is not rare for patients with JMML and CBL germline mutation to display no abnormalities at all [26,41,42].
Noonan syndrome (NS), the most common rasopathy with an incidence of 1 in 1000–2500 children [43], bears clinical similarities with Turner syndrome. Patients with NS exhibit a short statue, facial dysmorphism, congenital heart defects, skeletal defects, a webbed neck, mental retardation, and cryptorchidism. The genetic basis is a germline mutation in PTPN11 (around 50% of NS cases), SOS1, RAF1, KRAS, BRAF, NRAS or other members of the RAS pathway [5,22,44,45]. Children with NS may experience a polyclonal myeloproliferative disorder (MPD) at a very young age, sometimes shortly after birth [4,46]. Although the condition is indistinguishable from JMML by clinical and hematological features, it has a self-limiting course in the vast majority of cases. Only a small fraction of children with NS/MPD progress to JMML, presumably after the acquisition of additional genetic changes [5,47]. Although the landscape of PTPN11 mutations is not identical in JMML and NS/MPD [31], there is considerable overlap, and it is not well understood how the same mutation elicits a transient disorder when present in the germline and a fatal disorder when acquired somatically. Obviously, the occurrence of germline and somatic Ras pathway mutations in the same clinical context requires analysis of non-hematopoietic tissue (e.g., hair follicles or skin fibroblasts) to differentiate these conditions [9].
Systematic exome sequencing studies revealed that JMML is generally characterized by a paucity of somatic mutations in the neoplastic clone when compared to most other types of cancer [48]. However, subclonal secondary gene mutations can be found in up to half of the cases [23,24,48]. These mutations primarily target the SETBP1, JAK3, SH2B3, or ASXL1 genes. Not infrequently, the secondary mutations affect the Ras pathway itself (“Ras double mutants”). In addition, a role for subclonal mutations in the Polycomb Repressive Complex 2 network was highlighted [23]. Several studies have linked the presence of secondary mutations with an aggressive clinical course or disease progression [23,49]. Furthermore, an association with an increased risk of recurrence after allogeneic HSCT was demonstrated [23,49].
Less than 10% of JMML cases are negative for the five canonical driver mutations. Rarely, these children harbor germline or somatic activating RRAS mutations [23,50]. Recently, a CCDC88C-FLT3 fusion responsive to sorafenib was described in a pediatric patient with clinical features of JMML and monosomy 7 [51]. Other fusions detected in children with myeloproliferative disease include ALK [52,53], ROS1 [52,53], FIP1L1-RARA [54], HCMOGT-1-PDGFRB [55], NDEL1-PDGFRB [56], and NUP98-HOXA11 [57]. Although kinase fusion-positive cases without Ras pathway mutation may fulfill the clinical and diagnostic criteria of JMML, they likely represent a genetically distinct myeloproliferative neoplasm in childhood. When identified, these tyrosine kinase fusions offer an attractive target for personalized therapies [51,53].
3. Clinical and Hematological Features of JMML
JMML occurs in 1.2 children per million per year, accounting for 2% of pediatric hematopoietic malignancies [58]. One half of the children with JMML are diagnosed below the age of two years and two-thirds are male [59]. Clinical signs at diagnosis include non-specific symptoms such as infections, fatigue, or failure to thrive. Splenomegaly is noted in nearly all cases, often accompanied by hepatomegaly and lymphadenopathy. Pulmonary infiltration by leukemic cells manifests with dry cough, tachypnea and, radiologically, interstitial infiltrates [59,60]. Abdominal symptoms may arise in patients with intestinal infiltration [59]. Variable cutaneous features may be present, ranging from eczematous lesions to erythematous papules or nodules and/or petechiae [60]. In contrast to other pediatric leukemias, JMML does not usually invade the central nervous system. As a substantial proportion of JMML cases arise on the basis of an underlying predisposition syndrome, the clinician needs to examine the patient carefully, paying attention to growth, facial dysmorphism, congenital heart defects, skeletal anomalies, developmental status, and skin lesions such as café-au-lait macules or juvenile xanthogranulomas [45,61,62,63].
The PB smear typically shows mild to pronounced leukocytosis with monocytosis without a significantly increased number of blasts (median 2% myeloblasts) [60,64]. Immature precursor cells of the granulocytic lineage (myelocytes, metamyelocytes), immature monocytes and nucleated erythropoietic cells are found, giving the blood film examination a pivotal diagnostic role [59]. Platelet and erythrocyte counts are usually decreased, whereas the absolute monocyte count is increased to >1 × 109 G/L in all but exceptional cases [1]. Bone marrow examination is necessary to exclude acute leukemia, but is per se insufficient to confirm a suspected diagnosis of JMML. BM findings in JMML include hypercellularity from myelomonocytic proliferation, reduction of megakaryocytes and moderate increase of blasts (<20% myeloblasts) [59].
The combination of young age, splenomegaly, skin lesions, appearance of myeloid and erythroid precursors in the PB, and/or elevated levels of HbF should prompt the pediatric oncologist to suspect JMML and initiate specific tests. First of all, this involves the molecular analysis of driver mutations in the PTPN11, KRAS, NRAS, and CBL genes, and a meticulous search for features of NF-1 including family history. Genetic analysis of NF1 can be added, but it is laborious, and the interpretation of findings is not always straightforward. On cytogenetics, two-thirds of cases exhibit a normal karyotype. Monosomy 7 is the most frequent aberration [33,53,65], occurring in combination with PTPN11 and KRAS mutations, but rarely with NF1, NRAS, or CBL. A traditional hallmark of clonogenic JMML cells is their hypersensitivity to GM-CSF in vitro [66,67]. However, laboratory tests of this feature are poorly standardized and not widely available. In the era of efficient mutational analysis, GM-CSF hypersensitivity has become largely dispensable, but may potentially be helpful in occasional cases without a canonical driver mutation. The direct antiglobulin test may come back positive due to autoantibodies, but this is usually not accompanied by clinical or laboratory hemolysis [59]. Similarly, increased levels of IgG, IgM and IgA can be observed [59]. It was suggested that flow cytometric analysis of STAT5 hyperphosphorylation after stimulation with GM-CSF may aid in distinguishing JMML from other conditions [68].
Bacterial [69] and viral (e.g., Epstein–Barr virus [70], cytomegalovirus [71], and herpesvirus 6 [72]) infections can mimic the clinical and laboratory findings of JMML in infants, including fever, splenomegaly, leukocytosis with monocytosis, hypersensitivity to GM-CSF, and STAT5 hyperphosphorylation. Genetic or non-leukemic hematological disorders, such as infantile malignant osteopetrosis, leukocyte adhesion deficiency, Wiskott–Aldrich syndrome, or Ras-related autoimmune lymphoproliferative disease (RALD), must also be differentiated from JMML [69,73,74]. The latter is a non-malignant, chronic condition induced by an apoptosis defect in lymphocytes [74,75]. RALD is characterized by monocytosis, lymphoproliferation and autoimmune phenomena. Blood leukocytes exhibit similar somatic NRAS and KRAS mutations as in JMML [74], but these patients do not require aggressive treatment. Two cases of JMML evolving from RALD were described in the literature [75,76], highlighting the need for close observation. Differentiation of both entities can be difficult in the absence of monosomy 7. Functional apoptosis assays might be helpful to diagnose RALD [75].
4. The Emerging Role of Epigenetics
The genetic subtypes discussed above account for the phenotypic diversity of JMML only incompletely. For example, long-identified prognostic parameters, such as age of the patient, sex, platelet count, or elevated levels of fetal hemoglobin [6,40,59], do not correspond to a specific Ras genotype. Further molecular factors related to the course of the disease were observed in JMML, including micro and long non-coding RNA expression [77,78], AML-like expression profile [79], secondary mutations [48,49] and alterations of the fetal hematopoietic regulator gene LIN28B [80]. In addition, the previous observation of epigenetic dysregulation during Ras-mediated transformation suggested aberrant DNA methylation as a potential disease modifier [81,82].
The first study examining the role DNA methylation changes in a large European series of 127 children with JMML revealed CpG island hypermethylation of a candidate gene set in up to half of the cases [81]. Importantly, CpG hypermethylation at diagnosis was an independent risk factor for poor overall survival (OS) and risk of relapse after HSCT [81]. The conclusions were largely confirmed in a Japanese study investigating a similar candidate gene set [83]. Both studies observed that hypermethylation in JMML affected a narrow subset of gene promoters, as opposed to broad random distribution across all genetic regions examined, suggesting that high-risk JMML is characterized by a CpG island hypermethylation phenotype, as discovered previously in other specific cancer types [82,83,84]. Several follow-up studies corroborated this concept at the candidate gene level [85,86,87].
Extending these findings to a genome-wide scope using array-based methods, study groups in Europe [33], Japan [53] and United States [65] analyzed independent JMML cohorts with the aim to establish a methylation based risk-stratification. Comparing the methylome patterns of 167 children with JMML, the European Working Group of Myelodysplastic Syndromes in Childhood (EWOG-MDS) discriminated 3 distinct methylation groups, again highlighting epigenetic dysregulation as a strong prognostic risk factor [33]. Factors associated with hypermethylation were repressed chromatin, Ras pathway double mutants and upregulation of methyltransferases DNMT1 and DNMT3B. This supported the emergence of DNA hypermethylation as a consequence of hyperactive Ras signaling [81,83]. Several associations between genetic driver mutation and DNA methylation pattern were noted. The group of patients with highest hypermethylation was dominated by somatic PTPN11 mutation and older children, both known factors for inferior clinical course, whereas the low-methylation group was enriched for patients with NS/MPD, CBL cases, and young children with somatic NRAS mutations. The group with intermediate hypermethylation was characterized by somatic KRAS mutations and occurrence of monosomy 7 [33]. The Japanese cohort, consisting of 106 JMML cases, was split into two methylation groups [53]. In addition to known clinical risk factors, the high-methylation group involved cases with NF1 or PTPN11 mutations, secondary mutations, LIN28B overexpression and AML-like expression profile [53]. The North American study defined three similar methylation classes in 39 patients [65]. Interestingly, some JMML patients with good transplantation-free outcome and all patients with NS/MPD exhibited a DNA methylation signature closer to healthy controls than to other JMML cases [65,88], again underlining the significance of disrupted epigenetic control for the biology of JMML. The fact that all three methylome studies had used a comparable technical platform provided the unique opportunity for a comprehensive overarching meta-analysis. These collaborative efforts succeeded in developing and validating an international standard classifier of three different methylation categories matching those above and correlating with disease biology and outcome [88]. The prospective use of methylation analysis as a biomarker in JMML will aid in adapting treatment strategies, e.g., use of pretransplant therapy or low-intensity graft-versus-host disease (GVHD) prophylaxis, and support the generation of internationally comparable JMML study data.
5. Current Recommendations for the Management of JMML
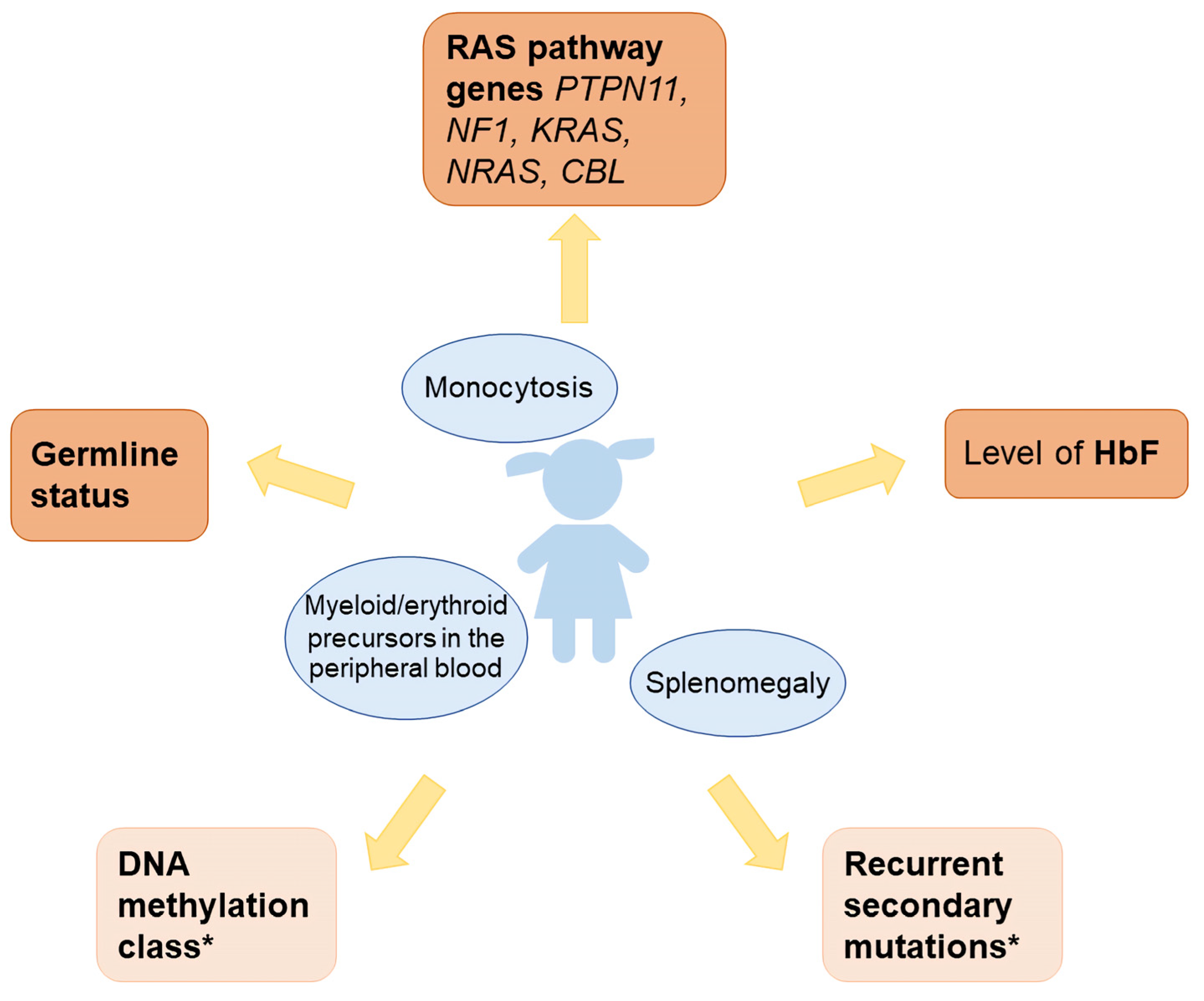
With extensive molecular diagnostic work-up of JMML established in major international study groups and large centers around the world, it has become evident that there can no longer be a uniform one-size-fits-all approach for this disorder (Table 1). The authors recommend that therapeutic decisions in a newly diagnosed case of JMML be based on the following diagnostic information (Figure 1):
- -
- Level of fetal hemoglobin (measured in a blood sample taken prior to erythrocyte transfusion; levels far above the age-adjusted reference value are also meaningful if sampled after transfusion)
- -
- Panel sequence analysis of the five primordial Ras pathway genes (PTPN11, NF1, KRAS, NRAS, CBL)
- -
- Presence of the primordial mutation in non-hematopoietic tissue (indicating germline status)
- -
- Where available, the following additional information will aid in clinical decision-making, though it is not indispensable for adequate management in most cases:
- -
- DNA methylation class
- -
- Panel sequence analysis of recurrent secondary mutations. The assessment should include SETBP1, JAK3, RRAS, RRAS2, and ASXL1; other targets are rare.
5.1. Somatic NRAS Mutation
The disease course in this group is remarkably heterogeneous. In older children with severe thrombocytopenia, increased HbF and high methylation class, a rapidly progressive course with a considerable risk of recurrence after HSCT is to be expected, likening the disorder to PTPN11- or NF1-driven JMML. On the other hand, a group of patients exist who are clinically well and have low HbF. Here, spontaneous clinical regression of the disease can occur in the long run without therapy. The search for an unrelated stem cell donor may be deferred in these cases. In between these two ends of the spectrum, the prospective identification of patients who benefit from watchful waiting is the real challenge. Factors suggesting surveillance without therapy include infant age, clinical status, age-appropriate levels of HbF, and low methylation class [41,89,90]. However, this must be balanced with the concern that delaying HSCT may compromise the outcome in some patients. EWOG-MDS data shows that the survival curve of JMML patients without HSCT keeps dropping during the first ten years and then plateaus at 25%.
It is very rare for a germline NRAS or KRAS mutation to be detected in a suspected JMML case. Most of these children have additional syndromic features of the rasopathy spectrum [91,92] or correspond in phenotype to Noonan syndrome [93]. Anecdotal observations contradict the paradigm that the canonical tumor-associated Ras mutations in codons 12, 13 or 61 are not tolerated in the germline; such cases are sometimes based on mosaicism [94,95]. Because of their rarity, no general recommendation can be given for the treatment of these highly individual cases.
5.2. Somatic KRAS Mutation
Children diagnosed with KRAS-JMML are typically very young, often infants. Concurrent monosomy 7 in the neoplastic clone is often observed (see below). Autoimmune phenomena (hyperimmunoglobulinemia, autoantibodies) should be searched for, and a diagnostic differentiation from RALD [74,96,97] should be kept in mind. The clinical presentation of KRAS-driven JMML tends to be aggressive, requiring rapid intervention. In the past, long-term survival without HSCT has not been reported for this group, but the picture is now changing with the introduction of azacitidine. KRAS-mutated JMML responds particularly well to low-dose azacitidine with long-lasting clinical and molecular remissions [98,99]. Regimens use 100 mg/m2/day on five consecutive days or 75 mg/m2/day on seven consecutive days, repeated every 28 days; due to instability, immediate intravenous or subcutaneous application of the cold reconstituted solution must be observed. Azacitidine has a favorable toxicity profile in children with JMML, mainly including lower-grade cytopenias, gastrointestinal discomfort, and infections [64,98]. It is variable how many cycles of azacitidine are necessary to achieve a response; between 6 and 9 cycles are usually administered. Frequently, the earliest sign of response to azacitidine is the improvement of thrombocytopenia. The spleen size diminishes after three to six cycles. Possibly, long-term cure with azacitidine alone will be achievable in KRAS-mutated JMML with a low risk profile.
5.3. Somatic PTPN11 Mutation
This is the numerically largest group that also carries the highest risk of rapid progression and early death. As no curative chemotherapy regimen is known, expeditious planning of allogeneic HSCT is mandatory. Within this group, risk factors for an unfavorable course are: age at diagnosis ≥2 years, significantly increased level of HbF, presence of secondary mutations, and/or high methylation class. In these cases, the aim should be to perform HSCT within a period of 3 months after diagnosis (recommendations for implementation see below). Low-dose azacitidine is the preferable option for upfront therapy until HSCT in non-high risk cases, with the goal to achieve a more favorable biological status of the leukemia. In many affected children, this treatment leads to a clinical partial remission or at least sufficient disease control until HSCT [64,98,99,100] However, high-risk cases carry the potential of immediate progression under azacitidine and may therefore be better off with more instant cytoreduction using 6-mercaptopurine (50 mg/m2/day, to be adjusted according to clinical course) and/or cytarabine (40 mg/m2/day × 5 days) [7]. If a patient presents in a critically ill condition that requires rapid reduction of tumor burden, the use of intensive chemotherapy with cytarabine (2 g/m2/day × 5 days) and fludarabine (30 mg/m2/day × 5 days) may also be considered [7]. However, this involves a substantial risk of organ toxicity and life-threatening infections. Removal of the spleen, which is often grossly enlarged, can be justified in individual cases to control respiratory impairment. A systematic beneficial effect of splenectomy on the further course of the disease has not been proven [40,101]. It is not yet clear whether treatment with azacitidine before HSCT also improves the long-term prognosis in this patient group. A recent non-randomized prospective study using a matched historical cohort as control group supports this assumption [98]. Until more precise evidence is available, the authors emphasize that treatment with azacitidine must not delay urgent HSCT in patients with PTPN11 mutation.
5.4. Germline Mutation in PTPN11
Germline mutations in PTPN11 cause Noonan syndrome, a condition of the rasopathy spectrum predisposing to a myeloproliferative disorder imitating JMML. For this reason, genetic diagnostics in suspected cases of JMML must always be carried out in both hematopoietic and non-hematopoietic material. Cultivated fibroblasts from a skin biopsy are ideal; hair follicles are less invasive, but more challenging to analyze. An oral mucous membrane swab has a high probability of contamination with hematopoietic cells, even if it is macroscopically not bloody, and should therefore only be scored if the result is negative.
In cases of suspected JMML in very young patients or with clinical evidence of Noonan syndrome, the diagnostic test for PTPN11 should not just encompass the hotspot exons 3 and 13 because Noonan syndrome mutations may also be found in exons 4 or 8. The spectrum of somatic PTPN11 mutations in nonsyndromic JMML and germline PTPN11 mutations in Noonan-associated MPD overlaps to a great extent but not completely [31].
The distinction between non-syndromic JMML and Noonan-associated MPD is important, as the latter is usually self-limiting. However, these patients must be monitored carefully, as there can be relevant clinical compromise from cell infiltrates, making it necessary to begin cytoreductive therapy. In individual cases, a clonal development towards a bona fide neoplastic disease is possible [47,102,103].
5.5. Neurofibromatosis Type 1
If not already recognized in the patient, the syndrome can usually be diagnosed clinically and/or through family history at the time of onset of JMML. In younger children, only café-au-lait spots but not the other typical signs of neurofibromatosis may be present. Six or more café-au-lait spots as stipulated in the NIH criteria are noted in the majority of children, but in exceptional cases there may be none or fewer. Genetic analysis of NF1 usually confirms the clinical diagnosis in children with JMML/NF-1 [104]. The typical finding is an NF1-inactivating heterozygous variation in the germline of the patient that arose de novo (~50% of cases) or was inherited. These lesions are often, but not always, deletions or truncating missense mutations reported as recurrent aberrations in the NF-1 literature. In addition, the neoplastic clone exhibits somatic loss of heterozygosity at the NF1 locus or an independent second NF1 mutation, leading to biallelic NF1 inactivation [21,104,105]. The judgment is more difficult if no clinical signs of neurofibromatosis are present and the genetic findings correspond to the above paradigm only incompletely (for example, in case of monoallelic lesions, variants of unclear significance, no germline findings or low allelic frequency). In such cases, a myeloid disorder with a secondary NF1 lesion, but driven by an unrelated event, may be present. With careful work-up, however, such dilemmas are rare.
JMML on the basis of NF-1 manifests more frequently at an older age than the other groups and typically does not involve a drastically reduced platelet count. Some children initially show little clinical impairment. However, long-term survival without HSCT has not yet been observed in this group, so that proceeding to transplant and inception of therapy with azacitidine is advisable as in patients with somatic PTPN11 mutation. Consistent with this recommendation, JMML/NF-1 cases almost always have an intermediate or high methylation profile [88].
5.6. CBL Mutation
The typical configuration of CBL mutations in JMML is a heterozygous missense point mutation in CBL exons 8 or 9 in the germline, accompanied by uniparental isodisomy of the 11q chromosome arm as a somatic event in hematopoietic cells, leading to loss of heterozygosity [26,36,37]. Many, but not all, children show syndromic rasopathy features, such as facial dysmorphia and growth retardation. A particular phenomenon in this patient group is the frequent occurrence of autoimmunity and vasculitis. Some children with CBL-JMML have massive organ enlargement and may require splenectomy for symptom relief. Most patients do not require swift HSCT but can be managed with watchful waiting; many of these experience spontaneous resolution of the myeloproliferation. The homozygous CBL-mutant status in hematopoietic cells may persist until adulthood even in the absence of hematologic abnormalities [106]. Patients who undergo transplant often revert to stable mixed chimerism with sufficient disease control [6,26,36]. It is still unclear if the allograft also prevents the later development of symptoms related to autoimmune vasculitis. A recent report highlighted the role of somatic-only CBL inactivation in five patients with a clinical course that required HSCT [42], again illustrating the need for proper germline analysis in the diagnostic evaluation of JMML.
5.7. None of the Above
Suspected cases of JMML with negative panel sequencing for all five primordial genes and no clinical evidence of NF-1 are called “quintuple-negative” or “all-negative”. In a third of these cases, in-depth multimodal genetic analysis uncovered a driving role of the NF1 gene in the absence of clinical NF-1 features [104]. In other cases, RNA sequencing identified activating fusions involving ALK, ROS1, or FLT3 [51,53]. Some authors argue in favor of diagnosing such myeloproliferative disorders as JMML due to the indistinguishable clinical and hematologic presentation. Excluding non-neoplastic causes of myelomonocytic proliferation, a maximum of 5–10% of suspected JMML cases remain genetically unexplained. It is advisable to refer these patients to an extended rasopathy work-up, especially if additional syndromic stigmata are present.
5.8. Monosomy 7
The significance of monosomy 7 for the biology of JMML is unclear, and there seems to be no association with clinical features or relevance for outcome [40]. Interestingly, monosomy 7 is observed more frequently in European compared to Japanese patients with JMML [33]. In a large international series, all patients with monosomy 7 and intermediate methylation class carried KRAS mutations, in contrast to an association between monosomy 7 and PTPN11 or NF1 in the high methylation group, and the absence of monosomy 7 in patients with low methylation pattern [88]. The mechanistic connection between this particular chromosomal lesion and aberrant DNA methylation patterns is not understood. Overall, it is likely that the presence of monosomy 7 plays a supportive role in JMML rather than being an independent pathogenetic factor [107]. This concept is also supported by the observation of secondary monosomy 7 in a watch-and-wait patient with Noonan syndrome and neonatal myeloproliferative disorder [102].
5.9. Allogeneic HSCT
Busulfan-based myeloablative conditioning regimens are commonly chosen and achieve 55–73% OS with a moderate 10–15% rate of transplant-related mortality but significant probability of leukemia relapse in the order of 25–35% [7,40,101,108,109,110,111,112]. The EWOG-MDS currently recommends a three-alkylator regimen consisting of busulfan (0.8–1.2 mg/kg/dose given 4 doses per day, day 7 to day 4), cyclophosphamide (60 mg/kg/d, day 3 to day 2), and melphalan (125–140 mg/m2/d on day 1) [40]. In an attempt to reduce toxicity, a recent prospective randomized trial compared busulfan, cyclophosphamide, and melphalan with busulfan and fludarabine alone but terminated early due to excessive disease recurrence in the latter arm [113]. Matched sibling donors (MSD) or matched/1-antigen-disparate unrelated donors (MUD) are considered the most suitable stem cell sources [40]. Matched cord blood units are a viable alternative, especially for smaller patients [114,115,116,117]. Although haploidentical relatives are readily available for urgent transplant and highly motivated, this should still be viewed as an approach with limited experience [101,112,118]. A recent study from China in 47 JMML patients suggested a lower relapse incidence in mismatched/haploidentical donor transplants compared to matched donors with similar rates of acute/chronic graft-versus-host disease and non-relapse mortality [101].
The North American group noted better post-HSCT outcome of patients with JMML who experienced molecular response to pretransplant chemotherapy [119], similar to other pediatric leukemias [120,121]. However, a limitation was that only a minority of patients responded to chemotherapy, conceivably those with favorable disease biology. Biomarkers predicting response to chemotherapy are lacking. Therefore, it cannot be generalized that pretransplant chemotherapy benefits survival in JMML, and the risk of unwarranted organ damage remains a concern [122].
Beside the leukemia biology factors discussed above, the way the transplant procedure is handled significantly influences the risk of relapse. It is likely that it is not so much the conditioning regimen but rather the establishment of a graft-versus-leukemia effect that is decisive for the success of allogeneic HSCT in JMML [101,109,110,111]. For this reason, EWOG-MDS recommends keeping immunosuppressive therapy with cyclosporine A at low levels (trough levels around 80 µg/L) and tapering early (from day +40 in the absence of grade II-IV GVHD). It is advisable to determine the recipient-donor chimerism at very close intervals (up to weekly in high-risk patients), as the reappearance of even small autologous cell populations mandates immediate withdrawal of the immunosuppressive therapy [123,124,125].
Age at diagnosis ≥2 years, NF1 or somatic PTPN11 mutation, and high DNA methylation define a patient group whose risk of JMML recurrence after HSCT is even higher than 50%, bringing up the question of post-transplant prophylaxis. On the basis of favorable data for other myeloid neoplasms [126,127,128,129,130,131] and in the absence of better alternatives, the authors consider it appropriate to recommend azacitidine (started as soon as safe and tolerable after engraftment; 32 mg/m2/day for five consecutive days, every 28 days) plus donor lymphocyte infusions (started after 3 cycles of azacitidine and 4 weeks after discontinuation of immunosuppressive prophylaxis, CD3+ cell dose 1–5 × 106/kg, repeated every 8 weeks with increasing cell dose up to 1–5 × 107/kg). However, we emphasize that there are no systematic data for this approach in JMML.
5.10. Experimental Agents and Targeted Therapy
Despite the prominent role of the Ras/MAPK network, attempts to target this complex signal cascade have shown limited therapeutic benefit in JMML [10,132]. The Children’s Oncology Group is currently recruiting patients for a phase II trial to examine the safety and efficacy of oral trametinib, a MEK1/2 inhibitor, in refractory or relapsed JMML (NCT03190915). In vitro data from induced pluripotent stem cell lines suggests mutation-specific sensitivity to kinase inhibition, with a preferential sensitivity of PTPN11-driven JMML to trametinib [133]. BCL2 inhibition gave impressive results when combined with azacitidine in elderly AML patients [134,135] and early results argue for a benefit in pediatric patients with advanced MDS/AML [136,137]. With an upregulation of the macrophage immune checkpoint CD47 in myeloid malignancies, ongoing preclinical and clinical trials test CD47-directed agents in MDS/AML, with encouraging efficacy results in combination with azacitidine [138,139,140].
Author Contributions
Conceptualization, C.M., C.M.N. and C.F.; writing—original draft preparation, C.M.; writing—review and editing, C.M.N. and C.F.; funding acquisition, C.M.N. and C.F. All authors have read and agreed to the published version of the manuscript.
Funding
This manuscript was supported by Bundesministerium für Bildung und Forschung MyPred 01GM1911A (to C.M.N. and C.F.) and Deutsche Forschungsgemeinschaft CRC992-C05 (to C.F.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
The authors wish to thank all physicians and scientists collaborating in the European Working Group of Myelodysplastic Syndromes in Childhood for their valuable contributions.
Conflicts of Interest
C.M.N. has a consultancy with Bristol-Myers Squibb. The other authors declare no conflict of interest. The funders had no role in the writing of the manuscript.
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Harris, N.L.; Jaffe, E.S.; Diebold, J.; Flandrin, G.; Muller-Hermelink, H.K.; Vardiman, J.; Lister, T.A.; Bloomfield, C.D. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: Report of the Clinical Advisory Committee meeting-Airlie House, Virginia, November 1997. J. Clin. Oncol. 1999, 17, 3835–3849. [Google Scholar] [CrossRef] [Green Version]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemeyer, C.M. JMML genomics and decisions. Hematol. Am. Soc. Hematol. Educ. Prog. 2018, 2018, 307–312. [Google Scholar] [CrossRef] [Green Version]
- Niemeyer, C.M. RAS diseases in children. Haematologica 2014, 99, 1653–1662. [Google Scholar] [CrossRef]
- Locatelli, F.; Niemeyer, C.M. How I treat juvenile myelomonocytic leukemia. Blood 2015, 125, 1083–1090. [Google Scholar] [CrossRef]
- Dvorak, C.C.; Loh, M.L. Juvenile myelomonocytic leukemia: Molecular pathogenesis informs current approaches to therapy and hematopoietic cell transplantation. Front. Pediatr. 2014, 2, 25. [Google Scholar] [CrossRef] [Green Version]
- Loh, M.L. Recent advances in the pathogenesis and treatment of juvenile myelomonocytic leukaemia. Br. J. Haematol. 2011, 152, 677–687. [Google Scholar] [CrossRef]
- Niemeyer, C.M.; Flotho, C. Juvenile myelomonocytic leukemia: Who’s the driver at the wheel? Blood 2019, 133, 1060–1070. [Google Scholar] [CrossRef] [Green Version]
- Stephen, A.G.; Esposito, D.; Bagni, R.K.; McCormick, F. Dragging ras back in the ring. Cancer Cell 2014, 25, 272–281. [Google Scholar] [CrossRef] [Green Version]
- Ward, A.F.; Braun, B.S.; Shannon, K.M. Targeting oncogenic Ras signaling in hematologic malignancies. Blood 2012, 120, 3397–3406. [Google Scholar] [CrossRef] [Green Version]
- Brummer, T.; McInnes, C. RAF kinase dimerization: Implications for drug discovery and clinical outcomes. Oncogene 2020, 39, 4155–4169. [Google Scholar] [CrossRef]
- Warne, P.H.; Viciana, P.R.; Downward, J. Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature 1993, 364, 352–355. [Google Scholar] [CrossRef]
- Pelech, S.L.; Sanghera, J.S. MAP kinases: Charting the regulatory pathways. Science 1992, 257, 1355–1356. [Google Scholar] [CrossRef]
- Boulton, T.G.; Nye, S.H.; Robbins, D.J.; Ip, N.Y.; Radzlejewska, E.; Morgenbesser, S.D.; DePinho, R.A.; Panayotatos, N.; Cobb, M.H.; Yancopoulos, G.D. ERKs: A family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell 1991, 65, 663–675. [Google Scholar] [CrossRef]
- Khosravi-Far, R.; White, M.A.; Westwick, J.K.; Solski, P.A.; Chrzanowska-Wodnicka, M.; van Aelst, L.; Wigler, M.H.; Der, C.J. Oncogenic Ras activation of Raf/mitogen-activated protein kinase-independent pathways is sufficient to cause tumorigenic transformation. Mol. Cell. Biol. 1996, 16, 3923–3933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joneson, T.; White, M.A.; Wigler, M.H.; Bar-Sagi, D. Stimulation of membrane ruffling and MAP kinase activation by distinct effectors of RAS. Science 1996, 271, 810–812. [Google Scholar] [CrossRef]
- Dinner, S.; Platanias, L.C. Targeting the mTOR Pathway in Leukemia. J. Cell. Biochem. 2016, 117, 1745–1752. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Yordy, J.S.; Muise-Helmericks, R.C. Signal transduction and the Ets family of transcription factors. Oncogene 2000, 19, 6503–6513. [Google Scholar] [CrossRef]
- Steinemann, D.; Arning, L.; Praulich, I.; Stuhrmann, M.; Hasle, H.; Stary, J.; Schlegelberger, B.; Niemeyer, C.M.; Flotho, C. Mitotic recombination and compound-heterozygous mutations are predominant NF1-inactivating mechanisms in children with juvenile myelomonocytic leukemia and neurofibromatosis type 1. Haematologica 2010, 95, 320–323. [Google Scholar] [CrossRef] [Green Version]
- Tartaglia, M.; Niemeyer, C.M.; Fragale, A.; Song, X.; Buechner, J.; Jung, A.; Hählen, K.; Hasle, H.; Licht, J.D.; Gelb, B.D. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat. Genet. 2003, 34, 148–150. [Google Scholar] [CrossRef]
- Caye, A.; Strullu, M.; Guidez, F.; Cassinat, B.; Gazal, S.; Fenneteau, O.; Lainey, E.; Nouri, K.; Nakhaei-Rad, S.; Dvorsky, R.; et al. Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat. Genet. 2015, 47, 1334–1340. [Google Scholar] [CrossRef]
- Stieglitz, E.; Taylor-Weiner, A.N.; Chang, T.Y.; Gelston, L.C.; Wang, Y.-D.; Mazor, T.; Esquivel, E.; Yu, A.; Seepo, S.; Olsen, S.; et al. The genomic landscape of juvenile myelomonocytic leukemia. Nat. Genet. 2015, 47, 1326–1333. [Google Scholar] [CrossRef] [PubMed]
- Flotho, C.; Valcamonica, S.; Mach-Pascual, S.; Schmahl, G.; Corral, L.; Ritterbach, J.; Hasle, H.; Aricò, M.; Biondi, A.; Niemeyer, C.M. RAS mutations and clonality analysis in children with juvenile myelomonocytic leukemia (JMML). Leukemia 1999, 13, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Niemeyer, C.M.; Kang, M.W.; Shin, D.H.; Furlan, I.; Erlacher, M.; Bunin, N.J.; Bunda, S.; Finklestein, J.Z.; Gorr, T.A.; Mehta, P.; et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat. Genet. 2010, 42, 794–800. [Google Scholar] [CrossRef]
- Caye, A.; Rouault-Pierre, K.; Strullu, M.; Lainey, E.; Abarrategi, A.; Fenneteau, O.; Arfeuille, C.; Osman, J.; Cassinat, B.; Pereira, S.; et al. Despite mutation acquisition in hematopoietic stem cells, JMML-propagating cells are not always restricted to this compartment. Leukemia 2020, 34, 1658–1668. [Google Scholar] [CrossRef]
- Louka, E.; Povinelli, B.; Rodriguez-Meira, A.; Buck, G.; Wen, W.X.; Wang, G.; Sousos, N.; Ashley, N.; Hamblin, A.; Booth, C.A.G.; et al. Heterogeneous disease-propagating stem cells in juvenile myelomonocytic leukemia. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef]
- Busque, L.; Gilliland, D.G.; Prchal, J.T.; Sieff, C.A.; Weinstein, H.J.; Sokol, J.M.; Belickova, M.; Wayne, A.S.; Zuckerman, K.S.; Sokol, L. Clonality in juvenile chronic myelogenous leukemia. Blood 1995, 85, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, K.; Sakashita, K.; Taira, C.; Tanaka-Yanagisawa, M.; Yanagisawa, R.; Shiohara, M.; Kanegane, H.; Hasegawa, D.; Kawasaki, K.; Endo, M.; et al. Quantitative assessment of PTPN11 or RAS mutations at the neonatal period and during the clinical course in patients with juvenile myelomonocytic leukaemia. Br. J. Haematol. 2010, 148, 593–599. [Google Scholar] [CrossRef]
- Kratz, C.P.; Niemeyer, C.M.; Castleberry, R.P.; Cetin, M.; Bergsträsser, E.; Emanuel, P.D.; Hasle, H.; Kardos, G.; Klein, C.; Kojima, S.; et al. The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Blood 2005, 106, 2183–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tartaglia, M.; Mehler, E.L.; Goldberg, R.; Zampino, G.; Brunner, H.G.; Kremer, H.; van der Burgt, I.; Crosby, A.H.; Ion, A.; Jeffery, S.; et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 2001, 29, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Lipka, D.B.; Witte, T.; Toth, R.; Yang, J.; Wiesenfarth, M.; Nöllke, P.; Fischer, A.; Brocks, D.; Gu, Z.; Park, J.; et al. RAS-pathway mutation patterns define epigenetic subclasses in juvenile myelomonocytic leukemia. Nat. Commun. 2017, 8, 2126. [Google Scholar] [CrossRef] [Green Version]
- Stiller, C.A.; Chessells, J.M.; Fitchett, M. Neurofibromatosis and childhood leukaemia/lymphoma: A population-based UKCCSG study. Br. J. Cancer 1994, 70, 969–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, K.M.; O’Connell, P.; Martin, G.A.; Paderanga, D.; Olson, K.; Dinndorf, P.; McCormick, F. Loss of the normal NF1 allele from the bone marrow of children with type 1 neurofibromatosis and malignant myeloid disorders. N. Engl. J. Med. 1994, 330, 597–601. [Google Scholar] [CrossRef]
- Pérez, B.; Mechinaud, F.; Galambrun, C.; Ben Romdhane, N.; Isidor, B.; Philip, N.; Derain-Court, J.; Cassinat, B.; Lachenaud, J.; Kaltenbach, S.; et al. Germline mutations of the CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic leukaemia. J. Med. Genet. 2010, 47, 686–691. [Google Scholar] [CrossRef]
- Loh, M.L.; Sakai, D.S.; Flotho, C.; Kang, M.; Fliegauf, M.; Archambeault, S.; Mullighan, C.G.; Chen, L.; Bergstraesser, E.; Bueso-Ramos, C.E.; et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood 2009, 114, 1859–1863. [Google Scholar] [CrossRef] [Green Version]
- Bollag, G.; Clapp, D.W.; Shih, S.; Adler, F.; Zhang, Y.Y.; Thompson, P.; Lange, B.J.; Freedman, M.H.; McCormick, F.; Jacks, T.; et al. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat. Genet. 1996, 12, 144–148. [Google Scholar] [CrossRef]
- Largaespada, D.A.; Brannan, C.I.; Jenkins, N.A.; Copeland, N.G. Nf1 deficiency causes Ras-mediated granulocyte/macrophage colony stimulating factor hypersensitivity and chronic myeloid leukaemia. Nat. Genet. 1996, 12, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Nöllke, P.; Zecca, M.; Korthof, E.; Lanino, E.; Peters, C.; Pession, A.; Kabisch, H.; Uderzo, C.; Bonfim, C.S.; et al. Hematopoietic stem cell transplantation (HSCT) in children with juvenile myelomonocytic leukemia (JMML): Results of the EWOG-MDS/EBMT trial. Blood 2005, 105, 410–419. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, K.; Yoshida, N.; Miura, S.; Nakazawa, Y.; Sakashita, K.; Hyakuna, N.; Saito, M.; Kato, F.; Ogawa, A.; Watanabe, A.; et al. Long-term haematological improvement after non-intensive or no chemotherapy in juvenile myelomonocytic leukaemia and poor correlation with adult myelodysplasia spliceosome-related mutations. Br. J. Haematol. 2012, 157, 647–650. [Google Scholar] [CrossRef]
- Hecht, A.; Meyer, J.A.; Behnert, A.; Wong, E.; Chehab, F.; Olshen, A.; Hechmer, A.; Aftandilian, C.; Bhat, R.; Choi, S.W.; et al. Molecular and phenotypic diversity of CBL-mutated juvenile myelomonocytic leukemia. Haematologica 2020. [Google Scholar] [CrossRef]
- Mendez, H.M.; Opitz, J.M. Noonan syndrome: A review. Am. J. Med. Genet. 1985, 21, 493–506. [Google Scholar] [CrossRef]
- Aoki, Y.; Niihori, T.; Inoue, S.; Matsubara, Y. Recent advances in RASopathies. J. Hum. Genet. 2016, 61, 33–39. [Google Scholar] [CrossRef]
- Roberts, A.E.; Allanson, J.E.; Tartaglia, M.; Gelb, B.D. Noonan syndrome. Lancet 2013, 381, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Bader-Meunier, B.; Tchernia, G.; Miélot, F.; Fontaine, J.L.; Thomas, C.; Lyonnet, S.; Lavergne, J.M.; Dommergues, J.P. Occurrence of myeloproliferative disorder in patients with Noonan syndrome. J. Pediatr. 1997, 130, 885–889. [Google Scholar] [CrossRef]
- Hofmans, M.; Schröder, R.; Lammens, T.; Flotho, C.; Niemeyer, C.; van Roy, N.; Decaluwe, W.; Philippé, J.; de Moerloose, B. Noonan syndrome-associated myeloproliferative disorder with somatically acquired monosomy 7: Impact on clinical decision making. Br. J. Haematol. 2019, 187, E83–E86. [Google Scholar] [CrossRef]
- Sakaguchi, H.; Okuno, Y.; Muramatsu, H.; Yoshida, K.; Shiraishi, Y.; Takahashi, M.; Kon, A.; Sanada, M.; Chiba, K.; Tanaka, H.; et al. Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat. Genet. 2013, 45, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Stieglitz, E.; Troup, C.B.; Gelston, L.C.; Haliburton, J.; Chow, E.D.; Yu, K.B.; Akutagawa, J.; Taylor-Weiner, A.N.; Liu, Y.L.; Wang, Y.-D.; et al. Subclonal mutations in SETBP1 confer a poor prognosis in juvenile myelomonocytic leukemia. Blood 2015, 125, 516–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flex, E.; Jaiswal, M.; Pantaleoni, F.; Martinelli, S.; Strullu, M.; Fansa, E.K.; Caye, A.; de Luca, A.; Lepri, F.; Dvorsky, R.; et al. Activating mutations in RRAS underlie a phenotype within the RASopathy spectrum and contribute to leukaemogenesis. Hum. Mol. Genet. 2014, 23, 4315–4327. [Google Scholar] [CrossRef] [Green Version]
- Chao, A.K.; Meyer, J.A.; Lee, A.G.; Hecht, A.; Tarver, T.; van Ziffle, J.; Koegel, A.K.; Golden, C.; Braun, B.S.; Sweet-Cordero, E.A.; et al. Fusion driven JMML: A novel CCDC88C-FLT3 fusion responsive to sorafenib identified by RNA sequencing. Leukemia 2020, 34, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Röttgers, S.; Gombert, M.; Teigler-Schlegel, A.; Busch, K.; Gamerdinger, U.; Slany, R.; Harbott, J.; Borkhardt, A. ALK fusion genes in children with atypical myeloproliferative leukemia. Leukemia 2010, 24, 1197–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, N.; Okuno, Y.; Yoshida, K.; Shiraishi, Y.; Nagae, G.; Suzuki, K.; Narita, A.; Sakaguchi, H.; Kawashima, N.; Wang, X.; et al. Integrated molecular profiling of juvenile myelomonocytic leukemia. Blood 2018, 131, 1576–1586. [Google Scholar] [CrossRef] [PubMed]
- Buijs, A.; Bruin, M. Fusion of FIP1L1 and RARA as a result of a novel t(4;17)(q12;q21) in a case of juvenile myelomonocytic leukemia. Leukemia 2007, 21, 1104–1108. [Google Scholar] [CrossRef]
- Morerio, C.; Acquila, M.; Rosanda, C.; Rapella, A.; Dufour, C.; Locatelli, F.; Maserati, E.; Pasquali, F.; Panarello, C. HCMOGT-1 is a novel fusion partner to PDGFRB in juvenile myelomonocytic leukemia with t(5;17)(q33;p11.2). Cancer Res. 2004, 64, 2649–2651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrgazov, K.; Kastner, R.; Gorna, M.; Hoermann, G.; Koenig, M.; Lucini, C.B.; Ulreich, R.; Benesch, M.; Strenger, V.; Lackner, H.; et al. NDEL1-PDGFRB fusion gene in a myeloid malignancy with eosinophilia associated with resistance to tyrosine kinase inhibitors. Leukemia 2017, 31, 237–240. [Google Scholar] [CrossRef] [Green Version]
- Mizoguchi, Y.; Fujita, N.; Taki, T.; Hayashi, Y.; Hamamoto, K. Juvenile myelomonocytic leukemia with t(7;11)(p15;p15) and NUP98-HOXA11 fusion. Am. J. Hematol. 2009, 84, 295–297. [Google Scholar] [CrossRef]
- Hasle, H.; Kerndrup, G.; Jacobsen, B.B. Childhood myelodysplastic syndrome in Denmark: Incidence and predisposing conditions. Leukemia 1995, 9, 1569–1572. [Google Scholar]
- Niemeyer, C.M.; Arico, M.; Basso, G.; Biondi, A.; Rajnoldi, A.C.; Creutzig, U.; Haas, O.; Harbott, J.; Hasle, H.; Kerndrup, G.; et al. Chronic myelomonocytic leukemia in childhood: A retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS). Blood 1997, 89, 3534–3543. [Google Scholar]
- Castro-malaspina, H.; Schaison, G.; Passe, S.; Pasquier, A.; Berger, R.; Bayle-Weisgerber, C.; Miller, D.; Seligmann, M.; Bernard, J. Subacute and chronic myelomonocytic leukemia in children (juvenile CML). Clinical and hematologic observations, and identification of prognostic factors. Cancer 1984, 54, 675–686. [Google Scholar] [CrossRef]
- So, N.; Liu, R.; Hogeling, M. Juvenile xanthogranulomas: Examining single, multiple, and extracutaneous presentations. Pediatr. Dermatol. 2020, 37, 637–644. [Google Scholar] [CrossRef]
- Tidyman, W.E.; Rauen, K.A. The RASopathies: Developmental syndromes of Ras/MAPK pathway dysregulation. Curr. Opin. Genet. Dev. 2009, 19, 230–236. [Google Scholar] [CrossRef] [Green Version]
- Aoki, Y.; Matsubara, Y. Ras/MAPK syndromes and childhood hemato-oncological diseases. Int. J. Hematol. 2013, 97, 30–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cseh, A.; Niemeyer, C.M.; Yoshimi, A.; Dworzak, M.; Hasle, H.; van den Heuvel-Eibrink, M.M.; Locatelli, F.; Masetti, R.; Schmugge, M.; Groß-Wieltsch, U.; et al. Bridging to transplant with azacitidine in juvenile myelomonocytic leukemia: A retrospective analysis of the EWOG-MDS study group. Blood 2015, 125, 2311–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stieglitz, E.; Mazor, T.; Olshen, A.B.; Geng, H.; Gelston, L.C.; Akutagawa, J.; Lipka, D.B.; Plass, C.; Flotho, C.; Chehab, F.F.; et al. Genome-wide DNA methylation is predictive of outcome in juvenile myelomonocytic leukemia. Nat. Commun. 2017, 8, 2127. [Google Scholar] [CrossRef] [Green Version]
- Bagby, G.C.; Dinarello, C.A.; Neerhout, R.C.; Ridgway, D.; McCall, E. Interleukin 1-dependent paracrine granulopoiesis in chronic granulocytic leukemia of the juvenile type. J. Clin. Investig. 1988, 82, 1430–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emanuel, P.D.; Bates, L.J.; Castleberry, R.P.; Gualtieri, R.J.; Zuckerman, K.S. Selective hypersensitivity to granulocyte-macrophage colony-stimulating factor by juvenile chronic myeloid leukemia hematopoietic progenitors. Blood 1991, 77, 925–929. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, D.; Bugarin, C.; Giordan, M.; Bresolin, S.; Longoni, D.; Micalizzi, C.; Ramenghi, U.; Bertaina, A.; Basso, G.; Locatelli, F.; et al. Validation of flow cytometric phospho-STAT5 as a diagnostic tool for juvenile myelomonocytic leukemia. Blood Cancer J. 2013, 3, e160. [Google Scholar] [CrossRef] [Green Version]
- Karow, A.; Baumann, I.; Niemeyer, C.M. Morphologic differential diagnosis of juvenile myelomonocytic leukemia—Pitfalls apart from viral infection. J. Pediatr. Hematol. Oncol. 2009, 31, 380. [Google Scholar] [CrossRef]
- Herrod, H.G.; Dow, L.W.; Sullivan, J.L. Persistent epstein-barr virus infection mimicking juvenile chronic myelogenous leukemia: Immunologic and hematologic studies. Blood 1983, 61, 1098–1104. [Google Scholar] [CrossRef] [Green Version]
- Nishio, N.; Takahashi, Y.; Tanaka, M.; Xu, Y.; Yoshida, N.; Sakaguchi, H.; Doisaki, S.; Hama, A.; Muramatsu, H.; Shimada, A.; et al. Aberrant phosphorylation of STAT5 by granulocyte-macrophage colony-stimulating factor in infant cytomegalovirus infection mimicking juvenile myelomonocytic leukemia. Leuk. Res. 2011, 35, 1261–1264. [Google Scholar] [CrossRef]
- Lorenzana, A.; Lyons, H.; Sawaf, H.; Higgins, M.; Carrigan, D.; Emanuel, P.D. Human herpesvirus 6 infection mimicking juvenile myelomonocytic leukemia in an infant. J. Pediatr. Hematol. Oncol. 2002, 24, 136–141. [Google Scholar] [CrossRef]
- Yoshimi, A.; Kamachi, Y.; Imai, K.; Watanabe, N.; Nakadate, H.; Kanazawa, T.; Ozono, S.; Kobayashi, R.; Yoshida, M.; Kobayashi, C.; et al. Wiskott-Aldrich syndrome presenting with a clinical picture mimicking juvenile myelomonocytic leukaemia. Pediatr. Blood Cancer 2013, 60, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Calvo, K.R.; Price, S.; Braylan, R.C.; Oliveira, J.B.; Lenardo, M.; Fleisher, T.A.; Rao, V.K. JMML and RALD (Ras-associated autoimmune leukoproliferative disorder): Common genetic etiology yet clinically distinct entities. Blood 2015, 125, 2753–2758. [Google Scholar] [CrossRef] [Green Version]
- Neven, Q.; Boulanger, C.; Bruwier, A.; de Ville Goyet, M.; Meyts, I.; Moens, L.; van Damme, A.; Brichard, B. Clinical Spectrum of Ras-Associated Autoimmune Leukoproliferative Disorder (RALD). J. Clin. Immunol. 2021, 41, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Lanzarotti, N.; Bruneau, J.; Trinquand, A.; Stolzenberg, M.-C.; Neven, B.; Fregeac, J.; Levy, E.; Jeremiah, N.; Suarez, F.; Mahlaoui, N.; et al. RAS-associated lymphoproliferative disease evolves into severe juvenile myelo-monocytic leukemia. Blood 2014, 123, 1960–1963. [Google Scholar] [CrossRef]
- Hofmans, M.; Lammens, T.; Helsmoortel, H.H.; Bresolin, S.; Cavé, H.; Flotho, C.; Hasle, H.; van den Heuvel-Eibrink, M.M.; Niemeyer, C.; Stary, J.; et al. The long non-coding RNA landscape in juvenile myelomonocytic leukemia. Haematologica 2018, 103, e501–e504. [Google Scholar] [CrossRef]
- Leoncini, P.P.; Bertaina, A.; Papaioannou, D.; Flotho, C.; Masetti, R.; Bresolin, S.; Menna, G.; Santoro, N.; Zecca, M.; Basso, G.; et al. MicroRNA fingerprints in juvenile myelomonocytic leukemia (JMML) identified miR-150-5p as a tumor suppressor and potential target for treatment. Oncotarget 2016, 7, 55395–55408. [Google Scholar] [CrossRef] [Green Version]
- Bresolin, S.; Zecca, M.; Flotho, C.; Trentin, L.; Zangrando, A.; Sainati, L.; Stary, J.; de Moerloose, B.; Hasle, H.; Niemeyer, C.M.; et al. Gene expression-based classification as an independent predictor of clinical outcome in juvenile myelomonocytic leukemia. J. Clin. Oncol. 2010, 28, 1919–1927. [Google Scholar] [CrossRef]
- Helsmoortel, H.H.; Bresolin, S.; Lammens, T.; Cavé, H.; Noellke, P.; Caye, A.; Ghazavi, F.; de Vries, A.; Hasle, H.; Labarque, V.; et al. LIN28B overexpression defines a novel fetal-like subgroup of juvenile myelomonocytic leukemia. Blood 2016, 127, 1163–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olk-Batz, C.; Poetsch, A.R.; Nöllke, P.; Claus, R.; Zucknick, M.; Sandrock, I.; Witte, T.; Strahm, B.; Hasle, H.; Zecca, M.; et al. Aberrant DNA methylation characterizes juvenile myelomonocytic leukemia with poor outcome. Blood 2011, 117, 4871–4880. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.-P.J.; Kantarjian, H.M.; Kirkpatrick, P. Azacitidine. Nat. Rev. Drug Discov. 2005, 4, 275–276. [Google Scholar] [CrossRef]
- Abe, M.; Ohira, M.; Kaneda, A.; Yagi, Y.; Yamamoto, S.; Kitano, Y.; Takato, T.; Nakagawara, A.; Ushijima, T. CpG island methylator phenotype is a strong determinant of poor prognosis in neuroblastomas. Cancer Res. 2005, 65, 828–834. [Google Scholar] [PubMed]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef] [Green Version]
- Fluhr, S.; Boerries, M.; Busch, H.; Symeonidi, A.; Witte, T.; Lipka, D.B.; Mücke, O.; Nöllke, P.; Krombholz, C.F.; Niemeyer, C.M.; et al. CREBBP is a target of epigenetic, but not genetic, modification in juvenile myelomonocytic leukemia. Clin. Epigenet. 2016, 8, 50. [Google Scholar] [CrossRef] [Green Version]
- Poetsch, A.R.; Lipka, D.B.; Witte, T.; Claus, R.; Nöllke, P.; Zucknick, M.; Olk-Batz, C.; Fluhr, S.; Dworzak, M.; de Moerloose, B.; et al. RASA4 undergoes DNA hypermethylation in resistant juvenile myelomonocytic leukemia. Epigenetics 2014, 9, 1252–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, T.; Lipka, D.B.; Witte, T.; Wierzbinska, J.A.; Fluhr, S.; Helf, M.; Mücke, O.; Claus, R.; Konermann, C.; Nöllke, P.; et al. Epigenetic silencing of AKAP12 in juvenile myelomonocytic leukemia. Epigenetics 2016, 11, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönung, M.; Meyer, J.; Nöllke, P.; Olshen, A.B.; Hartmann, M.; Murakami, N.; Wakamatsu, M.; Okuno, Y.; Plass, C.; Loh, M.L.; et al. International Consensus Definition of DNA Methylation Subgroups in Juvenile Myelomonocytic Leukemia. Clin. Cancer Res. 2021, 27, 158–168. [Google Scholar] [CrossRef]
- Matsuda, K.; Shimada, A.; Yoshida, N.; Ogawa, A.; Watanabe, A.; Yajima, S.; Iizuka, S.; Koike, K.; Yanai, F.; Kawasaki, K.; et al. Spontaneous improvement of hematologic abnormalities in patients having juvenile myelomonocytic leukemia with specific RAS mutations. Blood 2007, 109, 5477–5480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flotho, C.; Kratz, C.P.; Bergsträsser, E.; Hasle, H.; Starý, J.; Trebo, M.; van den Heuvel-Eibrink, M.M.; Wójcik, D.; Zecca, M.; Locatelli, F.; et al. Genotype-phenotype correlation in cases of juvenile myelomonocytic leukemia with clonal RAS mutations. Blood 2008, 111, 966–967. [Google Scholar] [CrossRef] [Green Version]
- de Filippi, P.; Zecca, M.; Lisini, D.; Rosti, V.; Cagioni, C.; Carlo-Stella, C.; Radi, O.; Veggiotti, P.; Mastronuzzi, A.; Acquaviva, A.; et al. Germ-line mutation of the NRAS gene may be responsible for the development of juvenile myelomonocytic leukaemia. Br. J. Haematol. 2009, 147, 706–709. [Google Scholar] [CrossRef]
- Kratz, C.P.; Schubbert, S.; Bollag, G.; Niemeyer, C.M.; Shannon, K.M.; Zenker, M. Germline mutations in components of the Ras signaling pathway in Noonan syndrome and related disorders. Cell Cycle 2006, 5, 1607–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kratz, C.P.; Franke, L.; Peters, H.; Kohlschmidt, N.; Kazmierczak, B.; Finckh, U.; Bier, A.; Eichhorn, B.; Blank, C.; Kraus, C.; et al. Cancer spectrum and frequency among children with Noonan, Costello, and cardio-facio-cutaneous syndromes. Br. J. Cancer 2015, 112, 1392–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourdeaut, F.; Hérault, A.; Gentien, D.; Pierron, G.; Ballet, S.; Reynaud, S.; Paris, R.; Schleiermacher, G.; Baumann, C.; Philippe-Chomette, P.; et al. Mosaicism for oncogenic G12D KRAS mutation associated with epidermal nevus, polycystic kidneys and rhabdomyosarcoma. J. Med. Genet. 2010, 47, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Hafner, C.; Toll, A.; Real, F.X. HRAS mutation mosaicism causing urothelial cancer and epidermal nevus. N. Engl. J. Med. 2011, 365, 1940–1942. [Google Scholar] [CrossRef] [Green Version]
- Niemela, J.E.; Lu, L.; Fleisher, T.A.; Davis, J.; Caminha, I.; Natter, M.; Beer, L.A.; Dowdell, K.C.; Pittaluga, S.; Raffeld, M.; et al. Somatic KRAS mutations associated with a human nonmalignant syndrome of autoimmunity and abnormal leukocyte homeostasis. Blood 2011, 117, 2883–2886. [Google Scholar] [CrossRef] [Green Version]
- Takagi, M.; Piao, J.; Lin, L.; Kawaguchi, H.; Imai, C.; Ogawa, A.; Watanabe, A.; Akiyama, K.; Kobayashi, C.; Mori, M.; et al. Autoimmunity and persistent RAS-mutated clones long after the spontaneous regression of JMML. Leukemia 2013, 27, 1926–1928. [Google Scholar] [CrossRef] [Green Version]
- Niemeyer, C.M.; Flotho, C.; Lipka, D.B.; Stary, J.; Rössig, C.; Baruchel, A.; Klingebiel, T.; Micalizzi, C.; Michel, G.; Nysom, K.; et al. Response to upfront azacitidine in juvenile myelomonocytic leukemia in the AZA-JMML-001 trial. Blood Adv. 2021, in press.. [Google Scholar]
- Furlan, I.; Batz, C.; Flotho, C.; Mohr, B.; Lübbert, M.; Suttorp, M.; Niemeyer, C.M. Intriguing response to azacitidine in a patient with juvenile myelomonocytic leukemia and monosomy 7. Blood 2009, 113, 2867–2868. [Google Scholar] [CrossRef]
- Fabri, O.; Horakova, J.; Bodova, I.; Svec, P.; Laluhova Striezencova, Z.; Bubanska, E.; Cermak, M.; Galisova, V.; Skalicka, K.; Vaska, A.; et al. Diagnosis and treatment of juvenile myelomonocytic leukemia in Slovak Republic: Novel approaches. Neoplasma 2019, 66, 818–824. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Luo, C.-J.; Miao, Y.; Wang, J.-M.; Luo, C.-Y.; Qin, X.; Cai, J.-Y.; Li, B.-S.; Chen, J. Human leukocyte antigen disparities reduce relapse after hematopoietic stem cell transplantation in children with juvenile myelomonocytic leukemia: A single-center retrospective study from China. Pediatr. Transplant. 2020, 25, e13825. [Google Scholar] [CrossRef]
- O’Halloran, K.; Ritchey, A.K.; Djokic, M.; Friehling, E. Transient juvenile myelomonocytic leukemia in the setting of PTPN11 mutation and Noonan syndrome with secondary development of monosomy 7. Pediatr. Blood Cancer 2017, 64. [Google Scholar] [CrossRef]
- Strullu, M.; Caye, A.; Lachenaud, J.; Cassinat, B.; Gazal, S.; Fenneteau, O.; Pouvreau, N.; Pereira, S.; Baumann, C.; Contet, A.; et al. Juvenile myelomonocytic leukaemia and Noonan syndrome. J. Med. Genet. 2014, 51, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, S.; Lebrecht, D.; Schanze, D.; Schanze, I.; Wieland, I.; Albert, M.H.; Borkhardt, A.; Bresters, D.; Büchner, J.; Catala, A.; et al. NF1 Tumor Suppressor Gene Inactivation in Juvenile Myelomonocytic Leukemia: New Genetic Evidence and Consequences for Diagnostic Work-up. Blood 2020, 136, 2181. [Google Scholar] [CrossRef]
- Flotho, C.; Steinemann, D.; Mullighan, C.G.; Neale, G.; Mayer, K.; Kratz, C.P.; Schlegelberger, B.; Downing, J.R.; Niemeyer, C.M. Genome-wide single-nucleotide polymorphism analysis in juvenile myelomonocytic leukemia identifies uniparental disomy surrounding the NF1 locus in cases associated with neurofibromatosis but not in cases with mutant RAS or PTPN11. Oncogene 2007, 26, 5816–5821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, H.; Yoshida, K.; Blagitko-Dorfs, N.; Claus, R.; Pantic, M.; Abdelkarim, M.; Niemöller, C.; Greil, C.; Hackanson, B.; Shiraishi, Y.; et al. Tracing the development of acute myeloid leukemia in CBL syndrome. Blood 2014, 123, 1883–1886. [Google Scholar] [CrossRef]
- Inaba, T.; Honda, H.; Matsui, H. The enigma of monosomy 7. Blood 2018, 131, 2891–2898. [Google Scholar] [CrossRef] [PubMed]
- Yabe, M.; Sako, M.; Yabe, H.; Osugi, Y.; Kurosawa, H.; Nara, T.; Tokuyama, M.; Adachi, S.; Kobayashi, C.; Yanagimachi, M.; et al. A conditioning regimen of busulfan, fludarabine, and melphalan for allogeneic stem cell transplantation in children with juvenile myelomonocytic leukemia. Pediatr. Transplant. 2008, 12, 862–867. [Google Scholar] [CrossRef]
- Manabe, A.; Okamura, J.; Yumura-Yagi, K.; Akiyama, Y.; Sako, M.; Uchiyama, H.; Kojima, S.; Koike, K.; Saito, T.; Nakahata, T. Allogeneic hematopoietic stem cell transplantation for 27 children with juvenile myelomonocytic leukemia diagnosed based on the criteria of the International JMML Working Group. Leukemia 2002, 16, 645–649. [Google Scholar] [CrossRef] [Green Version]
- Smith, F.O.; King, R.; Nelson, G.; Wagner, J.E.; Robertson, K.A.; Sanders, J.E.; Bunin, N.; Emaunel, P.D.; Davies, S.M. Unrelated donor bone marrow transplantation for children with juvenile myelomonocytic leukaemia. Br. J. Haematol. 2002, 116, 716–724. [Google Scholar] [CrossRef]
- Yoshida, N.; Sakaguchi, H.; Yabe, M.; Hasegawa, D.; Hama, A.; Hasegawa, D.; Kato, M.; Noguchi, M.; Terui, K.; Takahashi, Y.; et al. Clinical Outcomes after Allogeneic Hematopoietic Stem Cell Transplantation in Children with Juvenile Myelomonocytic Leukemia: A Report from the Japan Society for Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2020, 26, 902–910. [Google Scholar] [CrossRef]
- Yabe, M.; Ohtsuka, Y.; Watanabe, K.; Inagaki, J.; Yoshida, N.; Sakashita, K.; Kakuda, H.; Yabe, H.; Kurosawa, H.; Kudo, K.; et al. Transplantation for juvenile myelomonocytic leukemia: A retrospective study of 30 children treated with a regimen of busulfan, fludarabine, and melphalan. Int. J. Hematol. 2015, 101, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, C.C.; Satwani, P.; Stieglitz, E.; Cairo, M.S.; Dang, H.; Pei, Q.; Gao, Y.; Wall, D.; Mazor, T.; Olshen, A.B.; et al. Disease burden and conditioning regimens in ASCT1221, a randomized phase II trial in children with juvenile myelomonocytic leukemia: A Children’s Oncology Group study. Pediatr. Blood Cancer 2018, 65, e27034. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Crotta, A.; Ruggeri, A.; Eapen, M.; Wagner, J.E.; MacMillan, M.L.; Zecca, M.; Kurtzberg, J.; Bonfim, C.; Vora, A.; et al. Analysis of risk factors influencing outcomes after cord blood transplantation in children with juvenile myelomonocytic leukemia: A EUROCORD, EBMT, EWOG-MDS, CIBMTR study. Blood 2013, 122, 2135–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eapen, M.; Klein, J.P.; Ruggeri, A.; Spellman, S.; Lee, S.J.; Anasetti, C.; Arcese, W.; Barker, J.N.; Baxter-Lowe, L.A.; Brown, M.; et al. Impact of allele-level HLA matching on outcomes after myeloablative single unit umbilical cord blood transplantation for hematologic malignancy. Blood 2014, 123, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Locatelli, F. Improving cord blood transplantation in children. Br. J. Haematol. 2009, 147, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Flotho, C.; Vraetz, T.; Lang, P.; Niemeyer, C.M.; Strahm, B. Successful double umbilical cord blood transplantation for relapsed juvenile myelomonocytic leukemia. Leukemia 2013, 27, 988–989. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Xiaoqin, F.; Yuelin, H.; Yuchen, L.; Jianyun, L.; Xuedong, W.; Jiaqi, C.; Chunfu, L. Hypomethylation of Decitabine Improved Outcomes of Hematopoietic Stem Cell Transplantation in Children with Juvenile Myelomonocytic Leukemia. Blood 2017, 130, 3232. [Google Scholar] [CrossRef]
- Hecht, A.; Meyer, J.; Chehab, F.F.; White, K.L.; Magruder, K.; Dvorak, C.C.; Loh, M.L.; Stieglitz, E. Molecular assessment of pretransplant chemotherapy in the treatment of juvenile myelomonocytic leukemia. Pediatr. Blood Cancer 2019, 66, e27948. [Google Scholar] [CrossRef] [PubMed]
- Bader, P.; Kreyenberg, H.; Henze, G.H.R.; Eckert, C.; Reising, M.; Willasch, A.; Barth, A.; Borkhardt, A.; Peters, C.; Handgretinger, R.; et al. Prognostic value of minimal residual disease quantification before allogeneic stem-cell transplantation in relapsed childhood acute lymphoblastic leukemia: The ALL-REZ BFM Study Group. J. Clin. Oncol. 2009, 27, 377–384. [Google Scholar] [CrossRef]
- van der Velden, V.H.J.; van der Sluijs-Geling, A.; Gibson, B.E.S.; te Marvelde, J.G.; Hoogeveen, P.G.; Hop, W.C.J.; Wheatley, K.; Bierings, M.B.; Schuurhuis, G.J.; de Graaf, S.S.N.; et al. Clinical significance of flowcytometric minimal residual disease detection in pediatric acute myeloid leukemia patients treated according to the DCOG ANLL97/MRC AML12 protocol. Leukemia 2010, 24, 1599–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergstraesser, E.; Hasle, H.; Rogge, T.; Fischer, A.; Zimmermann, M.; Noellke, P.; Niemeyer, C.M. Non-hematopoietic stem cell transplantation treatment of juvenile myelomonocytic leukemia: A retrospective analysis and definition of response criteria. Pediatr. Blood Cancer 2007, 49, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, J.; Fukano, R.; Nishikawa, T.; Nakashima, K.; Sawa, D.; Ito, N.; Okamura, J. Outcomes of immunological interventions for mixed chimerism following allogeneic stem cell transplantation in children with juvenile myelomonocytic leukemia. Pediatr. Blood Cancer 2013, 60, 116–120. [Google Scholar] [CrossRef]
- Yoshimi, A.; Niemeyer, C.M.; Bohmer, V.; Duffner, U.; Strahm, B.; Kreyenberg, H.; Dilloo, D.; Zintl, F.; Claviez, A.; Wössmann, W.; et al. Chimaerism analyses and subsequent immunological intervention after stem cell transplantation in patients with juvenile myelomonocytic leukaemia. Br. J. Haematol. 2005, 129, 542–549. [Google Scholar] [CrossRef]
- Locatelli, F.; Lucarelli, B. Treatment of disease recurrence after allogeneic hematopoietic stem cell transplantation in children with juvenile myelomonocytic leukemia: A great challenge still to be won. Pediatr. Blood Cancer 2013, 60, 1–2. [Google Scholar] [CrossRef]
- Guillaume, T.; Malard, F.; Magro, L.; Labopin, M.; Tabrizi, R.; Borel, C.; Chevallier, P.; Vigouroux, S.; Peterlin, P.; Garnier, A.; et al. Prospective phase II study of prophylactic low-dose azacitidine and donor lymphocyte infusions following allogeneic hematopoietic stem cell transplantation for high-risk acute myeloid leukemia and myelodysplastic syndrome. Bone Marrow Transplant. 2019, 54, 1815–1826. [Google Scholar] [CrossRef]
- Craddock, C.; Slade, D.; de Santo, C.; Wheat, R.; Ferguson, P.; Hodgkinson, A.; Brock, K.; Cavenagh, J.; Ingram, W.; Dennis, M.; et al. Combination Lenalidomide and Azacitidine: A Novel Salvage Therapy in Patients Who Relapse After Allogeneic Stem-Cell Transplantation for Acute Myeloid Leukemia. J. Clin. Oncol. 2019, 37, 580–588. [Google Scholar] [CrossRef]
- Craddock, C.; Jilani, N.; Siddique, S.; Yap, C.; Khan, J.; Nagra, S.; Ward, J.; Ferguson, P.; Hazlewood, P.; Buka, R.; et al. Tolerability and Clinical Activity of Post-Transplantation Azacitidine in Patients Allografted for Acute Myeloid Leukemia Treated on the RICAZA Trial. Biol. Blood Marrow Transplant. 2016, 22, 385–390. [Google Scholar] [CrossRef] [Green Version]
- Schmid, C.; Labopin, M.; Schaap, N.; Veelken, H.; Schleuning, M.; Stadler, M.; Finke, J.; Hurst, E.; Baron, F.; Ringden, O.; et al. Prophylactic donor lymphocyte infusion after allogeneic stem cell transplantation in acute leukaemia—A matched pair analysis by the Acute Leukaemia Working Party of EBMT. Br. J. Haematol. 2019, 184, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Oran, B.; de Lima, M.; Garcia-Manero, G.; Thall, P.F.; Lin, R.; Popat, U.; Alousi, A.M.; Hosing, C.; Giralt, S.; Rondon, G.; et al. A phase 3 randomized study of 5-azacitidine maintenance vs. observation after transplant in high-risk AML and MDS patients. Blood Adv. 2020, 4, 5580–5588. [Google Scholar] [CrossRef]
- Schroeder, T.; Rautenberg, C.; Haas, R.; Germing, U.; Kobbe, G. Hypomethylating agents for treatment and prevention of relapse after allogeneic blood stem cell transplantation. Int. J. Hematol. 2018, 107, 138–150. [Google Scholar] [CrossRef] [Green Version]
- Stieglitz, E.; Ward, A.F.; Gerbing, R.B.; Alonzo, T.A.; Arceci, R.J.; Liu, Y.L.; Emanuel, P.D.; Widemann, B.C.; Cheng, J.W.; Jayaprakash, N.; et al. Phase II/III trial of a pre-transplant farnesyl transferase inhibitor in juvenile myelomonocytic leukemia: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2015, 62, 629–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasian, S.K.; Casas, J.A.; Posocco, D.; Gandre-Babbe, S.; Gagne, A.L.; Liang, G.; Loh, M.L.; Weiss, M.J.; French, D.L.; Chou, S.T. Mutation-specific signaling profiles and kinase inhibitor sensitivities of juvenile myelomonocytic leukemia revealed by induced pluripotent stem cells. Leukemia 2019, 33, 181–190. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollyea, D.A.; Pratz, K.; Letai, A.; Jonas, B.A.; Wei, A.H.; Pullarkat, V.; Konopleva, M.; Thirman, M.J.; Arellano, M.; Becker, P.S.; et al. Venetoclax with azacitidine or decitabine in patients with newly diagnosed acute myeloid leukemia: Long term follow-up from a phase 1b study. Am. J. Hematol. 2021, 96, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Winters, A.C.; Maloney, K.W.; Treece, A.L.; Gore, L.; Franklin, A.K. Single-center pediatric experience with venetoclax and azacitidine as treatment for myelodysplastic syndrome and acute myeloid leukemia. Pediatr. Blood Cancer 2020, 67, e28398. [Google Scholar] [CrossRef]
- Karol, S.E.; Alexander, T.B.; Budhraja, A.; Pounds, S.B.; Canavera, K.; Wang, L.; Wolf, J.; Klco, J.M.; Mead, P.E.; Das Gupta, S.; et al. Venetoclax in combination with cytarabine with or without idarubicin in children with relapsed or refractory acute myeloid leukaemia: A phase 1, dose-escalation study. Lancet Oncol. 2020, 21, 551–560. [Google Scholar] [CrossRef]
- Chao, M.P.; Takimoto, C.H.; Feng, D.D.; McKenna, K.; Gip, P.; Liu, J.; Volkmer, J.-P.; Weissman, I.L.; Majeti, R. Therapeutic Targeting of the Macrophage Immune Checkpoint CD47 in Myeloid Malignancies. Front. Oncol. 2019, 9, 1380. [Google Scholar] [CrossRef]
- Zeidan, A.M.; DeAngelo, D.J.; Palmer, J.M.; Seet, C.S.; Tallman, M.S.; Wei, X.; Li, Y.F.; Hock, N.; Burgess, M.R.; Hege, K.; et al. A Phase I Study of CC-90002, a Monoclonal Antibody Targeting CD47, in Patients with Relapsed and/or Refractory (R/R) Acute Myeloid Leukemia (AML) and High-Risk Myelodysplastic Syndromes (MDS): Final Results. Blood 2019, 134, 1320. [Google Scholar] [CrossRef]
- Sallman, D.A.; Asch, A.S.; Al Malki, M.M.; Lee, D.J.; Donnellan, W.B.; Marcucci, G.; Kambhampati, S.; Daver, N.G.; Garcia-Manero, G.; Komrokji, R.S.; et al. The First-in-Class Anti-CD47 Antibody Magrolimab (5F9) in Combination with Azacitidine Is Effective in MDS and AML Patients: Ongoing Phase 1b Results. Blood 2019, 134, 569. [Google Scholar] [CrossRef]
Figure 1.
Diagnostic approach for children with JMML. * Helpful for clinical decision making, but not mandatory.
Figure 1.
Diagnostic approach for children with JMML. * Helpful for clinical decision making, but not mandatory.


{kind=link}
Table 1.
Management of JMML according to driver mutation. HSCT, hematopoietic stem cell transplantation; GVHD, graft-versus-host disease; DLI, donor lymphocyte infusions; NF-1, neurofibromatosis 1.
Table 1.
Management of JMML according to driver mutation. HSCT, hematopoietic stem cell transplantation; GVHD, graft-versus-host disease; DLI, donor lymphocyte infusions; NF-1, neurofibromatosis 1.
| Ras Pathway Mutation | Frequency in JMML | Features | DNA Methylation Profile | Recommendations for Treatment |
|---|---|---|---|---|
| Somatic NRAS | 10–15% | Diverse | Mostly low, occasional IM or HM | HSCT for many, careful selection of candidates for watch-and-wait |
| Somatic KRAS | 10–15% | Frequent monosomy 7, autoimmune phenomena | Intermediate or low | Azacitidine and/or HSCT |
| Somatic PTPN11 | 35% | Compromised clinical status at diagnosis, highest risk of unfavorable outcome | Mostly high | Swift HSCT (+pretransplant azacitidine) with low intensity GVHD prophylaxis, in absence of GVHD early withdrawal of prophylaxis, consider azacitidine plus DLI posttransplant |
| Germline NF1 | 10–15% | Café-au-lait spots, possibly positive family history, older age at diagnosis, less severe thrombocytopenia | High or intermediate | Swift HSCT (+pretransplant azacitidine) with low intensity GVHD prophylaxis, in absence of GVHD early withdrawal of prophylaxis |
| Germline CBL | 15% | Syndromic rasopathy features, autoimmunity and vasculitis | Low | Watch-and-wait. HSCT if disease progresses, patients after HSCT often revert to stable mixed chimerism |
| All negative | 5–10% | Rarely activating kinase fusions in RNA sequencing | Low or intermediate | Differentiate non-neoplastic disease, perform extended work-up for rasopathies. Most patients require HSCT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mayerhofer, C.; Niemeyer, C.M.; Flotho, C. Current Treatment of Juvenile Myelomonocytic Leukemia. J. Clin. Med. 2021, 10, 3084. https://doi.org/10.3390/jcm10143084
AMA Style
Mayerhofer C, Niemeyer CM, Flotho C. Current Treatment of Juvenile Myelomonocytic Leukemia. Journal of Clinical Medicine. 2021; 10(14):3084. https://doi.org/10.3390/jcm10143084
Chicago/Turabian StyleMayerhofer, Christina, Charlotte M. Niemeyer, and Christian Flotho. 2021. "Current Treatment of Juvenile Myelomonocytic Leukemia" Journal of Clinical Medicine 10, no. 14: 3084. https://doi.org/10.3390/jcm10143084
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.