
The Identification and Validation of Hub Genes Associated with Acute Myocardial Infarction Using Weighted Gene Co-Expression Network Analysis

Abstract
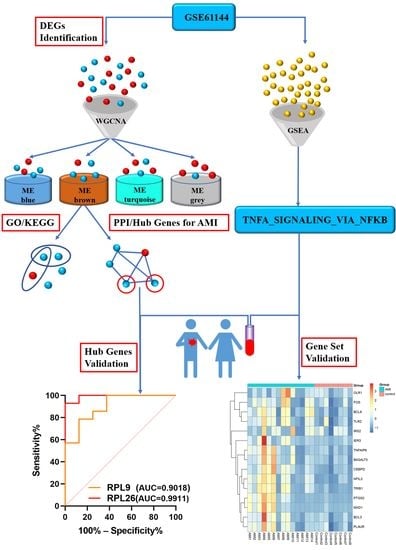
:
1. Introduction
2. Materials and Methods
2.1. Data Sources
2.2. Identification of Differentially Expressed Genes
2.3. Weighted Gene Co-Expression Network Analysis
2.4. Gene Ontology and Pathway Enrichment Analysis
2.5. Gene Set Enrichment Analysis
2.6. Protein–Protein Interaction Network and Hub Gene Identification
2.7. Putative Signaling Pathways Involving Hub Genes and GO Analysis
2.8. Sample Collection
2.9. RNA Extraction and Quantitative RT-PCR
2.10. Statistical Analysis
3. Results
3.1. Identifications of DEGs
3.2. GWCNA Analysis
3.3. Functional Enrichment Analysis
3.4. GSEA Analysis
3.5. PPI Network Construction, Modular Analysis, and Hub Gene Analysis
3.6. Construction of Putative RPL9 and RPL26 Protein–Protein Interaction Network and GO Analysis
3.7. Baseline Characteristics of Study Subjects
3.8. Validation of the Hub Genes
3.9. Validation of the Gene Set ‘TNFA_SIGNALING_VIA_NFKB’
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report from the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.; Johnson, C.; Abajobir, A.A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alam, K.; et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef]
- Hansson, G.K. Inflammation, Atherosclerosis, and Coronary Artery Disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [Green Version]
- Sreejit, G.; Abdel-Latif, A.; Athmanathan, B.; Annabathula, R.; Dhyani, A.; Noothi, S.K.; Quaife-Ryan, G.A.; Al-Sharea, A.; Pernes, G.; Dragoljevic, D.; et al. Neutrophil-Derived S100A8/A9 Amplify Granulopoiesis After Myocardial Infarction. Circulation 2020, 141, 1080–1094. [Google Scholar] [CrossRef]
- Henriksen, P.; Kotelevtsev, Y. Application of gene expression profiling to cardiovascular disease. Cardiovasc. Res. 2002, 54, 16–24. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; He, X.N.; Li, C.; Gong, L.; Liu, M. Identification of Candidate Genes and MicroRNAs for Acute Myocardial Infarction by Weighted Gene Coexpression Network Analysis. BioMed Res. Int. 2019, 2019, 5742608. [Google Scholar] [CrossRef]
- Niu, X.; Zhang, J.; Zhang, L.; Hou, Y.; Pu, S.; Chu, A.; Bai, M.; Zhang, Z. Weighted Gene Co-Expression Network Analysis Identifies Critical Genes in the Development of Heart Failure After Acute Myocardial Infarction. Front. Genet. 2019, 10, 1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Yin, Q.; Song, J.; Liu, C.; Chen, H.; Li, S. Identification of monocyte-associated genes as predictive biomarkers of heart failure after acute myocardial infarction. BMC Med Genom. 2021, 14, 44. [Google Scholar] [CrossRef]
- Park, H.-J.; Noh, J.H.; Eun, J.W.; Koh, Y.-S.; Seo, S.M.; Park, W.S.; Lee, J.Y.; Chang, K.; Seung, K.B.; Kim, P.-J.; et al. Assessment and diagnostic relevance of novel serum biomarkers for early decision of ST-elevation myocardial infarction. Oncotarget 2015, 6, 12970–12983. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W.V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2–27. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. S4), S11. [Google Scholar] [CrossRef] [Green Version]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D. The Executive Group on behalf of the Joint European Society of Cardiology (ESC)/American College of Cardiology (ACC)/American Heart Association (AHA)/World Heart Federation (WHF) Task Force for the Universal Definition of Myocardial Infarction Fourth Universal Definition of Myocardial Infarction (2018). Circulation 2018, 138, e618–e651. [Google Scholar] [CrossRef]
- Reed, G.W.; Rossi, J.E.; Cannon, C.P. Acute myocardial infarction. Lancet 2017, 389, 197–210. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, F.; Ma, X.; Adi, D.; Gai, M.-T.; Jin, X.; Yang, Y.-N.; Huang, Y.; Xie, X.; Li, X.-M.; et al. iTRAQ analysis of a mouse acute myocardial infarction model reveals that vitamin D binding protein promotes cardiomyocyte apoptosis after hypoxia. Oncotarget 2017, 9, 1969–1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danko, C.G.; Pertsov, A.M. Identification of gene co-regulatory modules and associated cis-elements involved in degenerative heart disease. BMC Med Genom. 2009, 2, 31. [Google Scholar] [CrossRef] [Green Version]
- Haselkorn, R.; Rothman-Denes, L.B. Protein synthesis. Annu. Rev. Biochem. 1973, 42, 397–438. [Google Scholar] [CrossRef]
- Ellsworth, D.L.; Bittman, B.; Croft, D.T., Jr.; Brinker, J.; Van Laar, R.; Vernalis, M.N. Recreational music-making alters gene expression pathways in patients with coronary heart disease. Med. Sci. Monit. 2013, 19, 139–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wang, C.; Zhang, X.; Liu, J.; Wang, Y.; Li, C.; Guo, D. Weighted gene co-expression network analysis revealed key biomarkers associated with the diagnosis of hypertrophic cardiomyopathy. Hereditas 2020, 157, 42. [Google Scholar] [CrossRef]
- Correia, L.C.; Andrade, B.B.; Borges, V.; Clarêncio, J.; Bittencourt, A.P.; Freitas, R.; Souza, A.C.; Almeida, M.C.; Leal, J.; Esteves, J.P.; et al. Prognostic value of cytokines and chemokines in addition to the GRACE Score in non-ST-elevation acute coronary syndromes. Clin. Chim. Acta 2010, 411, 540–545. [Google Scholar] [CrossRef]
- Hartman, M.H.; Groot, H.E.; Leach, I.M.; Karper, J.C.; van der Harst, P. Translational overview of cytokine inhibition in acute myocardial infarction and chronic heart failure. Trends Cardiovasc. Med. 2018, 28, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Tanno, M.; Gorog, D.A.; Bellahcene, M.; Cao, X.; Quinlan, R.A.; Marber, M.S. Tumor necrosis factor-induced protection of the murine heart is independent of p38-MAPK activation. J. Mol. Cell. Cardiol. 2003, 35, 1523–1527. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Atanasov, A.G. The microRNAs Regulating Vascular Smooth Muscle Cell Proliferation: A Minireview. Int. J. Mol. Sci. 2019, 20, 324. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Ma, C.; Gu, J.; Yu, M. Potential biomarkers of acute myocardial infarction based on weighted gene co-expression network analysis. Biomed. Eng. Online 2019, 18, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Chen, S.; Lu, R. Identification of Important Genes Related to Ferroptosis and Hypoxia in Acute Myocardial Infarction Based on WGCNA. Bioengineered 2021, 12, 7950–7963. [Google Scholar] [CrossRef]
- Zhang, B.; Li, B.; Sun, C.; Tu, T.; Xiao, Y.; Liu, Q. Identification of key gene modules and pathways of human platelet transcriptome in acute myocardial infarction patients through co-expression network. Am. J. Transl. Res. 2021, 13, 3890–3905. [Google Scholar]
- Xie, Y.; Wang, Y.; Zhao, L.; Wang, F.; Fang, J. Identification of potential biomarkers and immune cell infiltration in acute myocardial infarction (AMI) using bioinformatics strategy. Bioengineered 2021, 12, 2890–2905. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Duan, M.; Zhang, C.; Wang, K.; Feng, L.; Song, L.; Wu, S.; Chen, X. Identification of Potential Biomarkers Associated with Acute Myocardial Infarction by Weighted Gene Coexpression Network Analysis. Oxidative Med. Cell. Longev. 2021, 2021, 5553811. [Google Scholar] [CrossRef]
- De la Cruz, J.; Karbstein, K.; Woolford, J.L. Functions of Ribosomal Proteins in Assembly of Eukaryotic Ribosomes In Vivo. Annu. Rev. Biochem. 2015, 84, 93–129. [Google Scholar] [CrossRef] [Green Version]
- Warner, J.R.; McIntosh, K.B. How Common Are Extraribosomal Functions of Ribosomal Proteins? Mol. Cell 2009, 34, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casad, M.E.; Abraham, D.; Kim, I.-M.; Frangakis, S.; Dong, B.; Lin, N.; Wolf, M.J.; Rockman, H.A. Cardiomyopathy Is Associated with Ribosomal Protein Gene Haplo-Insufficiency in Drosophila melanogaster. Genetics 2011, 189, 861–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, S.J.; Woodling, N.S.; Yedvobnick, B.; Woodling, N. Insertional inactivation of the L13a ribosomal protein gene of Drosophila melanogaster identifies a new Minute locus. Gene 2006, 368, 46–52. [Google Scholar] [CrossRef]
- Smolock, E.M.; Korshunov, V.A.; Glazko, G.; Qiu, X.; Gerloff, J.; Berk, B. Ribosomal Protein L17, RpL17, is an Inhibitor of Vascular Smooth Muscle Growth and Carotid Intima Formation. Circulation 2012, 126, 2418–2427. [Google Scholar] [CrossRef] [Green Version]
- Pellegrino, S.; Yusupova, G. Eukaryotic Ribosome as a Target for Cardiovascular Disease. Cell Chem. Biol. 2016, 23, 1319–1321. [Google Scholar] [CrossRef] [Green Version]
- Lezzerini, M.; Penzo, M.; O’Donohue, M.-F.; Vieira, C.M.D.S.; Saby, M.; Elfrink, H.L.; Diets, I.J.; Hesse, A.-M.; Couté, Y.; Gastou, M.; et al. Ribosomal protein gene RPL9 variants can differentially impair ribosome function and cellular metabolism. Nucleic Acids Res. 2020, 48, 770–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz, V.; Olvera, J.; Chan, Y.-L.; Wool, I.G. The primary structure of rat ribosomal protein L26. FEBS Lett. 1989, 251, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Takagi, M.; Absalon, M.J.; McLure, K.G.; Kastan, M.B. Regulation of p53 Translation and Induction after DNA Damage by Ribosomal Protein L26 and Nucleolin. Cell 2005, 123, 49–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ofir-Rosenfeld, Y.; Boggs, K.; Michael, D.; Kastan, M.B.; Oren, M. Mdm2 Regulates p53 mRNA Translation through Inhibitory Interactions with Ribosomal Protein L26. Mol. Cell 2008, 32, 180–189. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Ge, M.; Yin, Y.; Luo, M.; Chen, D. Silencing expression of ribosomal protein L26 and L29 by RNA interfering inhibits proliferation of human pancreatic cancer PANC-1 cells. Mol. Cell. Biochem. 2012, 370, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, J.N. Receiver Operating Characteristic Curve in Diagnostic Test Assessment. J. Thorac. Oncol. 2010, 5, 1315–1316. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence (5′→3′) | |
|---|---|---|
| RPL26 | Forward | ACAACTGTCCACGTAGGCATTCAC |
| Reverse | TACTTGGCGAGATTTGGCTTTCCG | |
| RPL9 | Forward | TTACACTGGGCTTCCGTTACAAGATG |
| Reverse | GCAACACCTGGTCTCATCCGAAC | |
| TNFAIP6 | Forward | TTTCTCTTGCTATGGGAAGACAC |
| Reverse | GAGCTTGTATTTGCCAGACCG | |
| IRS2 | Forward | CGGTGAGTTCTACGGGTACAT |
| Reverse | TCAGGGTGTATTCATCCAGCG | |
| B4 GALT5 | Forward | TCCTCGCTGCTGTACTTCG |
| Reverse | AATGCCTTGGGCTTGCATCA | |
| OLR1 | Forward | TTGCCTGGGATTAGTAGTGACC |
| Reverse | GCTTGCTCTTGTGTTAGGAGGT | |
| FOS | Forward | CCGGGGATAGCCTCTCTTACT |
| Reverse | CCAGGTCCGTGCAGAAGTC | |
| NFIL3 | Forward | AAAATGCAGACCGTCAAAAAGGA |
| Reverse | TGACACTTCCGTTAAAGCAGAAT | |
| TRIB1 | Forward | GCTGCAAGGTGTTTCCCATTA |
| Reverse | TCCCCAAAGTCCTTCTCAAAGA | |
| BCL6 | Forward | GGAGTCGAGACATCTTGACTGA |
| Reverse | ATGAGGACCGTTTTATGGGCT | |
| TLR2 | Forward | ATCCTCCAATCAGGCTTCTCT |
| Reverse | GGACAGGTCAAGGCTTTTTACA | |
| PTGS2 | Forward | CTGGCGCTCAGCCATACAG |
| Reverse | CGCACTTATACTGGTCAAATCCC | |
| BCL3 | Forward | CCGGAGGCGCTTTACTACC |
| Reverse | TAGGGGTGTAGGCAGGTTCAC | |
| IER3 | Forward | CAGCCGCAGGGTTCTCTAC |
| Reverse | GATCTGGCAGAAGACGATGGT | |
| PLAUR | Forward | TGTAAGACCAACGGGGATTGC |
| Reverse | AGCCAGTCCGATAGCTCAGG | |
| CEBPD | Forward | GGAGAGACTCAGCAACGACC |
| Reverse | TTGCGCTCCTATGTCCCAAG | |
| MXD1 | Forward | CGGGCTCATCTTCGCTTGT |
| Reverse | GATTTGGTGAACGGCTTTTCTG | |
| ACTB | Forward | TCGTGCGTGACATTAAGGAGAAGC |
| Reverse | ATGGAGTTGAAGGTAGTTTCGTGGATG | |
| MNC | MCC | EPC | DMNC | Degree |
|---|---|---|---|---|
| RPS20 | RPS20 | RPS20 | GFM1 | RPS20 |
| RPS6 | RPS6 | RPS6 | PELP1 | RPS6 |
| RPS27 A | RPS18 | RPS18 | RPS17 | RPS27 A |
| SNRPD2 | RPL26 | RPL26 | CCT7 | SNRPD2 |
| RPL26 | RPL11 | RPL11 | RPL24 | RPL26 |
| RPL11 | RPLP0 | RPLP0 | RPS18 | RPL11 |
| RPL9 | RPL9 | RPL9 | EEF1 A1 | RPL9 |
| RPS2 | RPS2 | RPS2 | RPL26 | RPS2 |
| RPL3 | RPL3 | RPL3 | RPLP0 | RPL3 |
| NHP2 L1 | NHP2 L1 | NHP2 L1 | RPL9 | NHP2 L1 |
| Variables | AMI Group (n = 14) | Control (n = 8) | p-Value |
|---|---|---|---|
| Demographic features | |||
| Age (years) | 60.714 ± 3.010 | 60.571 ± 4.099 | 0.553 |
| Male/Female | 12/2 | 6/2 | 0.531 |
| Cardiovascular risk factors | |||
| Hypertension | 6 (42.86%) | 4 (50%) | 0.746 |
| Dyslipidemia | 1 (7.14%) | 0 | NA |
| Diabetes mellitus | 5 (35.71%) | 1 (12.5%) | NA |
| Current smoking | 8 (57.14%) | 2 (25%) | 0.145 |
| Vital signs on admission | |||
| SBP (mmHg) | 121.50 (114.75–140.50) | 120.00 (110.00–140.00) | 0.868 |
| DBP (mmHg) | 75.000 (69.500–94.000) | 72.000 (70.000–78.000) | 0.868 |
| Heart rate (bpm) | 78.000 (73.500–87.000) | 80.000 (75.000–84.000) | 0.973 |
| Echocardiographic finding | |||
| LVEF (%) | 52.500 ± 1.738 | 63.286 ± 1.848 | 0.001 |
| Laboratory findings | |||
| hs-cTnT (ng/mL) | 9.930 (8.138–10.000) | 0.007 (0.004–0.0100) | 0.000 |
| CKMB (U/L) | 342.00 (210.25–457.25) | 13.00 (9.00–15.00) | 0.000 |
| NT-pro-BNP (pg/mL) | 682.10 (266.33–894.50) | 68.00 (41.70–98.10) | 0.002 |
| TC (mmol/L) | 5.024 ± 0.199 | 3.556 ± 0.215 | 0.001 |
| TG (mmol/L) | 1.740 (0.758–2.395) | 2.280 (1.880–3.790) | 0.082 |
| LDL-C (mmol/L) | 3.161 ± 0.155 | 1.451 ± 0.217 | 0.000 |
| HDL-C (mmol/L) | 1.060 (0.833–1.365) | 0.860 (0.780–0.970) | 0.188 |
| Medications | |||
| Aspirin | 11 (78.57%) | 0 | NA |
| Clopidogrel | 2 (14.29%) | 0 | NA |
| Ticagrelor | 9 (64.29%) | 0 | NA |
| Statin | 7 (50%) | 0 | NA |
| ACEI/ARB | 2 (14.29%) | 3 (37.5%) | 0.211 |
| ß blocker | 3 (21.43%) | 2 (25%) | 0.848 |
| CCB | 6 (42.86%) | 0 | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, J.; Chen, L.; Cheng, H.; Song, X.; Shi, Y.; Li, L.; Xu, R.; Qin, Q.; Ma, J.; Ge, J. The Identification and Validation of Hub Genes Associated with Acute Myocardial Infarction Using Weighted Gene Co-Expression Network Analysis. J. Cardiovasc. Dev. Dis. 2022, 9, 30. https://doi.org/10.3390/jcdd9010030
Xue J, Chen L, Cheng H, Song X, Shi Y, Li L, Xu R, Qin Q, Ma J, Ge J. The Identification and Validation of Hub Genes Associated with Acute Myocardial Infarction Using Weighted Gene Co-Expression Network Analysis. Journal of Cardiovascular Development and Disease. 2022; 9(1):30. https://doi.org/10.3390/jcdd9010030
Chicago/Turabian StyleXue, Junqiang, Lu Chen, Hao Cheng, Xiaoyue Song, Yuekai Shi, Linnan Li, Rende Xu, Qing Qin, Jianying Ma, and Junbo Ge. 2022. "The Identification and Validation of Hub Genes Associated with Acute Myocardial Infarction Using Weighted Gene Co-Expression Network Analysis" Journal of Cardiovascular Development and Disease 9, no. 1: 30. https://doi.org/10.3390/jcdd9010030