
Transcriptomic Comparison of Human Peripartum and Dilated Cardiomyopathy Identifies Differences in Key Disease Pathways
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquisition and Sample Population
2.2. Data Processing and Identifying Differentially Expressed Genes
2.3. DEG Meta-Analysis of PPCM and DCM Patient LVs
2.4. Gene Set Enrichment Analysis
2.5. Cellular Deconvolution
2.6. Statistical Analysis
3. Results
3.1. Population Information
3.2. Differential Gene Expression Analysis in Heart Failure
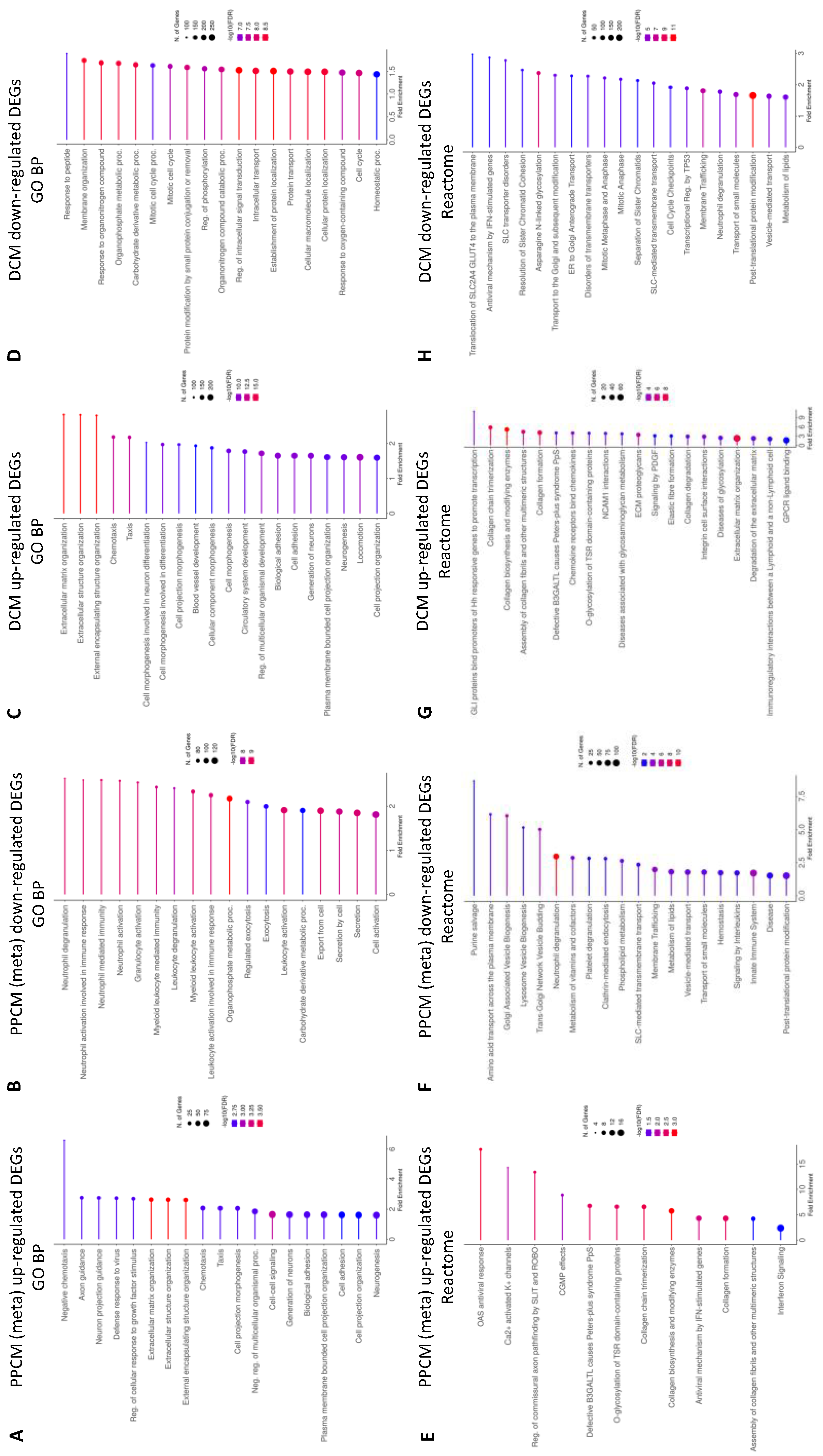
3.3. Enrichment Analysis of Heart Failure DEGs
3.4. Enrichment Analysis Comparing PPCMs and NF Donors
3.5. Enrichment Analysis Comparing DCMs and NF Donor Controls
3.6. Infiltration of Immune Cell Populations in the Failing Heart
3.7. Cell Types Annotated by DEG Signatures
4. Discussion
- Immune cell signatures of PPCM and DCM
- Metabolic pathways in PPCM and DCM
- Myocardial fibrosis is common to PPCM and DCM
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arany, Z.; Elkayam, U. Peripartum Cardiomyopathy. Circulation 2016, 133, 1397–1409. [Google Scholar] [CrossRef]
- Li, A.; Campbell, K.; Lal, S.; Ge, Y.; Keogh, A.; Macdonald, P.S.; Lau, P.; Lai, J.; Linke, W.A.; Van der Velden, J.; et al. Peripartum cardiomyopathy: A global effort to find the cause and cure for the rare and little understood disease. Biophys. Rev. 2022, 14, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Frantz, S.; Falcao-Pires, I.; Balligand, J.L.; Bauersachs, J.; Brutsaert, D.; Ciccarelli, M.; Dawson, D.; de Windt, L.J.; Giacca, M.; Hamdani, N.; et al. The innate immune system in chronic cardiomyopathy: A European Society of Cardiology (ESC) scientific statement from the Working Group on Myocardial Function of the ESC. Eur. J. Heart Fail. 2018, 20, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Goli, R.; Li, J.; Brandimarto, J.; Levine, L.D.; Riis, V.; McAfee, Q.; DePalma, S.; Haghighi, A.; Seidman, J.G.; Seidman, C.E.; et al. Genetic and Phenotypic Landscape of Peripartum Cardiomyopathy. Circulation 2021, 143, 1852–1862. [Google Scholar] [CrossRef] [PubMed]
- Ware, J.S.; Li, J.; Mazaika, E.; Yasso, C.M.; DeSouza, T.; Cappola, T.P.; Tsai, E.J.; Hilfiker-Kleiner, D.; Kamiya, C.A.; Mazzarotto, F.; et al. Shared Genetic Predisposition in Peripartum and Dilated Cardiomyopathies. N. Engl. J. Med. 2016, 374, 233–241. [Google Scholar] [CrossRef]
- Gerull, B.; Klaassen, S.; Brodehl, A. The Genetic Landscape of Cardiomyopathies. In Genetic Causes of Cardiac Disease; Erdmann, J., Moretti, A., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 45–91. [Google Scholar] [CrossRef]
- Sliwa, K.; Petrie, M.C.; van der Meer, P.; Mebazaa, A.; Hilfiker-Kleiner, D.; Jackson, A.M.; Maggioni, A.P.; Laroche, C.; Regitz-Zagrosek, V.; Schaufelberger, M.; et al. Clinical presentation, management, and 6-month outcomes in women with peripartum cardiomyopathy: An ESC EORP registry. Eur. Heart J. 2020, 41, 3787–3797. [Google Scholar] [CrossRef]
- Sliwa, K.; Mebazaa, A.; Hilfiker-Kleiner, D.; Petrie, M.C.; Maggioni, A.P.; Laroche, C.; Regitz-Zagrosek, V.; Schaufelberger, M.; Tavazzi, L.; van der Meer, P.; et al. Clinical characteristics of patients from the worldwide registry on peripartum cardiomyopathy (PPCM): EURObservational Research Programme in conjunction with the Heart Failure Association of the European Society of Cardiology Study Group on PPCM. Eur. J. Heart Fail. 2017, 19, 1131–1141. [Google Scholar] [CrossRef]
- Moulig, V.; Pfeffer, T.J.; Ricke-Hoch, M.; Schlothauer, S.; Koenig, T.; Schwab, J.; Berliner, D.; Pfister, R.; Michels, G.; Haghikia, A.; et al. Long-term follow-up in peripartum cardiomyopathy patients with contemporary treatment: Low mortality, high cardiac recovery, but significant cardiovascular co-morbidities. Eur. J. Heart Fail. 2019, 21, 1534–1542. [Google Scholar] [CrossRef]
- Halliday, B.P.; Wassall, R.; Lota, A.S.; Khalique, Z.; Gregson, J.; Newsome, S.; Jackson, R.; Rahneva, T.; Wage, R.; Smith, G.; et al. Withdrawal of pharmacological treatment for heart failure in patients with recovered dilated cardiomyopathy (TRED-HF): An open-label, pilot, randomised trial. Lancet 2019, 393, 61–73. [Google Scholar] [CrossRef]
- McTiernan, C.F.; Morel, P.; Cooper, L.T.; Rajagopalan, N.; Thohan, V.; Zucker, M.; Boehmer, J.; Bozkurt, B.; Mather, P.; Thornton, J.; et al. Circulating T-Cell Subsets, Monocytes, and Natural Killer Cells in Peripartum Cardiomyopathy: Results From the Multicenter IPAC Study. J. Card. Fail. 2018, 24, 33–42. [Google Scholar] [CrossRef]
- Koczo, A.; Marino, A.; Jeyabalan, A.; Elkayam, U.; Cooper, L.T.; Fett, J.; Briller, J.; Hsich, E.; Blauwet, L.; McTiernan, C.; et al. Breastfeeding, Cellular Immune Activation, and Myocardial Recovery in Peripartum Cardiomyopathy. JACC Basic Transl. Sci. 2019, 4, 291–300. [Google Scholar] [CrossRef]
- Zeng, Z.; Wang, K.; Li, Y.; Xia, N.; Nie, S.; Lv, B.; Zhang, M.; Tu, X.; Li, Q.; Tang, T.; et al. Down-regulation of microRNA-451a facilitates the activation and proliferation of CD4+ T cells by targeting Myc in patients with dilated cardiomyopathy. J. Biol. Chem. 2017, 292, 6004–6013. [Google Scholar] [CrossRef]
- Efthimiadis, I.; Skendros, P.; Sarantopoulos, A.; Boura, P. CD4+/CD25+ T-Lymphocytes and Th1/Th2 regulation in dilated cardiomyopathy. Hippokratia 2011, 15, 335–342. [Google Scholar] [PubMed]
- Nakayama, T.; Sugano, Y.; Yokokawa, T.; Nagai, T.; Matsuyama, T.A.; Ohta-Ogo, K.; Ikeda, Y.; Ishibashi-Ueda, H.; Nakatani, T.; Ohte, N.; et al. Clinical impact of the presence of macrophages in endomyocardial biopsies of patients with dilated cardiomyopathy. Eur. J. Heart Fail. 2017, 19, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Rurik, J.G.; Aghajanian, H.; Epstein, J.A. Immune Cells and Immunotherapy for Cardiac Injury and Repair. Circ. Res. 2021, 128, 1766–1779. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Sun, P.; Chen, Q.; Sun, Y.; Shi, M.; Mang, G.; Yu, S.; Zheng, Y.; Li, Z.; Sun, M.; et al. Metabolic reprogramming orchestrates CD4+ T-cell immunological status and restores cardiac dysfunction in autoimmune induced-dilated cardiomyopathy mice. J. Mol. Cell. Cardiol. 2019, 135, 134–148. [Google Scholar] [CrossRef]
- Afanasyeva, M.; Georgakopoulos, D.; Belardi, D.F.; Bedja, D.; Fairweather, D.; Wang, Y.; Kaya, Z.; Gabrielson, K.L.; Rodriguez, E.R.; Caturegli, P.; et al. Impaired up-regulation of CD25 on CD4+ T cells in IFN- γ knockout mice is associated with progression of myocarditis to heart failure. Proc. Natl. Acad. Sci. USA 2005, 102, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Koenig, A.L.; Shchukina, I.; Amrute, J.; Andhey, P.S.; Zaitsev, K.; Lai, L.; Bajpai, G.; Bredemeyer, A.; Smith, G.; Jones, C.; et al. Single-cell transcriptomics reveals cell-type-specific diversification in human heart failure. Nat. Cardiovasc. Res. 2022, 1, 263–280. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Lv, Z.; Yu, Z.; Wang, K.; Xu, C.; Li, Y.; Wang, Y. Exploration of dilated cardiomyopathy for biomarkers and immune microenvironment: Evidence from RNA-seq. BMC Cardiovasc. Disord. 2022, 22, 320. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef]
- Grabie, N.; Lichtman, A.H.; Padera, R. T cell checkpoint regulators in the heart. Cardiovasc. Res. 2019, 115, 869–877. [Google Scholar] [CrossRef]
- Kamphorst, A.O.; Wieland, A.; Nasti, T.; Yang, S.; Zhang, R.; Barber, D.L.; Konieczny, B.T.; Daugherty, C.Z.; Koenig, L.; Yu, K.; et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 2017, 355, 1423–1427. [Google Scholar] [CrossRef]
- Garcia-Rivas, G.; Castillo, E.C.; Gonzalez-Gil, A.M.; Maravillas-Montero, J.L.; Brunck, M.; Torres-Quintanilla, A.; Elizondo-Montemayor, L.; Torre-Amione, G. The role of B cells in heart failure and implications for future immunomodulatory treatment strategies. ESC Heart Fail. 2020, 7, 1387–1399. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, A.; Badr, M.E.S.G.; Miyauchi, K.; Ishihara, C.; Onishi, R.; Guo, Z.; Sasaki, Y.; Ike, H.; Takumi, A.; Tsuji, N.M.; et al. CRTAM determines the CD4+ cytotoxic T lymphocyte lineage. J. Exp. Med. 2015, 213, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Koczo, A.; Marino, A.; Rocco, J.; Ewald, G.; Givertz, M.M.; Rajagopalan, N.; Bozkurt, B.; Elkayam, U.; Cooper, L.T.; Fett, J.; et al. Proinflammatory TH17 cytokine activation, disease severity and outcomes in peripartum cardiomyopathy. Int. J. Cardiol. 2021, 339, 93–98. [Google Scholar] [CrossRef]
- Papa, S.; Choy, P.M.; Bubici, C. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene 2019, 38, 2223–2240. [Google Scholar] [CrossRef]
- Link, W.; Fernandez-Marcos, P.J. FOXO transcription factors at the interface of metabolism and cancer. Int. J. Cancer 2017, 141, 2379–2391. [Google Scholar] [CrossRef] [PubMed]
- Masui, K.; Tanaka, K.; Akhavan, D.; Babic, I.; Gini, B.; Matsutani, T.; Iwanami, A.; Liu, F.; Villa, G.R.; Gu, Y.; et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013, 18, 726–739. [Google Scholar] [CrossRef]
- Liu, L.X.; Arany, Z. Maternal cardiac metabolism in pregnancy. Cardiovasc. Res. 2014, 101, 545–553. [Google Scholar] [CrossRef]
- Ritterhoff, J.; Tian, R. Metabolism in cardiomyopathy: Every substrate matters. Cardiovasc. Res. 2017, 113, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Davila-Roman, V.G.; Vedala, G.; Herrero, P.; de las Fuentes, L.; Rogers, J.G.; Kelly, D.P.; Gropler, R.J. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 2002, 40, 271–277. [Google Scholar] [CrossRef]
- Hoes, M.F.; Bomer, N.; Ricke-Hoch, M.; de Jong, T.V.; Arevalo Gomez, K.F.; Pietzsch, S.; Hilfiker-Kleiner, D.; van der Meer, P. Human iPSC-Derived Cardiomyocytes of Peripartum Patients With Cardiomyopathy Reveal Aberrant Regulation of Lipid Metabolism. Circulation 2020, 142, 2288–2291. [Google Scholar] [CrossRef]
- Schulze, P.C.; Drosatos, K.; Goldberg, I.J. Lipid Use and Misuse by the Heart. Circ. Res. 2016, 118, 1736–1751. [Google Scholar] [CrossRef] [PubMed]
- Bankaitis, V.A.; Garcia-Mata, R.; Mousley, C.J. Golgi membrane dynamics and lipid metabolism. Curr. Biol. 2012, 22, R414–R424. [Google Scholar] [CrossRef]
- Lu, L.; Zhou, Q.; Chen, Z.; Chen, L. The significant role of the Golgi apparatus in cardiovascular diseases. J. Cell. Physiol. 2018, 233, 2911–2919. [Google Scholar] [CrossRef] [PubMed]
- Bollen, I.A.E.; Ehler, E.; Fleischanderl, K.; Bouwman, F.; Kempers, L.; Ricke-Hoch, M.; Hilfiker-Kleiner, D.; Dos Remedios, C.G.; Kruger, M.; Vink, A.; et al. Myofilament Remodeling and Function Is More Impaired in Peripartum Cardiomyopathy Compared with Dilated Cardiomyopathy and Ischemic Heart Disease. Am. J. Pathol. 2017, 187, 2645–2658. [Google Scholar] [CrossRef] [PubMed]
- Nicin, L.; Abplanalp, W.T.; Schanzer, A.; Sprengel, A.; John, D.; Mellentin, H.; Tombor, L.; Keuper, M.; Ullrich, E.; Klingel, K.; et al. Single Nuclei Sequencing Reveals Novel Insights into the Regulation of Cellular Signatures in Children with Dilated Cardiomyopathy. Circulation 2021, 143, 1704–1719. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ. Res. 2019, 125, 117–146. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.L.T.; Ismahil, M.A.; Jegga, A.G.; Zilliox, M.J.; Troidl, C.; Prabhu, S.D.; Sadayappan, S. Cardiac inflammation in genetic dilated cardiomyopathy caused by MYBPC3 mutation. J. Mol. Cell. Cardiol. 2017, 102, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Belke, D.D.; Garnett, L.; Martens, K.; Abdelfatah, N.; Rodriguez, M.; Diao, C.; Chen, Y.X.; Gordon, P.M.; Nygren, A.; et al. Transgenic mice overexpressing desmocollin-2 (DSC2) develop cardiomyopathy associated with myocardial inflammation and fibrotic remodeling. PLoS ONE 2017, 12, e0174019. [Google Scholar] [CrossRef] [PubMed]
- Sielemann, K.; Elbeck, Z.; Gärtner, A.; Brodehl, A.; Stanasiuk, C.; Fox, H.; Paluszkiewicz, L.; Tiesmeier, J.; Wlost, S.; Gummert, J.; et al. Distinct Myocardial Transcriptomic Profiles of Cardiomyopathies Stratified by the Mutant Genes. Genes 2020, 11, 1430. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Donor | PPCM | DCM | ||
|---|---|---|---|---|
| SHB | N | 6 | 6 | - |
| Age (years) | 34.8 ± 8.0 | 34.2 ± 6.9 | - | |
| Female (%) | 67 | 100 | - | |
| GSE141910 | N | 29 | 6 | 33 |
| Age (years) | 47.4 ± 9.8 | 34.7 ± 10.3 * | 42.5 ± 10.9 | |
| Female (%) | 100 | 100 | 100 | |
| Total N | 35 | 12 | 33 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taylor, J.; Yeung, A.C.Y.; Ashton, A.; Faiz, A.; Guryev, V.; Fang, B.; Lal, S.; Grosser, M.; dos Remedios, C.G.; Braet, F.; et al. Transcriptomic Comparison of Human Peripartum and Dilated Cardiomyopathy Identifies Differences in Key Disease Pathways. J. Cardiovasc. Dev. Dis. 2023, 10, 188. https://doi.org/10.3390/jcdd10050188
Taylor J, Yeung ACY, Ashton A, Faiz A, Guryev V, Fang B, Lal S, Grosser M, dos Remedios CG, Braet F, et al. Transcriptomic Comparison of Human Peripartum and Dilated Cardiomyopathy Identifies Differences in Key Disease Pathways. Journal of Cardiovascular Development and Disease. 2023; 10(5):188. https://doi.org/10.3390/jcdd10050188
Chicago/Turabian StyleTaylor, Jude, Anna C. Y. Yeung, Anthony Ashton, Alen Faiz, Victor Guryev, Bernard Fang, Sean Lal, Mark Grosser, Cristobal G. dos Remedios, Filip Braet, and et al. 2023. "Transcriptomic Comparison of Human Peripartum and Dilated Cardiomyopathy Identifies Differences in Key Disease Pathways" Journal of Cardiovascular Development and Disease 10, no. 5: 188. https://doi.org/10.3390/jcdd10050188