Genetic Differentiation of a New World Screwworm Fly Population from Uruguay Detected by SNPs, Mitochondrial DNA and Microsatellites in Two Consecutive Years

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Sampling Information
2.2. DNA Extraction
2.3. Single-Nucleotide Polymorphisms
2.4. mtDNA
2.5. Microsatellites
2.6. Genetic Diversity and Population Differentiation
2.7. Demographic Inferences
3. Results
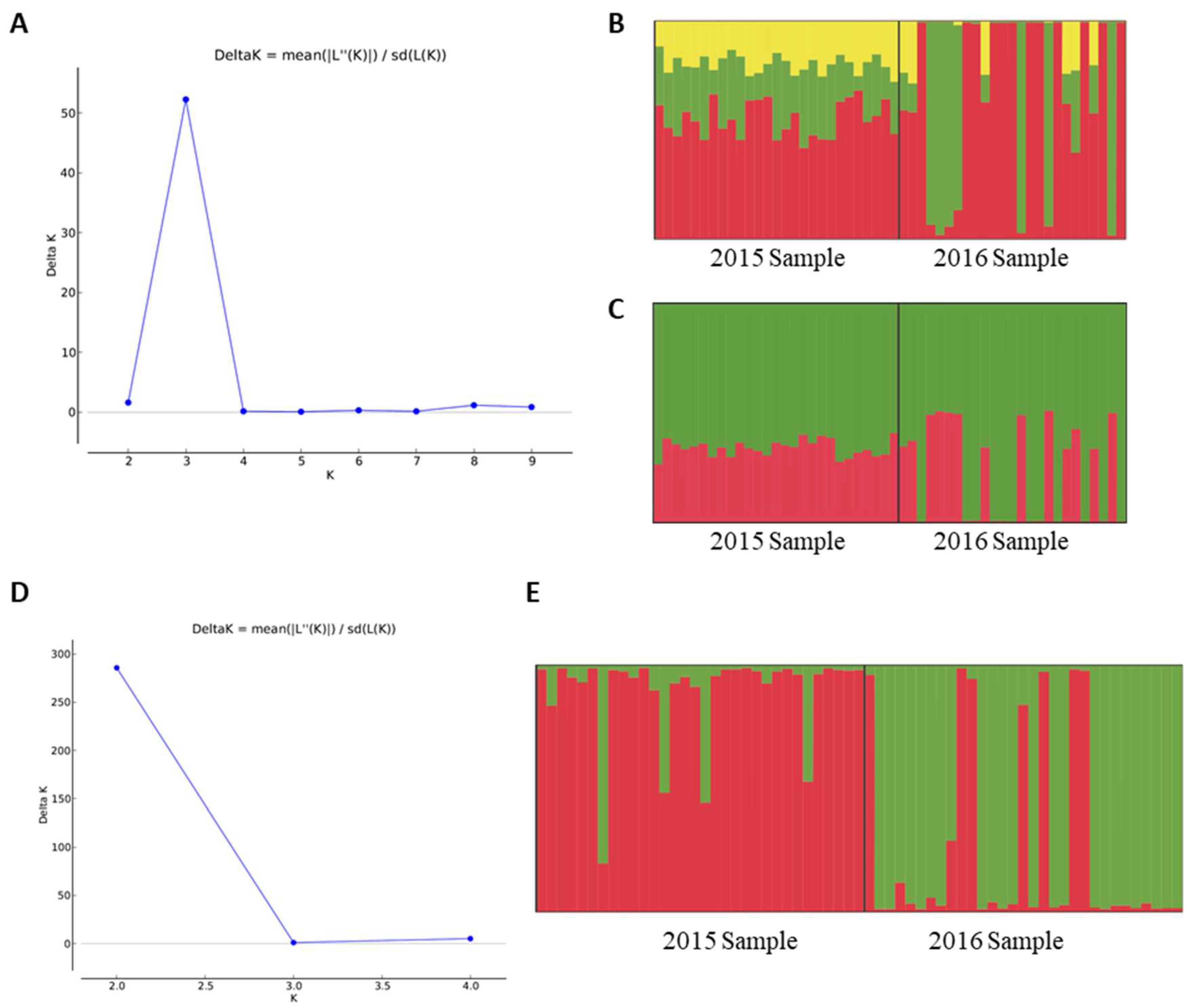
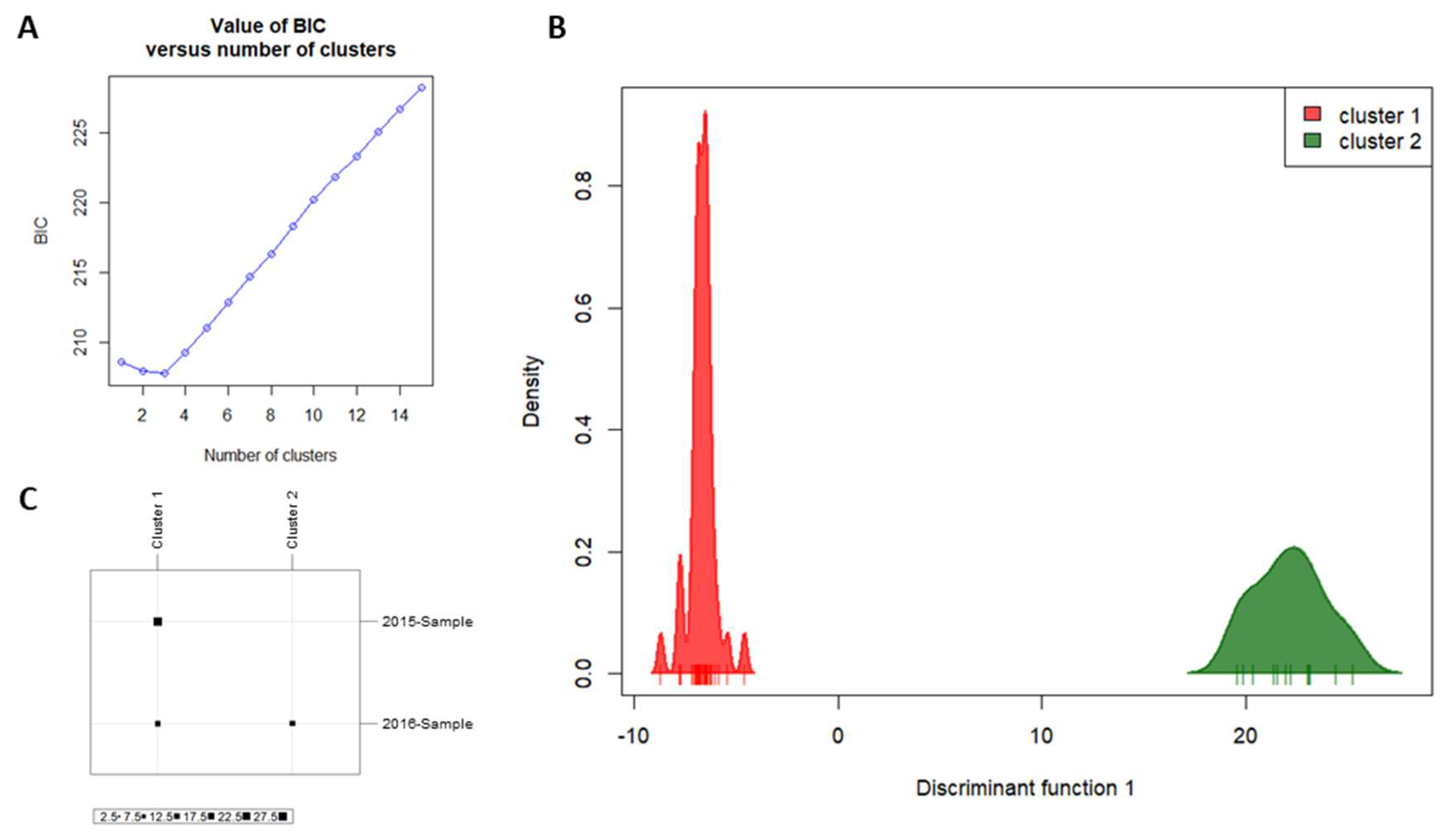
3.1. Single-Nucleotide Polymorphisms
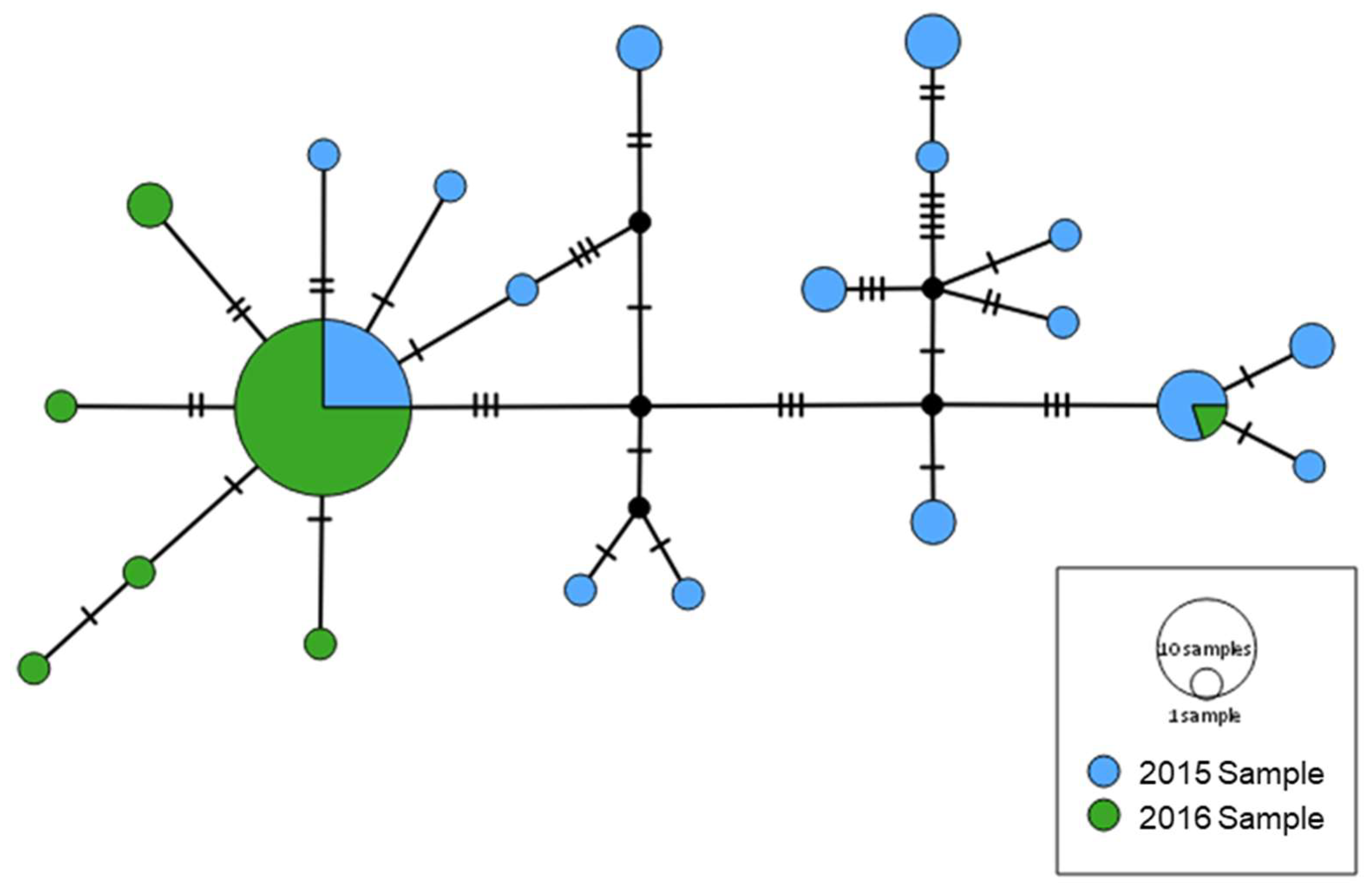
3.2. mtDNA
3.3. Microsatellites
3.4. Demographic Inferences
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability Statement
Additional Information
References
- Luikart, G.; England, P.R.; Tallmon, D.; Jordan, S.; Taberlet, P. The power and promise of population genomics: From genotyping to genome typing. Nat. Rev. Genet. 2003, 4, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M. Genome-Wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 2016, 17, 81–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poland, J.; Brown, P.J.; Sorrells, M.E.; Jannink, J.-L. Development of High-Density Genetic Maps for Barley and Wheat Using a Novel Two-Enzyme Genotyping-by-Sequencing Approach. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nosil, P.; Gompert, Z.; Farkas, T.E.; Comeault, A.A.; Feder, J.L.; Buerkle, C.A.; Parchman, T.L. Genomic consequences of multiple speciation processes in a stick insect. Proc. R. Soc. B Biol. Sci. 2012, 279, 5058–5065. [Google Scholar] [CrossRef] [Green Version]
- Gompert, Z.; Comeault, A.A.; Farkas, T.E.; Feder, J.L.; Parchman, T.L.; Buerkle, C.A.; Nosil, P. Experimental evidence for ecological selection on genome variation in the wild. Ecol. Lett. 2013, 17, 369–379. [Google Scholar] [CrossRef]
- Lozier, J.D. Revisiting comparisons of genetic diversity in stable and declining species: Assessing genome-wide polymorphism in North American bumble bees using RAD sequencing. Mol. Ecol. 2014, 23, 788–801. [Google Scholar] [CrossRef]
- Silva-Brandão, K.L.; E Silva, O.A.B.N.; Brandão, M.M.; Omoto, C.; Sperling, F.A. Genotyping-By-Sequencing approach indicates geographic distance as the main factor affecting genetic structure and gene flow in Brazilian populations of Grapholita molesta (Lepidoptera, Tortricidae). Evol. Appl. 2015, 8, 476–485. [Google Scholar] [CrossRef]
- Silva-Brandão, K.L.; Peruchi, A.; Seraphim, N.; Murad, N.F.; Carvalho, R.A.; Farias, J.R.; Omoto, C.; Cônsoli, F.L.; Figueira, A.; Brandão, M.M. Loci under selection and markers associated with host plant and host-related strains shape the genetic structure of Brazilian populations of Spodoptera frugiperda (Lepidoptera, Noctuidae). PLoS ONE 2018, 13, e0197378. [Google Scholar] [CrossRef]
- Anderson, C.; Tay, W.T.; McGaughran, A.; Gordon, K.H.; Walsh, T. Population structure and gene flow in the global pest, Helicoverpa armigera. Mol. Ecol. 2016, 25, 5296–5311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wachi, N.; Matsubayashi, K.W.; Maeto, K. Application of next-generation sequencing to the study of non-model insects. Entomol. Sci. 2017, 21, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Habel, J.C.; Husemann, M.; Finger, A.; Danley, P.D.; Zachos, F.E. The relevance of time series in molecular ecology and conservation biology. Biol. Rev. 2013, 89, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Nunziata, S.O.; Weisrock, D.W. Estimation of contemporary effective population size and population declines using RAD sequence data. Heredity 2017, 120, 196–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunziata, S.O.; Lance, S.L.; Scott, D.; Lemmon, E.M.; Weisrock, D.W. Genomic data detect corresponding signatures of population size change on an ecological time scale in two salamander species. Mol. Ecol. 2017, 26, 1060–1074. [Google Scholar] [CrossRef] [PubMed]
- Owings, C.G.; Spiegelman, C.; Tarone, A.M.; Tomberlin, J.K. Developmental variation among Cochliomyia macellaria Fabricius (Diptera: Calliphoridae) populations from three ecoregions of Texas, USA. Int. J. Leg. Med. 2014, 128, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, J.H.; Papavero, N.; Prado, Â.P.D. As miíases na região neotropical (identificação, biologia, bibliografia). Rev. Bras. Zool. 1982, 1, 239–416. [Google Scholar] [CrossRef] [Green Version]
- Adams, T.S. The Reproductive Physiology of the Screwworm, Cochliomyia Hominivorax (Diptera: Calliphoridae) II. Effect of constant temperatures on oogenesis. J. Med. Entomol. 1979, 15, 484–487. [Google Scholar] [CrossRef]
- Wyss, J.H. Screwworm Eradication in the Americas. Ann. N. Y. Acad. Sci. 2000, 916, 186–193. [Google Scholar] [CrossRef]
- Vargas-Terán, M.; Hofmann, H.C.; Tweddle, N.E. Impact of Screwworm Eradication Programmes Using the Sterile Insect Technique. In Sterile Insect Technique: Principles and Practice in Area-Wide Integrated Pest Management; Springer: Dordrecht, The Netherlands, 2005; pp. 629–650. [Google Scholar]
- Bergamo, L.W.; Fresia, P.; Azeredo-Espin, A.M.L. Phylogeography and Insecticide Resistance in the NWS in South America and Caribbean Regions. In Area-Wide Integrated Pest Management: Development and Field Application; Hendrichs, J., Pereira, R., Vreysen, M.J.B., Eds.; CRC Press: Boca Raton, FL, USA, in press.
- Lyra, M.L.; Klaczko, L.B.; Azeredo-Espin, A.M.L. Complex patterns of genetic variability in populations of the New World screwworm fly revealed by mitochondrial DNA markers. Med. Veter. Entomol. 2009, 23, 32–42. [Google Scholar] [CrossRef]
- Fresia, P.; Lyra, M.L.; Coronado, A.; De Azeredo-Espin, A.M.L. Genetic structure and demographic history of new world screwworm across its current geographic range. J. Med. Entomol. 2011, 48, 280–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyra, M.L.; Fresia, P.; Gama, S.; Cristina, J.; Klaczko, L.B.; de Azeredo-Espin, A.M.L. Analysis of Mitochondrial DNA Variability and Genetic Structure in Populations of New World Screwworm Flies (Diptera: Calliphoridae) from Uruguay. J. Med. Entomol. 2005, 42, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Torres, T.T.; Lyra, M.L.; Fresia, P.; Azeredo-Espin, A.M.L. Assessing Genetic Variation in New World Screwworm Cochliomyia Hominivorax Populations from Uruguay. In Area-Wide Control of Insect Pests; Vreysen, M.J.B., Robinson, A.S., Hendrichs, J., Eds.; Springer: Dordrecht, The Netherlands, 2007; pp. 183–191. [Google Scholar]
- Bergamo, L.W.; Fresia, P.; Azeredo-Espin, A.M.L. Incongruent Nuclear and Mitochondrial Genetic Structure of New World Screwworm Fly Populations Due to Positive Selection of Mutations Associated with Dimethyl- and Diethyl-Organophosphates Resistance. PLoS ONE 2015, 10, e0128441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergamo, L.W.; Fresia, P.; Lyra, M.L.; Azeredo-Espin, A.M.L. High Genetic Diversity and No Population Structure of the New World Screwworm Fly Cochliomyia hominivorax (Diptera: Calliphoridae) on a Microgeographic Scale: Implications for Management Units. J. Econ. Entomol. 2018, 111, 2476–2482. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Catchen, J.M.; Amores, A.; Hohenlohe, P.; Cresko, W.A.; Postlethwait, J.H. Stacks: Building and Genotyping Loci De Novo From Short-Read Sequences. G3 Genes Genom. Genet. 2011, 1, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [Green Version]
- Raymond, M.; Rousset, F. GENEPOP (Version 1.2): Population Genetics Software for Exact Tests and Ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Rousset, F. genepop’007: A complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef]
- Antao, T.; Lopes, A.; Lopes, R.J.; Beja-Pereira, A.; Luikart, G. LOSITAN: A workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinform. 2008, 9, 323. [Google Scholar] [CrossRef] [Green Version]
- Beaumont, M.A.; Nichols, R.A. Evaluating loci for use in the genetic analysis of population structure. Proc. R. Soc. B Biol. Sci. 1996, 263, 1619–1626. [Google Scholar] [CrossRef]
- Goetz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-Throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Lischer, H.E.L.; Excoffier, L. PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 2011, 28, 298–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lessinger, A.C.; Azeredo-Espin, A.M.L. Evolution and structural organisation of mitochondrial DNA control region of myiasis-causing flies. Med. Veter. Entomol. 2000, 14, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Torres, T.T.; Brondani, R.P.V.; Garcia, J.E.; Azeredo-Espin, A.M.L. Isolation and characterization of microsatellite markers in the new world screw-worm Cochliomyia hominivorax (Diptera: Calliphoridae). Mol. Ecol. Notes 2004, 4, 182–184. [Google Scholar] [CrossRef]
- Torres, T.T.; De Azeredo-Espin, A.M.L. Development of new polymorphic microsatellite markers for the New World screw-worm Cochliomyia hominivorax (Diptera: Calliphoridae). Mol. Ecol. Notes 2005, 5, 815–817. [Google Scholar] [CrossRef]
- Johathan Liu, C.S.; Hulce, D.; Li, X.; Snyder-Leiby, T. GeneMarker® genotyping software: Tools to increase the statistical power of DNA fragment analysis. J. Biomol. Tech. 2011, 22, S35–S36. [Google Scholar]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. Microchecker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Earl, D.A.; Vonholdt, B.M. Structure Harvester: A website and program for visualizing Structure output and implementing the Evanno method. Conserv. Genet. Resour. 2011, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, N.A. Distruct: A program for the graphical display of population structure. Mol. Ecol. Notes 2003, 4, 137–138. [Google Scholar] [CrossRef]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- Leigh, J.W.; Bryant, D. Popart: Full-Feature software for haplotype network construction. Method. Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Clement, M.; Snell, Q.; Walker, P.; Posada, D.; Crandall, K. TCS: Estimating Gene Genealogies. In Parallel and Distributed Processing Symposium, Proceedings International; IEEE: Fort Lauderdale, FL, USA, 2002; p. 184. [Google Scholar]
- Dieringer, D.; Schlötterer, C. Microsatellite analyser(MSA): A platform independent analysis tool for large microsatellite data sets. Mol. Ecol. Notes 2003, 3, 167–169. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.W.; Thompson, E.A. Performing the Exact Test of Hardy-Weinberg Proportion for Multiple Alleles. Biometrics 1992, 48, 361. [Google Scholar] [CrossRef] [PubMed]
- Jorde, P.E.; Ryman, N. Unbiased Estimator for Genetic Drift and Effective Population Size. Genetics 2007, 177, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Do, C.; Waples, R.S.; Peel, D.; Macbeth, G.M.; Tillett, B.J.; Ovenden, J.R. NeEstimatorv2: Re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol. Ecol. Resour. 2013, 14, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. The Effect of Change in Population Size on DNA Polymorphism. Genetics 1989, 123, 597–601. [Google Scholar] [PubMed]
- Fu, Y.X. Statistical Tests of Neutrality of Mutations Against Population Growth, Hitchhiking and Background Selection. Genetics 1997, 147, 915–925. [Google Scholar]
- Bravo, A.; Gill, S.S.; Soberón, M. Mode of action of Bacillus thuringiensis Cry and Cyt toxins and their potential for insect control. Toxicon 2007, 49, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Pardo-López, L.; Soberón, M.; Bravo, A. Bacillus thuringiensisinsecticidal three-domain Cry toxins: Mode of action, insect resistance and consequences for crop protection. FEMS Microbiol. Rev. 2013, 37, 3–22. [Google Scholar] [CrossRef] [Green Version]
- Kostaropoulos, I.; Papadopoulos, A.I.; Metaxakis, A.; Boukouvala, E.; Papadopoulou-Mourkidou, E. Glutathione S–Transferase in the defence against pyrethroids in insects. Insect Biochem. Mol. Biol. 2001, 31, 313–319. [Google Scholar] [CrossRef]
- Fang, S.-M. Insect Glutathione S-Transferase: A Review of Comparative Genomic Studies and Response to Xenobiotics. Bull. Insectol. 2012, 65, 265–271. [Google Scholar]
- Jombart, T.; Collins, C. A Tutorial for Discriminant Analysis of Principal Components (DAPC) Using Adegenet 2.0.0; Imperial College London–MRC Centre for Outbreak Analysis and Modelling: London, UK, 2015. [Google Scholar]
- Brumfield, R.T.; Beerli, P.; Nickerson, D.A.; Edwards, S.V. The Utility of Single Nucleotide Polymorphisms in Inferences of Population History. Trends Ecol. Evol. 2003, 18, 249–256. [Google Scholar] [CrossRef]
- Marandel, F.; Charrier, G.; Lamy, J.B.; Le Cam, S.; Lorance, P.; Trenkel, V.M. Estimating Effective Population Size Using RADseq: Effects of SNP Selection and Sample Size. Ecol. Evol. 2020, 10, 1929–1937. [Google Scholar] [CrossRef] [PubMed]
- Saarman, N.P.; Gloria-Soria, A.; Anderson, E.C.; Evans, B.R.; Pless, E.; Cosme, L.V.; Gonzalez-Acosta, C.; Kamgang, B.; Wesson, D.M.; Powell, J.R. Effective Population Sizes of a Major Vector of Human Diseases, Aedes Aegypti. Evol. Appl. 2017, 10, 1031–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shpak, M.; Wakeley, J.; Garrigan, D.; Lewontin, R.C. A Structured Coalescent Process for Seasonally Fluctuating Populations. Evolut. N. Y. 2009, 64, 1395–1409. [Google Scholar] [CrossRef] [PubMed]
- Mesnick, S.L.; Taylor, B.L.; Archer, F.I.; Martien, K.K.; Treviño, S.E.; Hancock-Hanser, B.L.; Medina, S.C.M.; Pease, V.L.; Robertson, K.M.; Straley, J.M.; et al. Sperm whale population structure in the eastern and central North Pacific inferred by the use of single-nucleotide polymorphisms, microsatellites and mitochondrial DNA. Mol. Ecol. Resour. 2011, 11, 278–298. [Google Scholar] [CrossRef] [PubMed]
- Mayer, D.G.; Atzeni, M.G. Estimation of Dispersal Distances for Cochliomyia hominivorax (Diptera: Calliphoridae). Environ. Entomol. 1993, 22, 368–374. [Google Scholar] [CrossRef]
- De Carvalho, R.A.; Torres, T.T.; De Azeredo-Espin, A.M.L. A survey of mutations in the Cochliomyia hominivorax (Diptera: Calliphoridae) esterase E3 gene associated with organophosphate resistance and the molecular identification of mutant alleles. Veter. Parasitol. 2006, 140, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.R.; De Azeredo-Espin, A.M.; Torres, T.T. Deep sequencing of New World screw-worm transcripts to discover genes involved in insecticide resistance. BMC Genom. 2010, 11, 695. [Google Scholar] [CrossRef] [Green Version]
- Ffrench-Constant, R.; Daborn, P.J.; Le Goff, G. The genetics and genomics of insecticide resistance. Trends Genet. 2004, 20, 163–170. [Google Scholar] [CrossRef]
- Tabashnik, B.E.; Brévault, T.; Carrière, Y. Insect resistance to Bt crops: Lessons from the first billion acres. Nat. Biotechnol. 2013, 31, 510–521. [Google Scholar] [CrossRef]
- Calla, B.; Demkovich, M.; Siegel, J.P.; Gomes Viana, J.P.; Walden, K.K.O.; Robertson, H.M.; Berenbaum, M.R. Selective Sweeps in a Nutshell; The Genomic Footprint of Rapid Insecticide Resistance Evolution in an Insect Introduction. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| 2015-Sample | 2016-Sample | |
|---|---|---|
| Total number of sequences | 145,692,719 | 136,389,952 |
| Reads containing adapter sequence | 9,343,260 | 6,878,774 |
| Ambiguous barcodes | 934,983 | 13,656,605 |
| Low quality reads | 51,524 | 164,258 |
| Ambiguous tags | 5,134,707 | 7,798,267 |
| Retained reads | 130,228,245 (89%) | 107,892,048 (79%) |
| Sample | N | Nh | Haplotypes (# Individuals) | Ĥ (SD) | π (SD) |
|---|---|---|---|---|---|
| 2015 | 32 | 19 | H1 (8), H2 (4), H3 (2), H4 (2), H5 (2), H6 (3), H8 (2), H9 to H17 (1) | 0.9375 (0.0286) | 0.004530 (0.002372) |
| 2016 | 31 | 8 | H1 (24), H2 (1), H7 (2), H18 to H21 (1) | 0.4538 (0.1109) | 0.001858 (0.001064) |
| Sample | Loci | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| CH01 | CH05 | CH09 | CH12 | CH14 | CH15 | CH24 | CH26 | ||
| 2015-Sample | N | 23 | 32 | 31 | 30 | 24 | 21 | 21 | 32 |
| (N = 32) | Na (AR) | 7 (7) | 5 (4.97) | 5 (5) | 11 (11) | 4 (4) | 8 (7.98) | 6 (6) | 11 (10.75) |
| Ho | 0.522 | 0.656 | 0.355 | 0.833 | 0.375 | 0.667 | 0.714 | 0.781 | |
| He | 0.693 | 0.665 | 0.361 | 0.804 | 0.533 | 0.811 | 0.787 | 0.815 | |
| FIS | 0.2677 | 0.0291 | 0.0337 | −0.0197 | 0.3157 | 0.2011 | 0.1163 | 0.0572 | |
| 2016-Sample | N | 31 | 31 | 31 | 31 | 31 | 19 | 31 | 30 |
| (N = 31) | Na (AR) | 6 (5.99) | 6 (6) | 5 (5) | 7 (6.97) | 5 (4.90) | 5 (5) | 4 (4) | 9 (9) |
| Ho | 0.548 | 0.645 | 0.613 | 0.710 | 0.645 | 0.579 | 0.613 | 0.833 | |
| He | 0.677 | 0.583 * | 0.624 | 0.729 | 0.616 * | 0.778 * | 0.670 | 0.852 * | |
| FIS | 0.2056 | −0.0909 | 0.0339 | 0.0435 | −0.0318 | 0.2813 | 0.1009 | 0.0391 | |
| Lowest Allele Frequency Used | |||||
|---|---|---|---|---|---|
| 0.05 | 0.02 | 0.01 | 0+ | ||
| Harmonic Mean Sample Size | 27.1 | 27 | 27.2 | 27.5 | |
| SNP | Fs | 0.07969 | 0.08043 | 0.07811 | 0.07739 |
| F′ | 0.03631 | 0.03710 | 0.03469 | 0.03435 | |
| Ne (95% CI, parametric) | 247.9 (211.4–287.2) | 242.6 (211.2–276.1) | 259.5 (233.3–287.0) | 262.0 (237.3–287.8) | |
| SSR | Fs | 0.17722 | 0.17167 | 0.17052 | 0.17003 |
| F′ | 0.13531 | 0.13013 | 0.12905 | 0.12859 | |
| Ne (95% CI, parametric) | 66.5 (39.5–100.5) | 69.2 (43.9–100.1) | 69.7 (45.1–99.6) | 70.0 (46.5–98.2) | |
| Tajima’s D | p-Value | Fu’s FS | p-Value | |
|---|---|---|---|---|
| 2015-Sample | −0.32833 | 0.416 | −2.77043 | 0.16600 |
| 2016-Sample | −2.38060 | <0.001 * | 1.10613 | 0.72700 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bergamo, L.W.; Silva-Brandão, K.L.; Vicentini, R.; Fresia, P.; Azeredo-Espin, A.M.L. Genetic Differentiation of a New World Screwworm Fly Population from Uruguay Detected by SNPs, Mitochondrial DNA and Microsatellites in Two Consecutive Years. Insects 2020, 11, 539. https://doi.org/10.3390/insects11080539
Bergamo LW, Silva-Brandão KL, Vicentini R, Fresia P, Azeredo-Espin AML. Genetic Differentiation of a New World Screwworm Fly Population from Uruguay Detected by SNPs, Mitochondrial DNA and Microsatellites in Two Consecutive Years. Insects. 2020; 11(8):539. https://doi.org/10.3390/insects11080539
Chicago/Turabian StyleBergamo, Luana Walravens, Karina Lucas Silva-Brandão, Renato Vicentini, Pablo Fresia, and Ana Maria Lima Azeredo-Espin. 2020. "Genetic Differentiation of a New World Screwworm Fly Population from Uruguay Detected by SNPs, Mitochondrial DNA and Microsatellites in Two Consecutive Years" Insects 11, no. 8: 539. https://doi.org/10.3390/insects11080539