NHI- and NHC-Supported Al(III) Hydrides for Amine–Borane Dehydrocoupling Catalysis


1
WACKER-Institute of Silicon Chemistry and Catalysis Research Center, Technische Universität München, Lichtenbergstraße 4, 85748 Garching bei München, Germany
2
Department of Chemistry and Chemical Biology, Gunma University, Kiryu 376-8515, Japan
*
Author to whom correspondence should be addressed.
Inorganics 2019, 7(8), 92; https://doi.org/10.3390/inorganics7080092
Submission received: 26 June 2019
/
Revised: 11 July 2019
/
Accepted: 12 July 2019
/
Published: 24 July 2019
(This article belongs to the Special Issue Organoaluminum Compounds)
Abstract
:The catalytic dehydrocoupling of amine–boranes has recently received a great deal of attention due to its potential in hydrogen storage applications. The use of aluminum catalysts for this transformation would provide an additional cost-effective and sustainable approach towards the hydrogen economy. Herein, we report the use of both N-heterocyclic imine (NHI)- and carbene (NHC)-supported Al(III) hydrides and their role in the catalytic dehydrocoupling of Me2NHBH3. Differences in the σ-donating ability of the ligand class resulted in a more stable catalyst for NHI-Al(III) hydrides, whereas a deactivation pathway was found in the case of NHC-Al(III) hydrides.
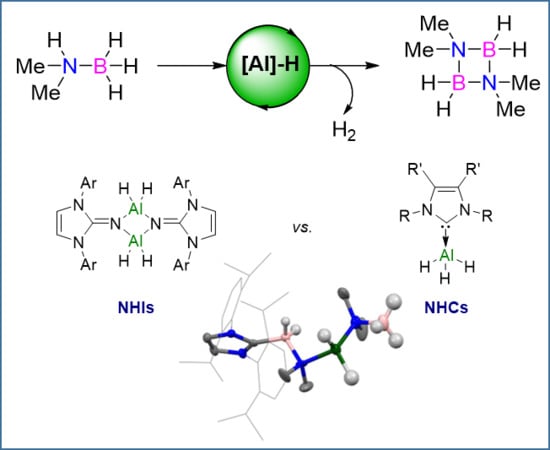
1. Introduction
Main group chemistry has seen a resurgence of interest in recent years, driven by the need for more economically viable and eco-friendly processes. Whilst catalytic transformations using transition metals are well established, the long-term sustainability of these naturally low-abundance metals is limited. In contrast, use of p-block elements such as aluminum allows for the use of earth-abundant and environmentally benign elements, with aluminum being the third most abundant element in the Earth’s crust. Owing to their high stability and Lewis acidity, Al(III) catalysts in the form of trialkyl or trihalide derivatives have traditionally been used in Zieglar–Natta [1] and Friedel–Crafts [2] reactions, respectively. In comparison, the chemistry of low-oxidation and/or coordinate Al complexes (and p-block complexes) is still in its infancy, and advances in past decades have shown that main group complexes can act as transition metals [3,4,5]. Few examples of low-oxidation and/or coordinate Al complexes in catalysis have been reported, such as the use of aluminum ions, R2Al+, in polymerization systems [6,7] and the recent example of dialumene—a compound with a neutral aluminum–aluminum double bond—for the catalytic reduction of CO2 [8].
At the forefront of main group catalysis, with the exception of frustrated Lewis pair (FLP) chemistry [9,10,11], is the use of metal hydrides, whereupon non-redox-based catalytic cycles have been proposed using a sequence of σ-bond metathesis and insertion reactions. Examples of both s- and p-block metal hydrides for the hydroelementation of unsaturated organic substrates, including CO2, have been reported in recent years [12,13]. In terms of aluminum hydride catalysis, the use of a β-diketiminate-supported Al(III) hydride (LAlH2) was made more reactive by increasing its Lewis acidity upon reaction with MeOTf (Tf = SO2CF3) to yield LAl(H)(OTf). The retention of the Al–H moiety and increased positive charge at Al drew comparisons to transition metal catalysts, as it showed excellent activity towards the hydroboration of carbonyl-containing substrates [14]. Other recent examples of Al hydrides in catalysis have shown that commercially available Al–H-containing reagents such as LiAlH4 and DIBAL-H are active towards the hydrogenation of imines using 1 bar of H2 [15] and hydroboration of alkynes [16], respectively.
Interest in dehydrocoupling reactions has recently risen, as it provides a clean and atom-efficient route to new element–element bonds, with the concomitant formation of dihydrogen, which has numerous applications in organic synthesis and materials chemistry [17,18,19]. The formation of dihydrogen in these types of reactions has gained the most attention due to the potential for hydrogen storage and therefore sustainable energy sources. In this regard, ammonia–borane (NH3BH3) has been earmarked as an ideal candidate for a future “hydrogen economy” due to its high weight percentage of hydrogen [20,21]. As such, catalysts from across the periodic table have shown their potential in the catalytic release of H2 from amine–boranes. Whilst transition metals dominate the field [22], examples from s-block [23,24,25,26], f-block [27,28], and metal-free [29,30,31] based systems have also contributed to furthering our understanding of these complex mechanisms. Central to most of the metal-based systems is the formation of metal hydrides [22] and their subsequent reactivity in enabling turnover. In terms of Al hydrides in dehydrocoupling catalysis, Wright and co-workers have reported the use of Al(III) amide pre-catalysts for amine–borane [32,33,34,35,36] and amine–silane [37] dehydrocoupling, in which the active Al hydride forms in situ.
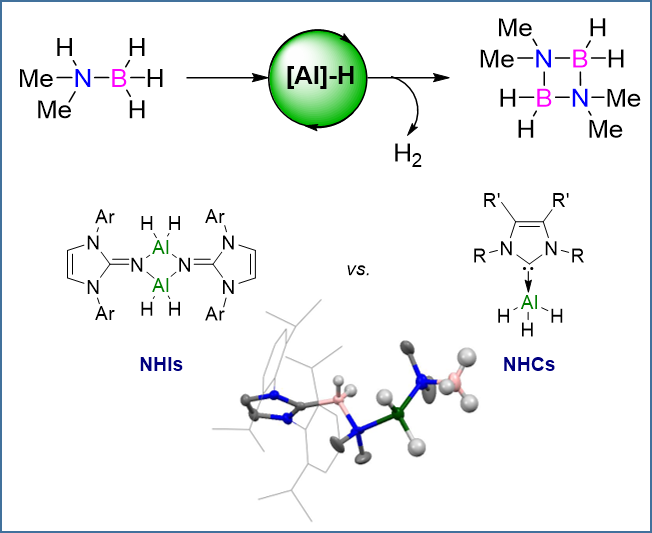
Recent work within our group has shown the use of N-heterocyclic imines (NHIs) for the stabilization of group-13 metal hydrides [38]. This class of ligand is comparable to that of N-heterocyclic carbenes (NHCs), which are widely established as ligands for many catalytic transformations [39], as they can both be considered to be strong 2σ-electron donors (Figure 1). The strong donating capabilities of NHI ligands has allowed for the stabilization and subsequent isolation of a variety of aluminum hydrides which have been used for a number of key transformations, such as the activation of elemental sulfur [40] and the dehydrogenative coupling and subsequent formation of a rare aluminum chalcogen double-bonded species [41]. Notably, the NHI-supported aluminum hydrides proved to be efficient catalysts in the hydroboration of terminal alkynes and carbonyl compounds [42]. Thus, our attention turned towards the use of NHI-supported aluminum hydrides for dehydrocoupling catalysis and comparisons with NHC aluminum hydrides.
2. Results and Discussion
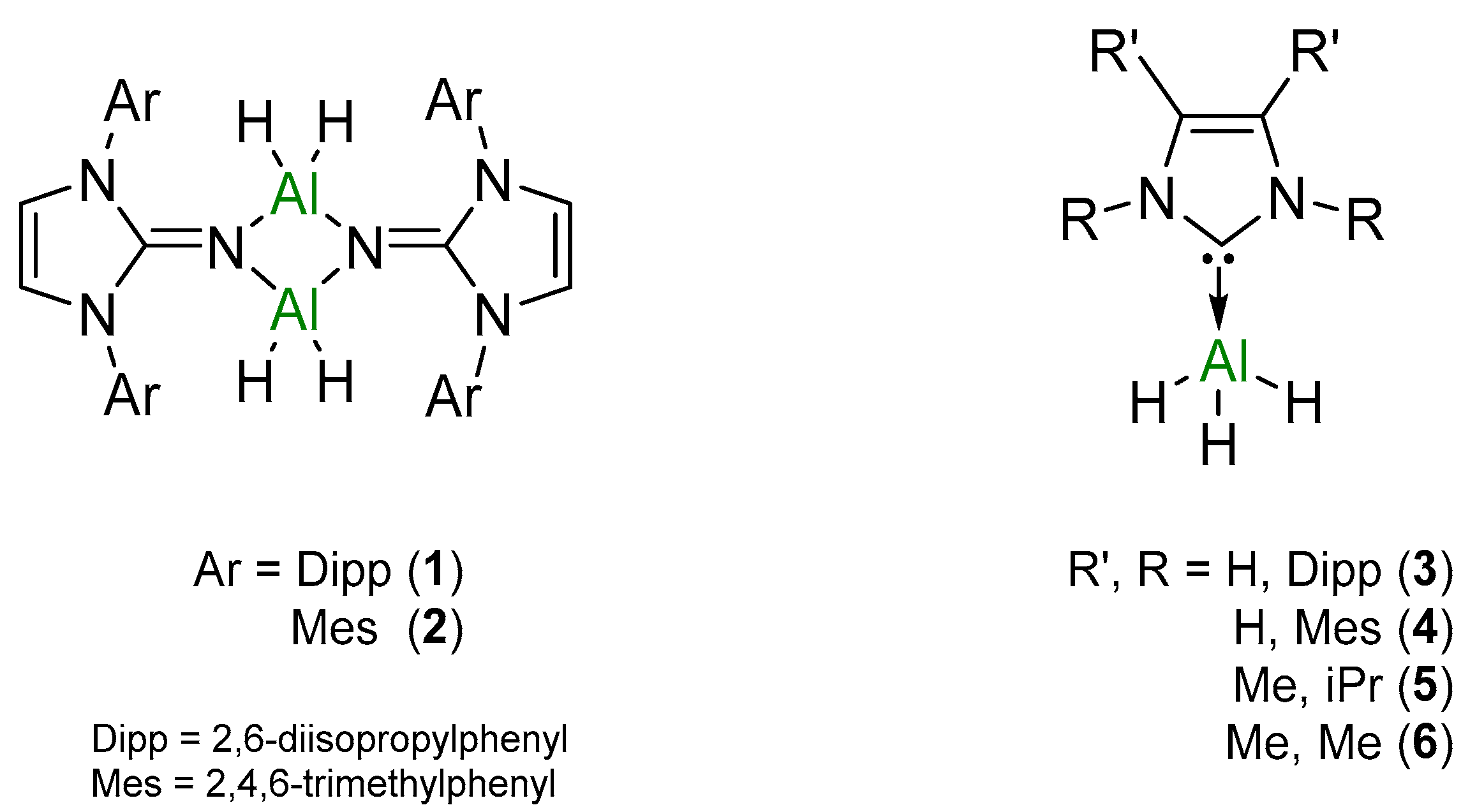
To compare the catalytic potential of NHI- and NHC-stabilized Al(III) hydrides, a direct comparison between the two ligand classes bearing the same substituents was targeted. For ease of synthesis, 2,6-diisopropylphenyl (Dipp) and 2,4,6-trimethylphenyl (Mes) substituents were used for both NHI and NHC ligands. Further comparisons within the NHC ligand class were drawn between aryl and alkyl groups. Synthesis of the six different Al(III) hydrides (1–6) used in this study (Figure 2) were prepared according to literature procedures and then subsequently trialed in dehydrocoupling catalysis.
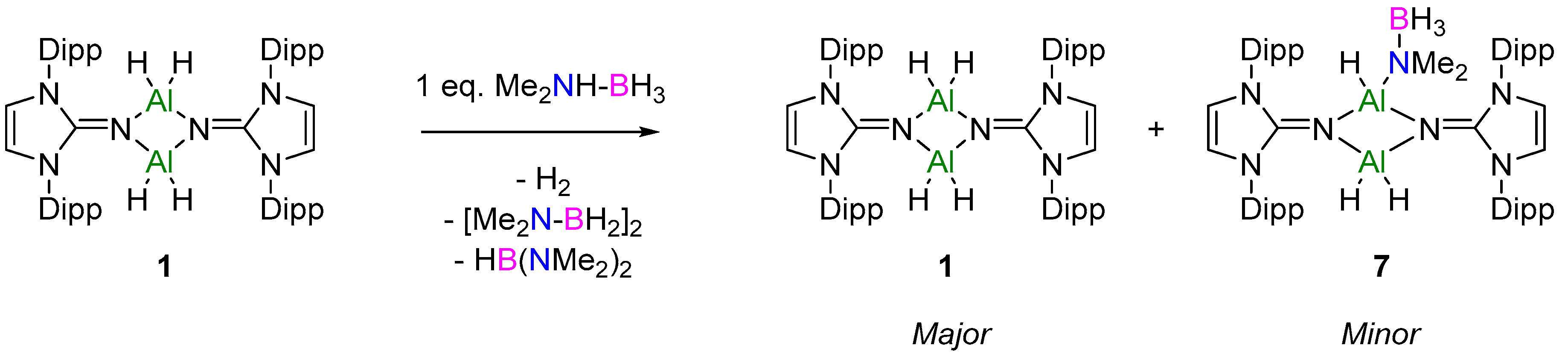
Notable differences between the two ligand classes are apparent upon comparison of the structures. Due to the presence of the imine moiety in NHIs, the steric protection is removed from the metal center, and as the NHI ligands can act as a multifunctional ligand, it forms both σ- and π-donor bonds to Al, resulting in a dimeric Al–H2 complex which prevails in solution and solid state. Initial attempts of using catalysts 1 and 3 in a series of silane–amine (PhSiH3 and HNiPr2) and silane–borane (PhSiH3 and HBpin) dehydrocoupling reactions did not result in turnover, even at elevated temperatures. However, the use of dimethylamine–borane (Me2NHBH3) resulted in turnover under mild conditions.
2.1. Catalysis
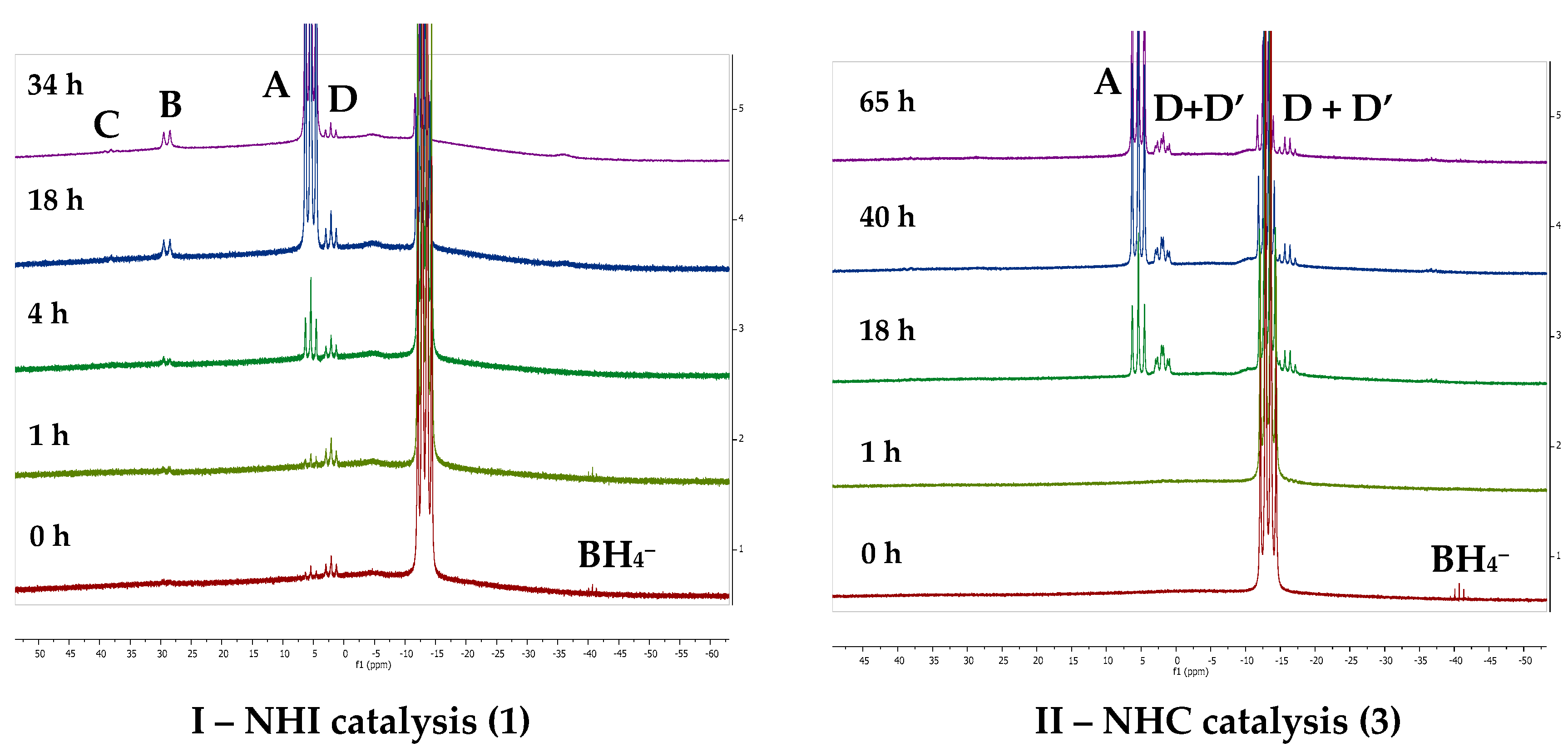
The initial reaction of 5 mol % of 1 with Me2NHBH3 showed a minor formation of H2 after 24 h at room temperature. Increasing the temperature to 50 °C for the same time period also resulted in minor consumption of Me2NHBH3. Upon increasing the temperature to 80 °C, visible gas evolution was noted to occur, and monitoring by both 1H and 11B NMR spectroscopy showed the formation of the cyclic dimer [Me2NBH2]2 (A) and minor formation of the borane species (HB(NMe2)2 (B)). In contrast, upon the use of NHC catalyst 3, turnover was achieved under milder conditions and the formation of A was noted to occur at 50 °C. Further screening using catalysts 1–6 (Table 1) also revealed lower but generally prolonged reaction times for NHC-supported Al(III) hydrides, whilst NHI-supported catalysts required shorter times but higher temperatures. Catalysts 1–6 performed at about the same rate in comparison to previous reports of Al(III) hydride catalysis of Me2NHBH3 [33,34].
Differences between the two ligand classes were found upon inspection of the product distribution (Figure 3 and Table S1), indicating likely subtle changes in the stability of reaction intermediates or different mechanisms. Both ligand classes resulted in the formation of the cyclic dimer (A) as the major species, proceeding with the initial formation of BH4− and [Me2NBH2N(Me)2BH3]− anions (D). However, only NHI-supported Al(III) hydrides (1,2) resulted in the formation of the borane (B) species and a trace amount of dimethylamino–borane, Me2N=BH2 (C), which can be considered an alkene analog. In the case of NHC catalysis, both the free and metal-bound [Me2NBH2N(Me)2BH3]− anions (D and D’, respectively) were observed. Differences within the ligand classes were marginal in the case of NHI ligands, with the steric demands being slightly removed from the Al center and thus minor differences in the TOFs were observed. However, in the case of NHC complexes (3–6), where the steric demands of the ligand substituents were in closer proximity to the Al center, differences in TOFs were apparent. The use of less-sterically demanding Mes (4) or methyl (6) substituents resulted in much higher rates of reaction in comparison to those bearing isopropyl substituents (3 and 5).
2.2. Mechanistic Studies
A series of stoichiometric reactions were undertaken in order to provide further insight into these reactions and the role of the supporting ligands.
2.2.1. NHI Mechanism, Catalysts 1 and 2
The initial reaction of 1 eq. of Me2NHBH3 with compound 1 resulted in no reaction at room temperature, in line with catalytic reduction. Upon heating to 80 °C, a small emergence of A was noted in the 11B NMR spectrum after 1 h, whilst the 1H NMR spectrum appeared unchanged (Figures S13 and S14). Further heating overnight led to the complete consumption of Me2NHBH3, as noted by the loss of Me2NHBH3 CH3 signal at δ 1.78 ppm in the 1H NMR spectrum. Two NHI-containing species were identified via 1H NMR spectroscopy, with the major species being catalyst 1 (85% by relative integrals). The 11B NMR spectrum showed the formation of compounds A and B, and a third species which had a minor shift of δ 0.5 ppm in comparison to Me2NHBH3 (Figure S15). We propose that this new minor NHI-containing species is the first step in the catalytic cycle, whereupon σ-bond metathesis of Al–H with the protic N–H bond of Me2NHBH3 occurs to yield 1 eq. of H2 and compound 7 (Figure 4). Unfortunately, attempts to isolate compound 7 through varying stoichiometries failed and resulted in the isolation of 1 along with dehydrocoupling products A and B.
Additional support for the proposed first step (i.e., formation of 7) is found from consideration of the formation of the dehydrocoupling product B. The formation of B is proposed to occur via β-hydride elimination from the metal-coordinated [Me2NBH2N(Me)2BH3]− anion along with BH3 elimination; this has been observed in both groups 2 and 3 d0 metal-based catalysis [23,43,44]. Alternatively, Wright and co-workers proposed that B is also the result of the deprotonation of Me2NHBH3 by the hydridic B–H rather than Al–H [33]. However, in the latter case B was observed to form at the start of the catalysis, with the concentration remaining static throughout the catalytic reaction [32]. On further inspection of the 11B NMR data obtained from the in situ monitoring of the catalytic reaction (Figure 3I, Figures S2 and S4), both NHI catalysts showed the initial formation of the [Me2NBH2N(Me)2BH3]− anion with the formation of B occurring in the latter stages of the catalysis. Therefore, it is highly likely that this mechanism proceeds via an intermediate akin to compound 7, as outlined in the proposed mechanism (Figure 5).
2.2.2. NHC Mechanism
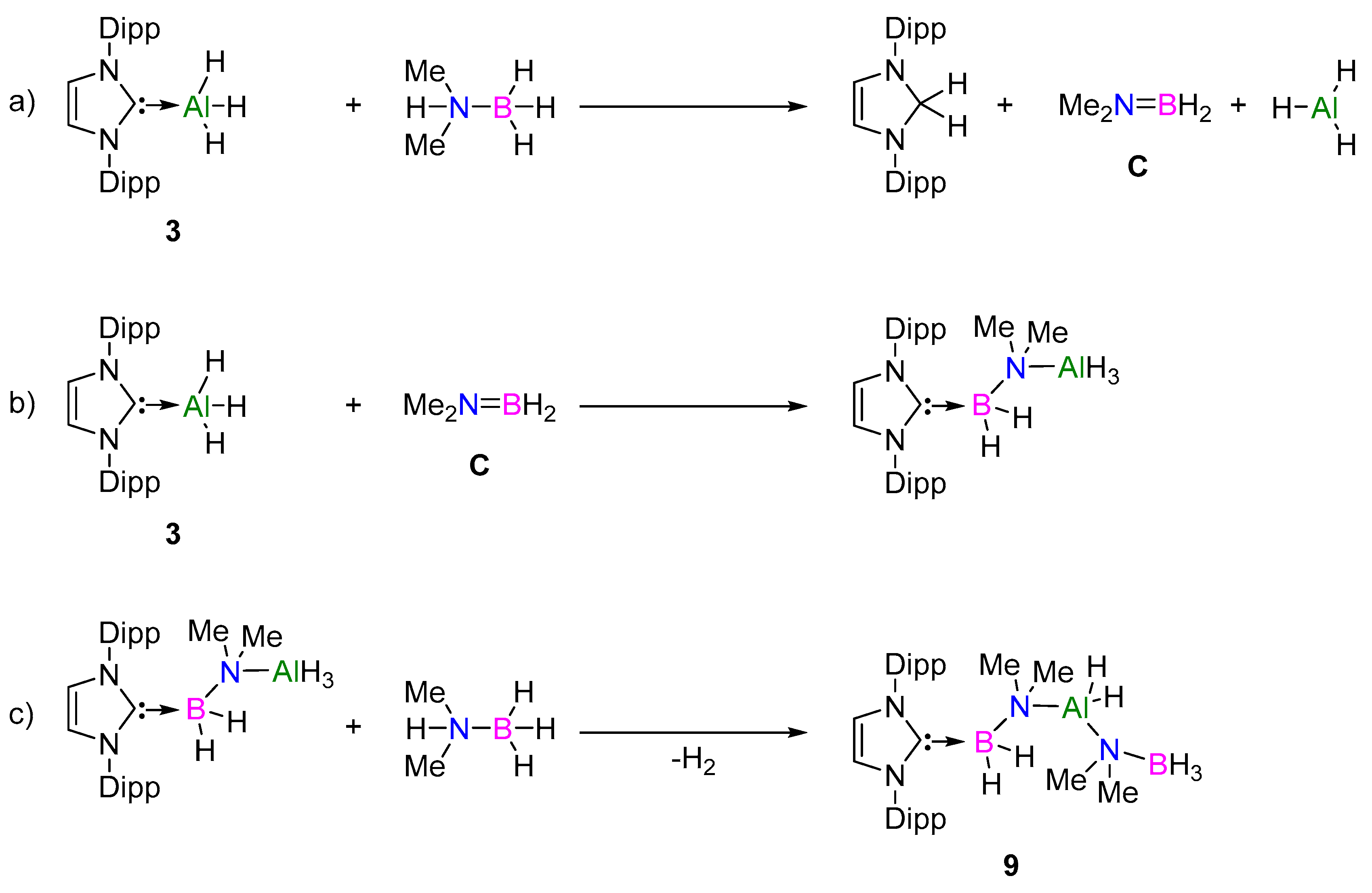
A similar investigation was carried out for the NHC-supported Al(III) hydrides, again using the diisopropylphenyl substituent (catalyst 3, IDippAlH3). Upon reaction of 3 with 1 eq. of Me2NHBH3, two new NHC-containing species were observed to form along with a notable lack of H2 formation in the 1H NMR spectrum. One of these species was identified as the dihydroaminal, IDippH2, which has been previously reported from the NHC-induced dehydrocoupling of MeNH2BH3 [30]. The 11B NMR spectrum again showed a minor shift of approximately δ 0.5 ppm, indicating the formation of the NHC-stabilized [Al]-N(Me)2BH3 complex 8, similar to that proposed for the NHI mechanism (7). Additional heating of this sample led to the increased formation of IDippH2 and a new NHC-containing compound in the 1H NMR spectrum. The 11B NMR spectrum did not show any dehydrocoupling products, but the formation of two broad signals at δ −13 and −17 ppm was evident. Subsequent scale-up of this stoichiometric reaction and XRD-analysis revealed the unexpected formation of compound 9 (Figure 6).
Compound 9 was identified as NHC–BH2–N(Me)2–AlH2–N(Me)2–BH3 complex, wherein one NHC–AlH3 reacted with two molecules of Me2NHBH3. The AlH2 unit is pseudo-tetrahedral, with the N(3)–Al(1) bond longer than N(4)–Al(1), suggesting some dative bonding character from the lone pair on N(3) to the empty orbital of Al(1). The NHC–BH2 bond lengths (1.628(3) Å) were in line with previously reported NHC–BH2-containing compounds, and also fit with the 11B NMR data matching the broad signal at δ −17 ppm [30,45,46]. Interestingly, the 27Al NMR spectrum showed a triplet (δ 85 ppm, JAl–H = 354 Hz), which has a comparable shift to that observed by Wright and co-workers for their Al–H-containing dehydrocoupling catalyst (δ 82.8 ppm, d, JAl–H = 288 Hz).
We propose that the formation of compound 9 (Figure 7) occurs in a similar sequence to that reported by Rivard and co-workers for the formation of NHC–BH2–N(H)(Me)–BH3 [30]. Firstly, upon reaction of 3 with Me2NHBH3, the reaction occurs at the NHC–Al bond rather than Al–H (Figure 7a). Here the NHC acts as a base, resulting in the formation of IDippH2 (confirmed by 1H NMR), dimethylamino–borane (C), and AlH3 species. In the second step (Figure 7b), C inserts into the NHC–AlH3 bond, resulting in NHC–BH2 formation. Finally, the second molecule of Me2NHBH3 undergoes σ-bond metathesis of its N–H with the terminal Al–H bond, resulting in the formation of compound 9 (Figure 7c).
The viability of compound 9 as a catalyst in the dehydrocoupling reaction was trialed, however only further decomposition to IDippH2 was noted to occur, suggesting that this is a deactivation pathway in the active catalyst cycle. This would also account for the increased reaction times with 3. To confirm the viability of the active cycle, that is, via compound 8 (NHC–Al(H)2NMe2BH3), the use of a more sterically demanding secondary amine–borane was tested. Reaction of 3 with 1 eq. of diisopropylamine–borane (iPr2NHBH3) resulted in the formation of 8iPr, with a reduced amount of IDippH2, thus confirming that this is the likely first step of the active catalytic cycle. It is further proposed that due to the increased steric congestion at the Al center with NHC ligands, the δ-hydride elimination is kinetically preferred over β-hydride elimination from the Al-coordinated [Me2NBH2N(Me)2BH3]− anion, therefore accounting for the absence of borane product (B) in the case of 3–6.
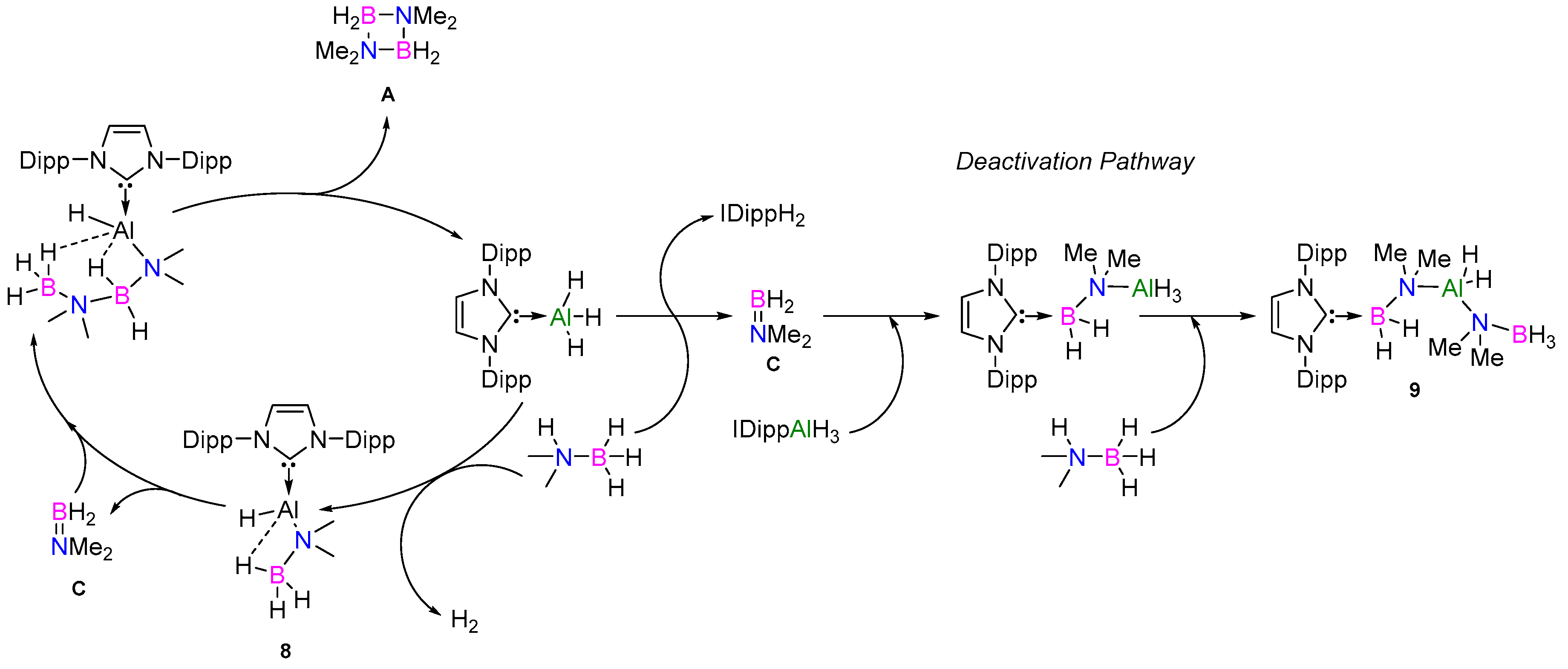
A proposed mechanism is outlined in Figure 8, taking both the active and deactivation pathways into account. Dimethylamino–borane (C) could either (i) react with compound 8 to form the metal-bound [Me2NBH2N(Me)2BH3]− anion (D’) (which was observed to form during the course of the catalysis) or (ii) insert into the NHC–AlH3 of the catalyst, leading to deactivation and formation of 9. Therefore, over the course of the catalysis, the remaining “active” species likely diminishes and results in prolonged reaction times. The differences in the reaction times observed with catalysts 3–6 were therefore likely due to the strength of the NHC–Al bond with strong donors such as IMe4 (6) resulting in the higher TOF due to the decreased likelihood of deactivation. This was also reflected in the use of the stronger σ-donating NHI ligands, as no deactivation pathways were observed.
3. Conclusions
In conclusion, differences in the use of NHI and NHC ligands for the stabilization of Al(III) hydrides and influence on catalytic activity were found. Whilst NHI-supported complexes required increased thermal activation, these systems were more stable in comparison to their NHC counterparts. Through a series of stoichiometric reactions, the isolation of compound 9 revealed a catalyst deactivation pathway that is prevalent in the NHC systems.
4. Materials and Methods
4.1. General Methods and Instruments
All manipulations were carried out under argon atmosphere using standard Schlenk or glovebox techniques. Glassware was heat-dried under vacuum prior to use. Unless otherwise stated, all chemicals were purchased from Sigma-Aldrich (Steinheim, Germany) and used as received. Me2NHBH3 was sublimed three times prior to use. Benzene, toluene, and n-hexane were refluxed over standard drying agents (benzene/hexane over sodium and benzophenone), distilled, and deoxygenated prior to use. Deuterated benzene (C6D6) was dried by storage over activated 4 Å molecular sieves. All NMR samples were prepared under argon in J. Young PTFE tubes. Catalysts 1–6 were synthesized according to procedures described in the literature [47]. NMR spectra were recorded on a Bruker AV-400 spectrometer (Rheinstetten, Germany) at ambient temperature (300 K). 1H, 11B, 13C, and 27Al NMR spectroscopic chemical shifts δ are reported in ppm relative to tetramethylsilane. δ(1H) and δ(13C) were referenced internally to the relevant residual solvent resonances. Details on XRD data are given in the Supplementary Materials.
4.2. General Catalytic Procedure for Catalysis of Me2NHBH3
5 mol % aluminum hydride catalyst (0.012 mmol) and Me2NHBH3 (0.24 mmol) in 0.5 mL of C6D6 were added to a J. Young PTFE NMR tube. The 1H and 11B NMR spectra were recorded and then placed in an oil bath at either 50 or 80 °C. Reaction progress was monitored by 1H/11B NMR spectroscopy until the consumption of Me2NHBH3.
4.3. General Procedure of Stoichiometric Reactions
Aluminum hydride catalyst (1: 40 mg, 0.046 mmol; 3: 40 mg, 0.095 mmol) and 1 eq. of Me2NHBH3 (1: 3 mg, 0.046 mmol; 3: 5.6 mg, 0.095 mmol) in 0.5 mL of C6D6 were added to a J. Young PTFE NMR tube. The 1H and 11B NMR spectra were recorded and then they were placed in an oil bath at either 50 or 80 °C. Reaction progress was monitored by 1H/11B NMR spectroscopy until consumption of Me2NHBH3.
4.4. Synthesis of Diisopropylamineborane, iPr2NHBH3
This synthesis was adapted from general literature procedures [48], with the exception that this was solely carried out in hexanes rather than THF. Diisopropyl amine (1.00 g, 1.3 mL, 9.88 mmol) was placed in a Schlenk flask followed by 10 mL of hexanes. The reaction mixture was cooled in an ice bath to approximately 0 °C. BH3·dms (dms = dimethylsulfide) (0.75g, 0.94 mL, 9.88 mmol) was added dropwise via syringe over 5 min. The reaction was allowed to stir for 10 min at 0 °C, before warming to room temperature and stirring overnight. Volatiles were removed under reduced pressure to leave a colorless oil (0.83 g, 73%).
1H NMR (400 MHz, 298 K, C6D6): δH 3.19 (1H, s, NH), 2.85 (2H, m, NCH(CH3)2), 2.16–1.48 (3H, br, q. BH3), 1.06 (6H, d, JHH = 6.6 Hz, CH(CH3)2), 1.00 (6H, d, JHH = 6.7 Hz, CH(CH3)2). 11B NMR (128 MHz, 298 K, C6D6): δB −20.49 (q, JBH = 96.5 Hz, BH3).
4.5. Synthesis of Compound 8iPr
IDippAlH3 (3: 100 mg, 0.24 mmol) was dissolved in 3 mL of toluene and transferred to a Schlenk flask, followed by the addition of 1 eq. of iPr2NHBH3 (35 μL, 0.24 mmol). This was allowed to stir at room temperature for 3 h. Solvent and volatiles were removed under reduced pressure to leave a pale-yellow gummy solid. This was subsequently washed with 3 × 10 mL of n-hexane to yield a colorless solid, 80 mg, 67% yield.
1H NMR (400 MHz, 298 K, C6D6): δH 7.25 (2H, m, Ar p-H), 7.12 (4H, m, Ar m-H), 6.48 (2H, s, NCH), 3.63 (2H, br.s. AlH2), 2.66 (6H, overlapping multiplets, CH(CH3)2 and NCH(CH3)2), 2.60–1.52 (3H, q, BH3), 1.43 (12H, d, JHH = 6.8 Hz, CH(CH3)2), 1.04 (12H, d, JHH = 6.9 Hz, CH(CH3)2), 0.96 (6H, d, JHH = 6.6 Hz, NCH(CH3)2), 0.82 (6H, d, JHH = 6.6 Hz, NCH(CH3)2). 13C{1H} NMR (100 MHz, 298 K, C6D6): δC 145.7 (Ar C), 134.8 (Ar C), 130.8 (Ar C), 124.3 (Ar C) 124.0 (NCH), 51.8 (NCH(CH3)2), 29.1 (CH(CH3)2), 25.1 (CH(CH3)2), 23.4 (CH(CH3)2), 20.7 (NCH(CH3)2), 18.8 (NCH(CH3)2). 11B NMR (128 MHz, 298 K, C6D6): δB −20.13 (q, JBH = 95.9 Hz, BH3). 27Al NMR (78 MHz, 298 K, C6D6): δAl 109.2 (br.s). Due to the presence of IDippH2, satisfactory elemental analysis results were not obtained.
4.6. Synthesis of Compound 9
IDippAlH3 (3: 200 mg, 0.48 mmol) and 2 eq. of Me2NHBH3 (56 mg, 0.96 mmol) were weighed in a Schlenk tube. To this, 5 mL of toluene was added, and the Schlenk tube was placed in an oil bath at 50 °C for 24 h. Solvent and volatiles were removed under reduced pressure to leave a pale-yellow gummy solid. This was subsequently washed with 3 × 10 mL of n-hexane to yield a colorless solid, 102 mg, 40% yield. Crystals suitable for single-crystal X-ray diffraction were grown from a concentrated toluene solution at −30 °C.
1H NMR (400 MHz, 298 K, C6D6): δH 7.19 (2H, m, Ar p-H), 7.11 (4H, m, Ar m-H), 6.53 (2H, s, NCH), 3.00 (4H, sept, JHH = 6.8 Hz, CH(CH3)2), 2.13 (12H, s, NCH3), 1.46 (12H, d, JHH = 6.7 Hz, CH(CH3)2), 0.99 (12H, d, JHH = 6.9 Hz, CH(CH3)2). 13C{1H} NMR (100 MHz, 298 K, C6D6): δC 146.2 (Ar-C), 135.4 (Ar-C), 130.7 (Ar-C), 124.4 (Ar m-C), 123.2 (NCH), 54.6 (NCH3), 29.1 (CH(CH3)2), 26.3 (CH(CH3)2), 22.5 (CH(CH3)2). 11B NMR (128 MHz, 298 K, C6D6): δB −13.5 (br.s. BH3), −17.0 (br.s. BH2). 27Al NMR (78 MHz, 298 K, C6D6): δAl 85 (t, JAl–H = 354 Hz, AlH2). Due to presence of IDippH2, satisfactory elemental analysis results were not obtained.
Supplementary Materials
The following are available online at https://www.mdpi.com/2304-6740/7/8/92/s1. Table S1 and Figures S1–S12: Catalysis data. Figures S13–S15: NHI mechanism NMR spectra. Figures S16–S20: NHC mechanism NMR spectra. Figures S21–S24: NMR spectra for compound 9. Table S2: Crystallographic details of 9 (CCDC 1936588). Copies of this information may be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif. Supplementary material includes the CIF and checkCIF files for 9.
Author Contributions
C.W. and N.I. performed the experiments. F.H. measured and solved the SC-XRD data. C.W. and S.I. supervised the complete project and wrote the manuscript. C.W., N.I., M.U., F.H. and S.I. discussed the results and commented on the manuscript.
Funding
This project received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No 754462 and TUM University Foundation (Fellowships CW), as well as the European Research Council (SILION 637394) and WACKER Chemie AG.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Boor, J., Jr. Zieglar-Natta Catalysts and Polymerisations; Academic Press Inc.: New York, NY, USA, 1979. [Google Scholar]
- Olah, G.A. Friedel-Crafts and Related Reactions; Wiley: New York, NY, USA, 1963. [Google Scholar]
- Power, P.P. Main-group elements as transition metals. Nature 2010, 463, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Weetman, C.; Inoue, S. The Road Travelled: After Main-Group Elements as Transition Metals. ChemCatChem 2018, 10, 4213–4228. [Google Scholar] [CrossRef]
- Melen, R.L. Frontiers in molecular p-block chemistry: From structure to reactivity. Science 2019, 363, 479. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-C.; Reed, C.A.; Long, G.S.; Sen, A. Et2Al+ Alumenium Ion-like Chemistry. Synthesis and Reactivity toward Alkenes and Alkene Oxides. J. Am. Chem. Soc. 2002, 124, 7662–7663. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, M.; Wehmschulte, R.J. Deoxygenative Reduction of Carbon Dioxide to Methane, Toluene, and Diphenylmethane with [Et2Al]+ as Catalyst. Angew. Chem. Int. Ed. 2012, 51, 7323–7326. [Google Scholar] [CrossRef] [PubMed]
- Weetman, C.; Bag, P.; Silvási, T.; Jandl, C.; Inoue, S. CO2 Fixation and Catalytic Reduction by a Neutral Aluminium Double bond. Angew. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.W.; Erker, G. Frustrated Lewis Pairs: Metal-free Hydrogen Activation and More. Angew. Chem. Int. Ed. 2009, 49, 46–76. [Google Scholar] [CrossRef]
- Stephan, D.W.; Erker, G. Frustrated Lewis Pair Chemistry: Development and Perspectives. Angew. Chem. Int. Ed. 2015, 54, 6400–6441. [Google Scholar] [CrossRef]
- Stephan, D.W. The broadening reach of frustrated Lewis pair chemistry. Science 2016, 354, aff7229. [Google Scholar] [CrossRef]
- Hill, M.S.; Liptrot, D.J.; Weetman, C. Alkaline earths as main group reagents in molecular catalysis. Chem. Soc. Rev. 2016, 45, 972–988. [Google Scholar] [CrossRef]
- Hadlington, T.J.; Driess, M.; Jones, C. Low-valent group 14 element hydride chemistry: Towards catalysis. Chem. Soc. Rev. 2018, 47, 4176–4197. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhong, M.; Ma, X.; De, S.; Anusha, C.; Parameswaran, P.; Roesky, H.W. An Aluminum Hydride That Functions like a Transition-Metal Catalyst. Angew. Chem. Int. Ed. 2015, 54, 10225–10229. [Google Scholar] [CrossRef] [PubMed]
- Elsen, H.; Färber, C.; Ballmann, G.; Harder, S. LiAlH4: From Stoichiometric Reduction to Imine Hydrogenation Catalysis. Angew. Chem. Int. Ed. 2018, 57, 7156–7160. [Google Scholar] [CrossRef] [PubMed]
- Bismuto, A.; Thomas Stephen, P.; Cowley Michael, J. Aluminum Hydride Catalyzed Hydroboration of Alkynes. Angew. Chem. Int. Ed. 2016, 55, 15356–15359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leitao, E.M.; Jurca, T.; Manners, I. Catalysis in service of main group chemistry offers a versatile approach to p-block molecules and materials. Nat. Chem. 2013, 5, 817. [Google Scholar] [CrossRef] [PubMed]
- Melen, R.L. Dehydrocoupling routes to element-element bonds catalysed by main group compounds. Chem. Soc. Rev. 2016, 45, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Slootweg, C.; Jupp, A.; Boom, D. Dehydrogenation of Amine–Boranes using p-block Compounds. Chem. Eur. J. 2019. [Google Scholar] [CrossRef]
- Keaton, R.J.; Blacquiere, J.M.; Baker, R.T. Base Metal Catalyzed Dehydrogenation of Ammonia−Borane for Chemical Hydrogen Storage. J. Am. Chem. Soc. 2007, 129, 1844–1845. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, C.W.; Baker, R.T.; Staubitz, A.; Manners, I. B–N compounds for chemical hydrogen storage. Chem. Soc. Rev. 2009, 38, 279–293. [Google Scholar] [CrossRef]
- Rossin, A.; Peruzzini, M. Ammonia–Borane and Amine–Borane Dehydrogenation Mediated by Complex Metal Hydrides. Chem. Rev. 2016, 116, 8848–8872. [Google Scholar] [CrossRef]
- Liptrot, D.J.; Hill, M.S.; Mahon, M.F.; MacDougall, D.J. Group 2 Promoted Hydrogen Release from NMe2H⋅BH3: Intermediates and Catalysis. Chem. Eur. J. 2010, 16, 8508–8515. [Google Scholar] [CrossRef] [PubMed]
- McLellan, R.; Kennedy, A.R.; Orr, S.A.; Robertson, S.D.; Mulvey, R.E. Lithium Dihydropyridine Dehydrogenation Catalysis: A Group 1 Approach to the Cyclization of Diamine Boranes. Angew. Chem. Int. Ed. 2017, 56, 1036–1041. [Google Scholar] [CrossRef] [PubMed]
- Bellham, P.; Hill, M.S.; Kociok-Köhn, G. Alkali metal-mediated dehydrocoupling of Me2NH·BH3. Dalton Trans. 2015, 44, 12078–12081. [Google Scholar] [CrossRef] [PubMed]
- Bellham, P.; Anker, M.D.; Hill, M.S.; Kociok-Köhn, G.; Mahon, M.F. The significance of secondary interactions during alkaline earth-promoted dehydrogenation of dialkylamine–boranes. Dalton Trans. 2016, 45, 13969–13978. [Google Scholar] [CrossRef] [PubMed]
- Erickson, K.A.; Kiplinger, J.L. Catalytic Dehydrogenation of Dimethylamine Borane by Highly Active Thorium and Uranium Metallocene Complexes. ACS Catal. 2017, 7, 4276–4280. [Google Scholar] [CrossRef]
- Cui, P.; Spaniol, T.P.; Maron, L.; Okuda, J. Dehydrogenation of Amine-Borane Me2NH⋅BH3 Catalyzed by a Lanthanum-Hydride Complex. Chem. Eur. J. 2013, 19, 13437–13444. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, N.E.; Robertson, A.P.M.; Leitao, E.M.; Manners, I. Amine–borane dehydrogenation chemistry: Metal-free hydrogen transfer, new catalysts and mechanisms, and the synthesis of polyaminoboranes. J. Organomet. Chem. 2013, 730, 84–89. [Google Scholar] [CrossRef]
- Sabourin, K.J.; Malcolm, A.C.; McDonald, R.; Ferguson, M.J.; Rivard, E. Metal-free dehydrogenation of amine–boranes by an N-heterocyclic carbene. Dalton Trans. 2013, 42, 4625–4632. [Google Scholar] [CrossRef]
- Lui Melanie, W.; Paisley Nathan, R.; McDonald, R.; Ferguson Michael, J.; Rivard, E. Metal-Free Dehydrogenation of Amine-Boranes by Tunable N-Heterocyclic Iminoboranes. Chem. Eur. J. 2016, 22, 2134–2145. [Google Scholar] [CrossRef]
- Hansmann, M.M.; Melen, R.L.; Wright, D.S. Group 13 BN dehydrocoupling reagents, similar to transition metal catalysts but with unique reactivity. Chem. Sci. 2011, 2, 1554–1559. [Google Scholar] [CrossRef]
- Cowley, H.J.; Holt, M.S.; Melen, R.L.; Rawson, J.M.; Wright, D.S. Catalytic dehydrocoupling of Me2NHBH3 with Al(NMe2)3. Chem. Commun. 2011, 47, 2682–2684. [Google Scholar] [CrossRef] [PubMed]
- Less, R.J.; Simmonds, H.R.; Dane, S.B.J.; Wright, D.S. Stoichiometric and catalytic reactions of LiAlH4 with Me2NHBH3. Dalton Trans. 2013, 42, 6337–6343. [Google Scholar] [CrossRef] [PubMed]
- Less, R.J.; Simmonds, H.R.; Wright, D.S. Reactivity and catalytic activity of tert-butoxy-aluminium hydride reagents. Dalton Trans. 2014, 43, 5785–5792. [Google Scholar] [CrossRef] [PubMed]
- Less, R.J.; García-Rodríguez, R.; Simmonds, H.R.; Allen, L.K.; Bond, A.D.; Wright, D.S. Use of crown ethers to isolate intermediates in ammonia–borane dehydrocoupling reactions. Chem. Commun. 2016, 52, 3650–3652. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.K.; García-Rodríguez, R.; Wright, D.S. Stoichiometric and catalytic Si–N bond formation using the p-block base Al(NMe2)3. Dalton Trans. 2015, 44, 12112–12118. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, T.; Franz, D.; Inoue, S. Applications of N-heterocyclic imines in main group chemistry. Chem. Soc. Rev. 2016, 45, 6327–6344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesterov, V.; Reiter, D.; Bag, P.; Frisch, P.; Holzner, R.; Porzelt, A.; Inoue, S. NHCs in Main Group Chemistry. Chem. Rev. 2018. [Google Scholar] [CrossRef]
- Franz, D.; Inoue, S. Activation of Elemental Sulfur by Aluminum Dihydride: Isolation of Mono-and Bis(hydrogensulfide) Complexes of Aluminum. Chem. Eur. J. 2014, 20, 10645–10649. [Google Scholar] [CrossRef]
- Franz, D.; Szilvasi, T.; Irran, E.; Inoue, S. A monotopic aluminum telluride with an Al=Te double bond stabilized by N-heterocyclic carbenes. Nat. Commun. 2015, 6, 10037–10042. [Google Scholar] [CrossRef]
- Franz, D.; Sirtl, L.; Pothig, A.; Inoue, S. Aluminum Hydrides Stabilized by N-Heterocyclic Imines as Catalysts for Hydroborations with Pinacolborane. Z. Anorg. Allg. Chem. 2016, 642, 1245–1250. [Google Scholar] [CrossRef]
- Spielmann, J.; Bolte, M.; Harder, S. Synthesis and structure of a magnesium–amidoborane complex and its role in catalytic formation of a new bis-aminoborane ligand. Chem. Commun. 2009, 6934–6936. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.S.; Kociok-Köhn, G.; Robinson, T.P. Group 3-centred dehydrocoupling of Me2NH·BH3. Chem. Comm. 2010, 46, 7587–7589. [Google Scholar] [CrossRef] [PubMed]
- Malcolm, A.C.; Sabourin, K.J.; McDonald, R.; Ferguson, M.J.; Rivard, E. Donor–Acceptor Complexation and Dehydrogenation Chemistry of Aminoboranes. Inorg. Chem. 2012, 51, 12905–12916. [Google Scholar] [CrossRef] [PubMed]
- Dübek, G.; Franz, D.; Eisenhut, C.; Altmann, P.J.; Inoue, S. Reactivity of an NHC-stabilized pyramidal hydrosilylene with electrophilic boron sources. Dalton Trans. 2019, 48, 5756–5765. [Google Scholar] [CrossRef] [PubMed]
- Francis, M.D.; Hibbs, D.E.; Hursthouse, M.B.; Jones, C.; Smithies, N.A. Carbene complexes of Group 13 trihydrides: Synthesis and characterisation of MH3{CN(Pri)C2Me2N(Pri)}, M = Al, Ga or In. J. Chem. Soc. Dalton Trans. 1998, 3249–3254. [Google Scholar] [CrossRef]
- Coles, N.T.; Mahon, M.F.; Webster, R.L. Phosphine– and Amine–Borane Dehydrocoupling Using a Three-Coordinate Iron(II) β-Diketiminate Precatalyst. Organometallics 2017, 36, 2262–2268. [Google Scholar] [CrossRef]
Figure 1.
Archetypal ligand systems comprising the five-membered N-heterocycle structural motif and resonance forms. NHC: N-heterocyclic carbene; NHI: N-heterocyclic imine.
Figure 1.
Archetypal ligand systems comprising the five-membered N-heterocycle structural motif and resonance forms. NHC: N-heterocyclic carbene; NHI: N-heterocyclic imine.

Figure 2.
Series of Al(III) hydrides utilized in the dehydrocoupling of amine–boranes.

Figure 3.
In situ reaction monitoring by 11B NMR spectroscopy for the catalytic dehydrocoupling of Me2NHBH3 with 1 (I, left) and 3 (II, right). A: [Me2NBH2]2; B: HB(NMe2)2; C: Me2N=BH2; D: [Me2NBH2N(Me)2BH3]−; D’: Al bound [Me2NBH2N(Me)2BH3]−.
Figure 3.
In situ reaction monitoring by 11B NMR spectroscopy for the catalytic dehydrocoupling of Me2NHBH3 with 1 (I, left) and 3 (II, right). A: [Me2NBH2]2; B: HB(NMe2)2; C: Me2N=BH2; D: [Me2NBH2N(Me)2BH3]−; D’: Al bound [Me2NBH2N(Me)2BH3]−.

Figure 4.
Stoichiometric reduction of Me2NHBH3 with catalyst 1.

Figure 5.
Proposed catalytic mechanism for the dehydrocoupling of Me2NHBH3 with NHI-supported Al(III) hydrides (1, 2).
Figure 5.
Proposed catalytic mechanism for the dehydrocoupling of Me2NHBH3 with NHI-supported Al(III) hydrides (1, 2).

Figure 6.
Molecular structure of compound 9 in the solid state with thermal ellipsoids set at the 50% probability level. Hydrogen atoms (except those on B(1), B(2), and Al(1)) are omitted for clarity, and diisopropylphenyl substituents on the NHC ligand are depicted in wireframe for simplicity. Selected bond lengths (Å) and angles (°): C(1)–B(1) 1.628(3), B(1)–N(3) 1.555(6), N(3)–Al(1) 2.026(5), N(4)–Al(1) 1.948(6), B(2)–N(4) 1.599(6), C(1)–B(1)–N(3) 115.6(2), B(1)–N(3)–Al(1) 108.0(3), N(3)–Al(1)–N(4) 112.89(2), Al(1)–N(4)–B(2) 95.7(3).
Figure 6.
Molecular structure of compound 9 in the solid state with thermal ellipsoids set at the 50% probability level. Hydrogen atoms (except those on B(1), B(2), and Al(1)) are omitted for clarity, and diisopropylphenyl substituents on the NHC ligand are depicted in wireframe for simplicity. Selected bond lengths (Å) and angles (°): C(1)–B(1) 1.628(3), B(1)–N(3) 1.555(6), N(3)–Al(1) 2.026(5), N(4)–Al(1) 1.948(6), B(2)–N(4) 1.599(6), C(1)–B(1)–N(3) 115.6(2), B(1)–N(3)–Al(1) 108.0(3), N(3)–Al(1)–N(4) 112.89(2), Al(1)–N(4)–B(2) 95.7(3).

Figure 7.
Proposed sequence of reactions which result in the formation of compound 9. (a) Initial deprotonation of Me2NHBH3 by NHC rather than Al–H. (b) Insertion of C into NHC–Al bond. (c) Deprotonation of Me2NHBH3 by Al–H bond for formation of compound 9.
Figure 7.
Proposed sequence of reactions which result in the formation of compound 9. (a) Initial deprotonation of Me2NHBH3 by NHC rather than Al–H. (b) Insertion of C into NHC–Al bond. (c) Deprotonation of Me2NHBH3 by Al–H bond for formation of compound 9.

Figure 8.
Proposed mechanism for the dehydrocoupling of Me2NHBH3 with NHC-supported Al(III) hydrides (3–6).
Figure 8.
Proposed mechanism for the dehydrocoupling of Me2NHBH3 with NHC-supported Al(III) hydrides (3–6).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Dehydrocoupling of Me2NHBH3 with 5 mol % Al(III) hydride catalysts (1–6).

| Catalyst | Temp/°C | Time/h | Conversion a/% | TOF b/h−1 |
|---|---|---|---|---|
| 1 | 80 | 34 | 90 | 0.53 |
| 2 | 80 | 34 | 84 | 0.49 |
| 3 | 50 | 65 | 80 | 0.25 |
| 4 | 50 | 26 | 92 | 0.74 |
| 5 | 50 | 65 | 93 | 0.29 |
| 6 | 50 | 40 | 92 | 0.46 |
Reaction conditions 5 mol % c 1–6 (0.012 mmol), Me2NHBH3 (0.24 mmol), 0.5 mL C6D6. a conversion of Me2NHBH3 determined by 11B NMR relative integrals; b TOF = (conversion/catalyst loading)/time. c Catalyst loading determined per molecule relative to amine–borane concentration.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Weetman, C.; Ito, N.; Unno, M.; Hanusch, F.; Inoue, S. NHI- and NHC-Supported Al(III) Hydrides for Amine–Borane Dehydrocoupling Catalysis. Inorganics 2019, 7, 92. https://doi.org/10.3390/inorganics7080092
AMA Style
Weetman C, Ito N, Unno M, Hanusch F, Inoue S. NHI- and NHC-Supported Al(III) Hydrides for Amine–Borane Dehydrocoupling Catalysis. Inorganics. 2019; 7(8):92. https://doi.org/10.3390/inorganics7080092
Chicago/Turabian StyleWeetman, Catherine, Nozomi Ito, Masafumi Unno, Franziska Hanusch, and Shigeyoshi Inoue. 2019. "NHI- and NHC-Supported Al(III) Hydrides for Amine–Borane Dehydrocoupling Catalysis" Inorganics 7, no. 8: 92. https://doi.org/10.3390/inorganics7080092
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.