
A Water-Soluble Chitosan Derivative for the Release of Bioactive Deferoxamine
 , , and
, , and 
Abstract
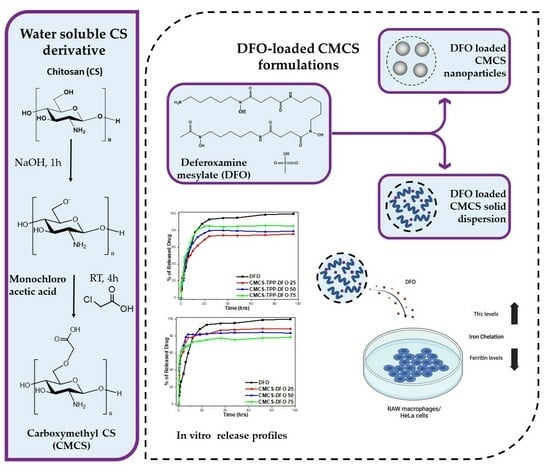
:
1. Introduction
2. Results and Discussion
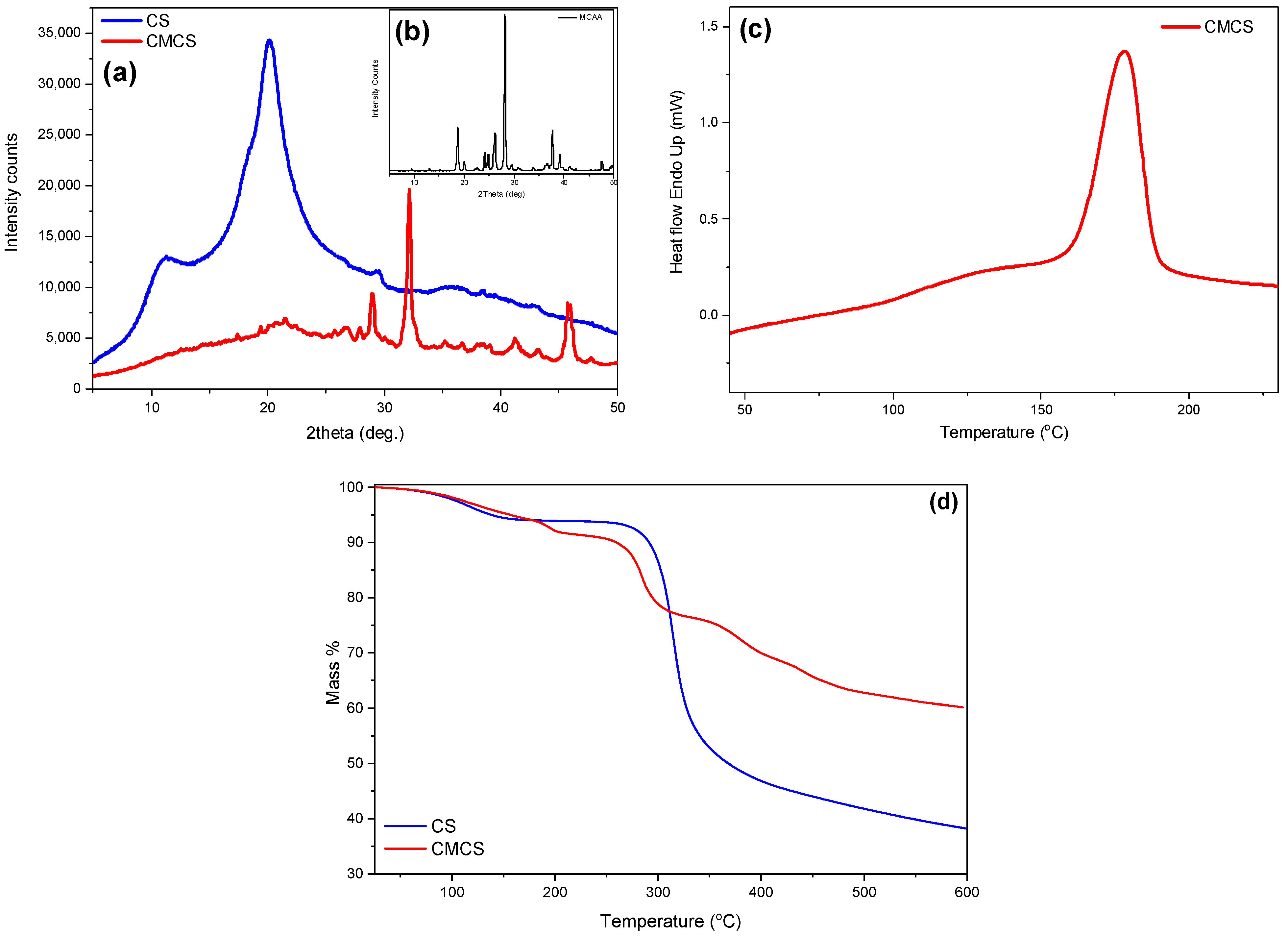
2.1. Characterization of the CMCS Material
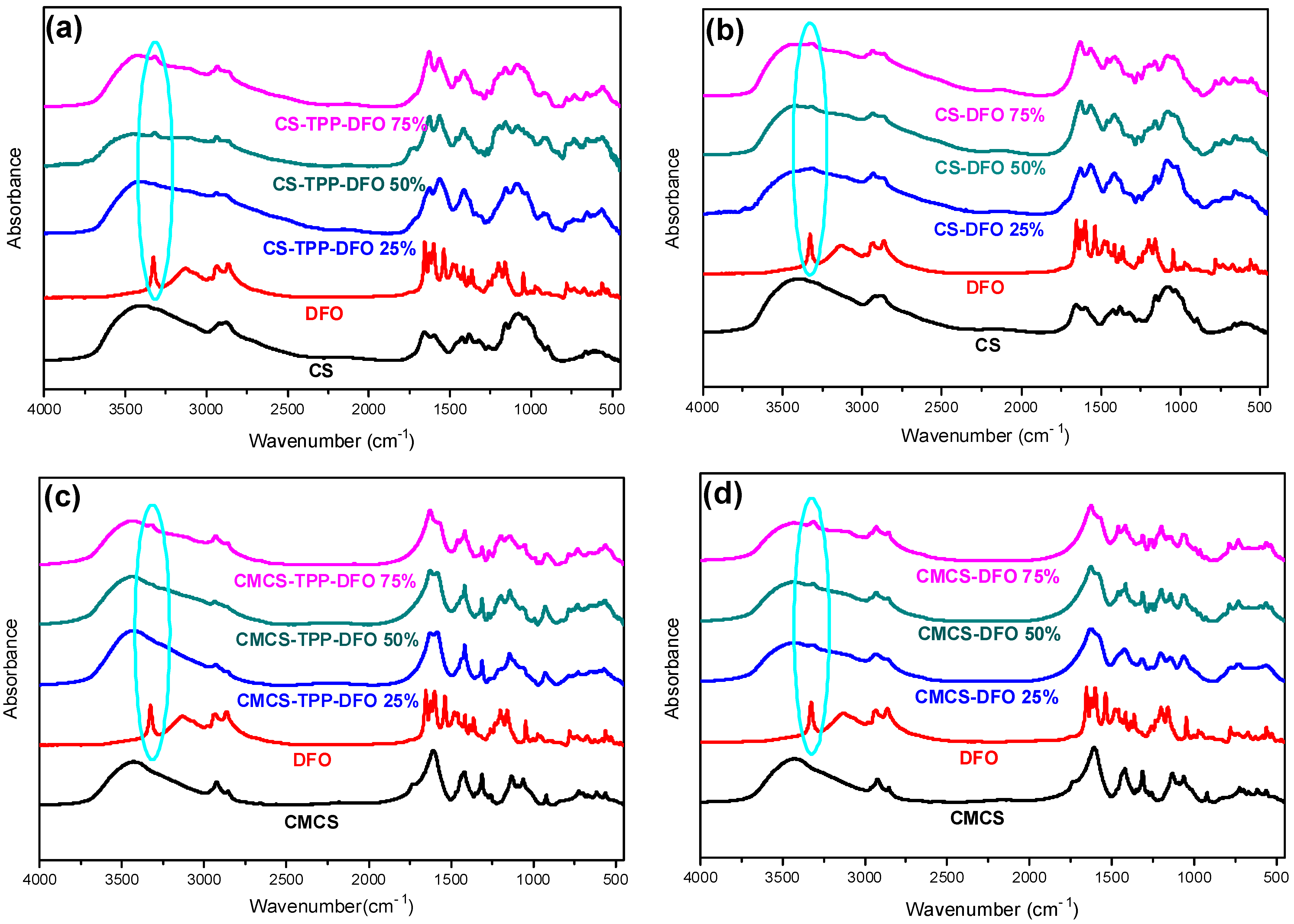
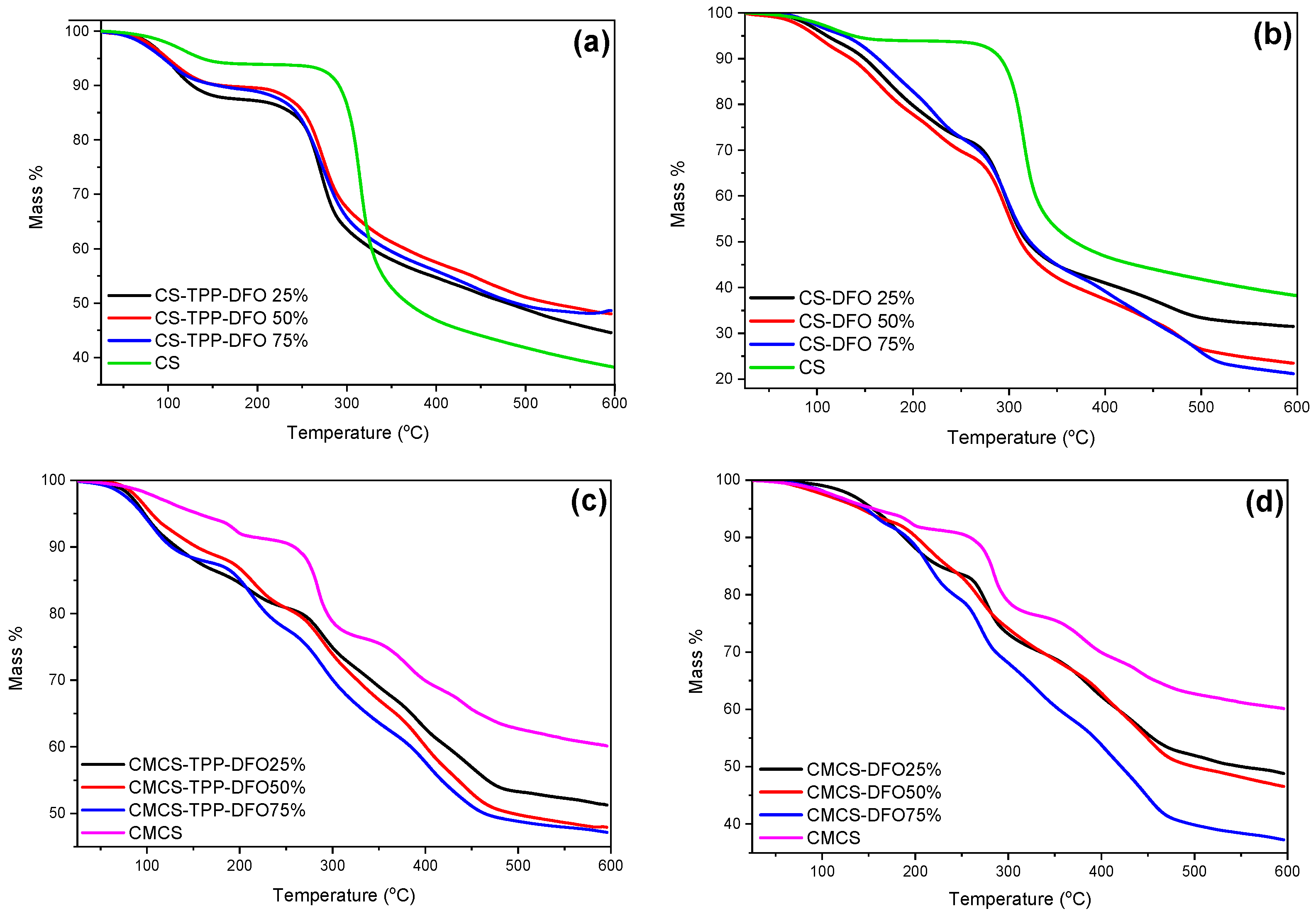
2.2. Characterization of the DFO-Loaded Nanoparticles
2.3. In Vitro Release Studies
2.4. Biological Activity of DFO Released from CMCS-DFO
3. Materials and Methods
3.1. Materials and Reagents
3.2. Synthesis of CMCS Derivative
3.3. Preparation of Nanoparticles
3.4. Preparation of Solid Dispersions
3.5. Characterization of Materials and DFO-Loaded Formulations
3.5.1. Fourier-Transformed Infrared Spectroscopy (FTIR)
3.5.2. Wide-Angle X-ray Scattering (XRD)
3.5.3. Dynamic Light Scattering (DLS)
3.5.4. Differential Scanning Calorimetry (DSC)
3.5.5. Thermogravimetric Analysis (TGA)
3.5.6. Scanning Electron Microscopy (SEM)
3.5.7. Contact Angle
3.5.8. High-Pressure Liquid Chromatography (HPLC)
3.5.9. In Vitro Dissolution Studies
3.5.10. Cell Culture
3.5.11. Quantitative Real-Time PCR (qPCR)
3.5.12. Western Blotting
3.5.13. Cell Viability
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chauhan, W.; Shoaib, S.; Fatma, R.; Zaka-ur-Rab, Z.; Afzal, M. β-thalassemia, and the advent of new Interventions beyond Transfusion and Iron chelation. Br. J. Clin. Pharmacol. 2022, 88, 3610–3626. [Google Scholar] [CrossRef] [PubMed]
- Piel, F.B.; Steinberg, M.H.; Rees, D.C. Sickle cell disease. N. Engl. J. Med. 2017, 376, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Kattamis, A.; Kwiatkowski, J.L.; Aydinok, Y. Thalassaemia. Lancet 2022, 399, 2310–2324. [Google Scholar] [CrossRef]
- De Simone, G.; Quattrocchi, A.; Mancini, B.; di Masi, A.; Nervi, C.; Ascenzi, P. Thalassemias: From gene to therapy. Mol. Asp. Med. 2022, 84, 101028. [Google Scholar] [CrossRef] [PubMed]
- Fucharoen, S.; Ketvichit, P.; Pootrakul, P.; Siritanaratkul, N.; Piankijagum, A.; Wasi, P. Clinical manifestation of β-thalassemia/hemoglobin e disease. Am. J. Pediatr. Hematol. Oncol. 2000, 22, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Fibach, E.; Rachmilewitz, E.A. Pathophysiology and treatment of patients with beta-thalassemia—An update. F1000Research 2017, 6, 2156. [Google Scholar] [CrossRef] [PubMed]
- Khandros, E.; Kwiatkowski, J.L. Beta Thalassemia: Monitoring and New Treatment Approaches. Hematol. Oncol. Clin. N. Am. 2019, 33, 339–353. [Google Scholar] [CrossRef]
- Cazzola, M. Ineffective erythropoiesis and its treatment. Blood 2022, 139, 2460–2470. [Google Scholar] [CrossRef]
- Brittenham, G.M.; Griffith, P.M.; Nienhuis, A.W.; McLaren, C.E.; Young, N.S.; Tucker, E.E.; Allen, C.J.; Farrell, D.E.; Harris, J.W. Efficacy of Deferoxamine in Preventing Complications of Iron Overload in Patients with Thalassemia Major. N. Engl. J. Med. 1994, 331, 567–573. [Google Scholar] [CrossRef]
- Reddy, P.S.; Locke, M.; Badawy, S.M. A systematic review of adherence to iron chelation therapy among children and adolescents with thalassemia. Ann. Med. 2022, 54, 326–342. [Google Scholar] [CrossRef]
- Jones, G.; Goswami, S.K.; Kang, H.; Choi, H.S.; Kim, J. Combating iron overload: A case for deferoxamine-based nanochelators. Nanomedicine 2020, 15, 1341–1356. [Google Scholar] [CrossRef] [PubMed]
- Lazaridou, M.; Christodoulou, E.; Nerantzaki, M.; Kostoglou, M.; Lambropoulou, D.A.; Katsarou, A.; Pantopoulos, K.; Bikiaris, D.N. Formulation and in-vitro characterization of chitosan-nanoparticles loaded with the iron chelator deferoxamine mesylate (DFO). Pharmaceutics 2020, 12, 238. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Liu, G.; Frazer, D.M.; Liu, T.; You, L.; Xu, J.; Wang, Y.; Anderson, G.J.; Nie, G. Polymeric Nanoparticles Enhance the Ability of Deferoxamine to Deplete Hepatic and Systemic Iron. Nano Lett. 2018, 18, 5782–5790. [Google Scholar] [CrossRef] [PubMed]
- Vignesh, S.; Sivashanmugam, A.; Annapoorna, M.; Janarthanan, R.; Subramania, I.; Shantikumar, V.N.; Jayakumar, R. Injectable deferoxamine nanoparticles loaded chitosan-hyaluronic acid coacervate hydrogel for therapeutic angiogenesis. Colloids Surf. B Biointerfaces 2018, 161, 129–138. [Google Scholar] [CrossRef]
- Li, S.; Wang, X.; Chen, J.; Guo, J.; Yuan, M.; Wan, G.; Yan, C.; Li, W.; Machens, H.G.; Rinkevich, Y.; et al. Calcium ion cross-linked sodium alginate hydrogels containing deferoxamine and copper nanoparticles for diabetic wound healing. Int. J. Biol. Macromol. 2022, 202, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Zhong, J.; Hu, J.; Yang, P.; Zhang, J.; Zhang, M.; Li, Y.; Gu, Z. Carrier-Free Deferoxamine Nanoparticles against Iron Overload in Brain. CCS Chem. 2022, 5, 257–270. [Google Scholar] [CrossRef]
- Michailidou, G.; Ainali, N.M.; Xanthopoulou, E.; Nanaki, S.; Kostoglou, M.; Koukaras, E.N.; Bikiaris, D.N. Effect of Poly(vinyl alcohol) on Nanoencapsulation of Budesonide in Chitosan Nanoparticles via Ionic Gelation and Its Improved Bioavailability. Polymers 2020, 12, 1101. [Google Scholar] [CrossRef] [PubMed]
- Sahariah, P.; Másson, M. Antimicrobial Chitosan and Chitosan Derivatives: A Review of the Structure-Activity Relationship. Biomacromolecules 2017, 18, 3846–3868. [Google Scholar] [CrossRef]
- Zamboulis, A.; Nanaki, S.; Michailidou, G.; Koumentakou, I.; Lazaridou, M.; Ainali, N.M.; Xanthopoulou, E.; Bikiaris, D.N. Chitosan and its derivatives for ocular delivery formulations: Recent advances and developments. Polymers 2020, 12, 1519. [Google Scholar] [CrossRef]
- le Dung, P.; Milas, M.; Rinaudo, M.; Desbrières, J. Water soluble derivatives obtained by controlled chemical modifications of chitosan. Carbohydr. Polym. 1994, 24, 209–214. [Google Scholar] [CrossRef]
- Jimtaisong, A.; Saewan, N. Utilization of carboxymethyl chitosan in cosmetics. Int. J. Cosmet. Sci. 2014, 36, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.G.; Park, H.J. Chemical characteristics of O-carboxymethyl chitosans related to the preparation conditions. Carbohydr. Polym. 2003, 53, 355–359. [Google Scholar] [CrossRef]
- Kong, X. Simultaneous determination of degree of deacetylation, degree of substitution and distribution fraction of -COONa in carboxymethyl chitosan by potentiometric titration. Carbohydr. Polym. 2012, 88, 336–341. [Google Scholar] [CrossRef]
- Chen, S.C.; Wu, Y.C.; Mi, F.L.; Lin, Y.H.; Yu, L.C.; Sung, H.W. A novel pH-sensitive hydrogel composed of N,O-carboxymethyl chitosan and alginate cross-linked by genipin for protein drug delivery. J. Control. Release 2004, 96, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Tzaneva, D.; Simitchiev, A.; Petkova, N.; Nenov, V.; Stoyanova, A.; Denev, P. Synthesis of carboxymethyl chitosan and its rheological behaviour in pharmaceutical and cosmetic emulsions. J. Appl. Pharm. Sci. 2017, 7, 70–78. [Google Scholar] [CrossRef]
- Kumar, S.; Koh, J. Physiochemical, optical and biological activity of chitosan-chromone derivative for biomedical applications. Int. J. Mol. Sci. 2012, 13, 6102–6116. [Google Scholar] [CrossRef]
- Oluwasina, O.O.; Olagboye, A.S.; Boboye, A.; Hassan, F.G. Carboxymethyl chitosan zinc supplement: Preparation, physicochemical, and preliminary antimicrobial analysis. Cogent Chem. 2017, 3, 1294470. [Google Scholar] [CrossRef]
- Chandra Dey, S.; Al-Amin, M.; Ur Rashid, T.; Zakir Sultan, M.; Ashaduzzaman, M.; Sarker, M.; Md Shamsuddin, S. Preparation, Characterization and Performance Evaluation of Chitosan As an Adsorbent for Remazol Red. Int. J. Latest Res. Eng. Technol. 2016, 2, 52–62. [Google Scholar]
- Corazzari, I.; Nisticò, R.; Turci, F.; Faga, M.G.; Franzoso, F.; Tabasso, S.; Magnacca, G. Advanced physico-chemical characterization of chitosan by means of TGA coupled on-line with FTIR and GCMS: Thermal degradation and water adsorption capacity. Polym. Degrad. Stab. 2015, 112, 1–9. [Google Scholar] [CrossRef]
- Lim, B.Y.; Poh, C.S.; Voon, C.H.; Salmah, H. Rheological and Thermal Study of Chitosan Filled Thermoplastic Elastomer Composites. Appl. Mech. Mater. 2015, 754–755, 34–38. [Google Scholar] [CrossRef]
- Ge, H.; Wang, S. Thermal preparation of chitosan-acrylic acid superabsorbent: Optimization, characteristic and water absorbency. Carbohydr. Polym. 2014, 113, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Camilo, P.-C.; Bolanos, G. Solubility of chitosan in aqueous acetic acid and pressurized carbon dioxide-water: Experimental equilibrium and solubilization kinetics. J. Supercrit. Fluids 2019, 151, 63–74. [Google Scholar] [CrossRef]
- Bidgoli, H.; Zamani, A.; Taherzadeh, M.J. Effect of carboxymethylation conditions on the water-binding capacity of chitosan-based superabsorbents. Carbohydr. Res. 2010, 345, 2683–2689. [Google Scholar] [CrossRef]
- Hubbe, M.A. Why, After All, Are Chitosan Films Hydrophobic? BioResources 2019, 14, 7630–7631. [Google Scholar] [CrossRef]
- Karava, A.; Lazaridou, M.; Nanaki, S.; Michailidou, G.; Christodoulou, E.; Kostoglou, M.; Iatrou, H.; Bikiaris, D.N. Chitosan Derivatives with Mucoadhesive and Antimicrobial Properties for Simultaneous Nanoencapsulation and Extended Ocular Release Formulations of Dexamethasone and Chloramphenicol Drugs. Pharmaceutics 2020, 12, 594. [Google Scholar] [CrossRef]
- Bikiaris, N.D.; Michailidou, G.; Lazaridou, M.; Christodoulou, E.; Gounari, E.; Ofrydopoulou, A.; Lambropoulou, D.A.; Vergkizi-Nikolakaki, S.; Lykidou, S.; Nikolaidis, N. Innovative skin product emulsions with enhanced antioxidant, antimicrobial and UV protection properties containing nanoparticles of pure and modified Chitosan with encapsulated fresh pomegranate juice. Polymers 2020, 12, 1542. [Google Scholar] [CrossRef]
- Fathi, M.; Samadi, M.; Rostami, H.; Parastouei, K. Encapsulation of ginger essential oil in chitosan-based microparticles with improved biological activity and controlled release properties. J. Food Process. Preserv. 2021, 45, e15373. [Google Scholar] [CrossRef]
- Jamil, B.; Abbasi, R.; Abbasi, S.; Imran, M.; Khan, S.U.; Ihsan, A.; Javed, S.; Bokhari, H. Encapsulation of cardamom essential oil in chitosan nano-composites: In-vitro efficacy on antibiotic-resistant bacterial pathogens and cytotoxicity studies. Front. Microbiol. 2016, 7, 1580. [Google Scholar] [CrossRef]
- Othman, N.; Masarudin, M.J.; Kuen, C.Y.; Dasuan, N.A.; Abdullah, L.C.; Jamil, S.N.A.M. Synthesis and optimization of chitosan nanoparticles loaded with l-ascorbic acid and thymoquinone. Nanomaterials 2018, 8, 920. [Google Scholar] [CrossRef]
- Lu, P.J.; Fu, W.E.; Huang, S.C.; Lin, C.Y.; Ho, M.L.; Chen, Y.P.; Cheng, H.F. Methodology for sample preparation and size measurement of commercial ZnO nanoparticles. J. Food Drug Anal. 2018, 26, 628–636. [Google Scholar] [CrossRef]
- Bootz, A.; Vogel, V.; Schubert, D.; Kreuter, J. Comparison of scanning electron microscopy, dynamic light scattering and analytical ultracentrifugation for the sizing of poly(butyl cyanoacrylate) nanoparticles. Eur. J. Pharm. Biopharm. 2004, 57, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Murugappan, R.; Karthikeyan, M.; Aravinth, A.; Alamelu, M. Siderophore-mediated iron uptake promotes yeast-bacterial symbiosis. Appl. Biochem. Biotechnol. 2012, 168, 2170–2183. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, S.; Bikiaris, D.; Avgoustakis, K.; Karavas, E.; Georgarakis, M. Chitosan nanoparticles loaded with dorzolamide and pramipexole. Carbohydr. Polym. 2008, 73, 44–54. [Google Scholar] [CrossRef]
- Zhihui, J.; Chun, Y.; Fangnan, Z.; Xiaolian, C.; Yuhi, L.; Huiping, X. One-Step Reinforcement and Deacidification of Paper. Coatings 2020, 10, 1226. [Google Scholar]
- Bhujbal, S.V.; Mitra, B.; Jain, U.; Gong, Y.; Agrawal, A.; Karki, S.; Taylor, L.S.; Kumar, S.; Zhou, Q.T. Pharmaceutical amorphous solid dispersion: A review of manufacturing strategies. Acta Pharm. Sin. B 2021, 11, 2505–2536. [Google Scholar] [CrossRef]
- Wesseling, P.; Ko, B.C.; Lewandowski, J.J. Quantitative evaluation of α-Al nano-particles in amorphous Al87Ni7Gd6—Comparison of XRD, DSC, and TEM. Scr. Mater. 2003, 48, 1537–1541. [Google Scholar] [CrossRef]
- Mattos, A.L.C.; Constantino, V.R.L.; de Couto, R.A.A.; Pinto, D.M.L.; Kaneko, T.M.; Espósito, B.P. Desferrioxamine-cadmium as a “Trojan horse” for the delivery of Cd to bacteria and fungi. J. Trace Elem. Med. Biol. 2013, 27, 103–108. [Google Scholar] [CrossRef]
- Ong, S.G.M.; Ming, L.C.; Lee, K.S.; Yuen, K.H. Influence of the encapsulation efficiency and size of liposome on the oral bioavailability of griseofulvin-loaded liposomes. Pharmaceutics 2016, 8, 25. [Google Scholar] [CrossRef]
- Qayoom, A.; Aneesha, V.A.; Anagha, S.; Dar, J.A.; Kumar, P.; Kumar, D. Lecithin-based deferoxamine nanoparticles accelerated cutaneous wound healing in diabetic rats. Eur. J. Pharmacol. 2019, 858, 172478. [Google Scholar] [CrossRef]
- Nieves, I.; Galbis, E.; Concepción, V.; De-Paz, M.-V.; Galbis, J.A. Reversible pH-Sensitive Chitosan-Based Hydrogels.Influence of Dispersion Composition on RheologicalProperties and Sustained Drug Delivery. Polymers 2018, 10, 392. [Google Scholar]
- Zha, S.; Utomo, Y.K.S.; Yang, L.; Liang, G.; Liu, W. Mechanic-Driven Biodegradable Polyglycolic Acid/Silk Fibroin Nanofibrous Scaffolds Containing Deferoxamine Accelerate Diabetic Wound Healing. Pharmaceutics 2022, 14, 601. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Liu, Y.; Selvaratnam, B.; Koodali, R.T.; Sun, H. Mesoporous Silicate Nanoparticles/3D Nanofibrous Scaffold- mediated Dual-drug Delivery for Bone Tissue Engineering. J. Control. Release 2018, 279, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Farr, A.C.; Xiong, M.P. Challenges and Opportunities of Deferoxamine Delivery for Treatment of Alzheimer’s Disease, Parkinson’s Disease, and Intracerebral Hemorrhage. Mol. Pharm. 2021, 18, 593–609. [Google Scholar] [CrossRef] [PubMed]
- Ran, Q.; Yu, Y.; Chen, W.; Shen, X.; Mu, C.; Yuan, Z.; Tao, B.; Hu, Y.; Yang, W.; Cai, K. Deferoxamine loaded titania nanotubes substrates regulate osteogenic and angiogenic differentiation of MSCs via activation of HIF-1α signaling. Mater. Sci. Eng. C 2018, 91, 44–54. [Google Scholar] [CrossRef]
- Marzban, A.; Akbarzadeh, A.; Ardestani, M.S.; Ardestani, F.; Akbari, M. Synthesis of nano-niosomal deferoxamine and evaluation of its functional characteristics to apply as an iron-chelating agent. Can. J. Chem. Eng. 2018, 96, 107–112. [Google Scholar] [CrossRef]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric Amorphous Solid Dispersions: A Review of Amorphization, Crystallization, Stabilization, Solid-State Characterization, and Aqueous Solubilization of Biopharmaceutical Classification System Class II Drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef]
- Strober, W. Trypan Blue Exclusion Test of Cell Viability. Curr. Protoc. Immunol. 2015, 111, A3.B.1–A3.B.3. [Google Scholar] [CrossRef] [PubMed]
- Saletta, F.; Rahmanto, Y.S.; Siafakas, A.R.; Richardson, D.R. Cellular iron depletion and the mechanisms involved in the iron-dependent regulation of the growth arrest and DNA damage family of genes. J. Biol. Chem. 2011, 286, 35396–35406. [Google Scholar] [CrossRef]
- You, L.; Wang, J.; Liu, T.; Zhang, Y.; Han, X.; Wang, T.; Guo, S.; Dong, T.; Xu, J.; Anderson, G.J.; et al. Targeted Brain Delivery of Rabies Virus Glycoprotein 29-Modified Deferoxamine-Loaded Nanoparticles Reverses Functional Deficits in Parkinsonian Mice. ACS Nano 2018, 12, 4123–4139. [Google Scholar] [CrossRef]
- Katsarou, A.; Gkouvatsos, K.; Fillebeen, C.; Pantopoulos, K. Tissue-Specific Regulation of Ferroportin in Wild-Type and Hjv-/- Mice Following Dietary Iron Manipulations. Hepatol. Commun. 2021, 5, 2139–2150. [Google Scholar] [CrossRef]
- Charlebois, E.; Pantopoulos, K. Iron overload inhibits BMP/SMAD and IL-6/STAT3 signaling to hepcidin in cultured hepatocytes. PLoS ONE 2021, 16, e0253475. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solubility (%) | Contact Angle, θ (°) | ||||
|---|---|---|---|---|---|
| pH = 3 | pH = 7 | pH = 10 | |||
| CS | 100 | 6.9 | 8.1 | 74.5 |  |
| CMCS | 92.4 | 95 | 97.5 | 56.1 |  |
| Sample Name | Z-Average (d.nm) | PdI | Zeta Potential (mV) |
|---|---|---|---|
| CS-TPP-DFO 25% | 194 | 0.227 | 33.7 |
| CS-TPP-DFO 50% | 210 | 0.185 | 30.8 |
| CS-TPP-DFO 75% | 340 | 0.348 | 32 |
| CMCS-TPP-DFO 25% | 335 | 0.561 | 14.9 |
| CMCS-TPP-DFO 50% | 265 | 0.580 | 14.6 |
| CMCS-TPP-DFO 75% | 205 | 0.581 | 14.8 |
| Sample | Drug Loading (%) |
|---|---|
| CS-TPP-DFO 25% CS-TPP-DFO 50% CS-TPP-DFO 75% | 24.13 27.01 56.64 |
| CS-DFO 25% CS-DFO 50% CS-DFO 75% | 41.67 16.13 31.19 |
| CMCS-TPP-DFO 25% CMCS-TPP-DFO 50% CMCS-TPP-DFO 75% | 30.00 66.95 92.20 |
| CMCS-DFO 25% CMCS-DFO 50% CMCS-DFO 75% | 38.35 42.56 67.45 |
| Gene | GenBank Accession No | Forward Primer Sequence | Reverse Primer Sequence |
|---|---|---|---|
| Tfrc | NM_011638.4 | AGCCAGATCAGCATTCTCTAACT | GCCTTCATGTTATTGTCGGCAT |
| Rpl19 | NM_009078.2 | AGGCATATGGGCATAGGGAAGAG | TTGACCTTCAGGTACAGGCTGTG |
| TFRC | NM_003234 | GCAAGTAGATGGCGATAACAG | GACGATCACAGCAATAGTCCC |
| ACTB | NM_001101.3 | AGGATGCAGAAGGAGATCACT | GGGTGTAACGCAACTAAGTCATAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michailidou, G.; Li, Y.; Zamboulis, A.; Karlioti, G.; Meimaroglou, D.; Pantopoulos, K.; Bikiaris, D.N. A Water-Soluble Chitosan Derivative for the Release of Bioactive Deferoxamine. Int. J. Mol. Sci. 2024, 25, 913. https://doi.org/10.3390/ijms25020913
Michailidou G, Li Y, Zamboulis A, Karlioti G, Meimaroglou D, Pantopoulos K, Bikiaris DN. A Water-Soluble Chitosan Derivative for the Release of Bioactive Deferoxamine. International Journal of Molecular Sciences. 2024; 25(2):913. https://doi.org/10.3390/ijms25020913
Chicago/Turabian StyleMichailidou, Georgia, Yupeng Li, Alexandra Zamboulis, Georgia Karlioti, Despoina Meimaroglou, Kostas Pantopoulos, and Dimitrios N. Bikiaris. 2024. "A Water-Soluble Chitosan Derivative for the Release of Bioactive Deferoxamine" International Journal of Molecular Sciences 25, no. 2: 913. https://doi.org/10.3390/ijms25020913