
Expression Quantitative Trait Locus of Wood Formation-Related Genes in Salix suchowensis

Abstract
:1. Introduction
2. Results
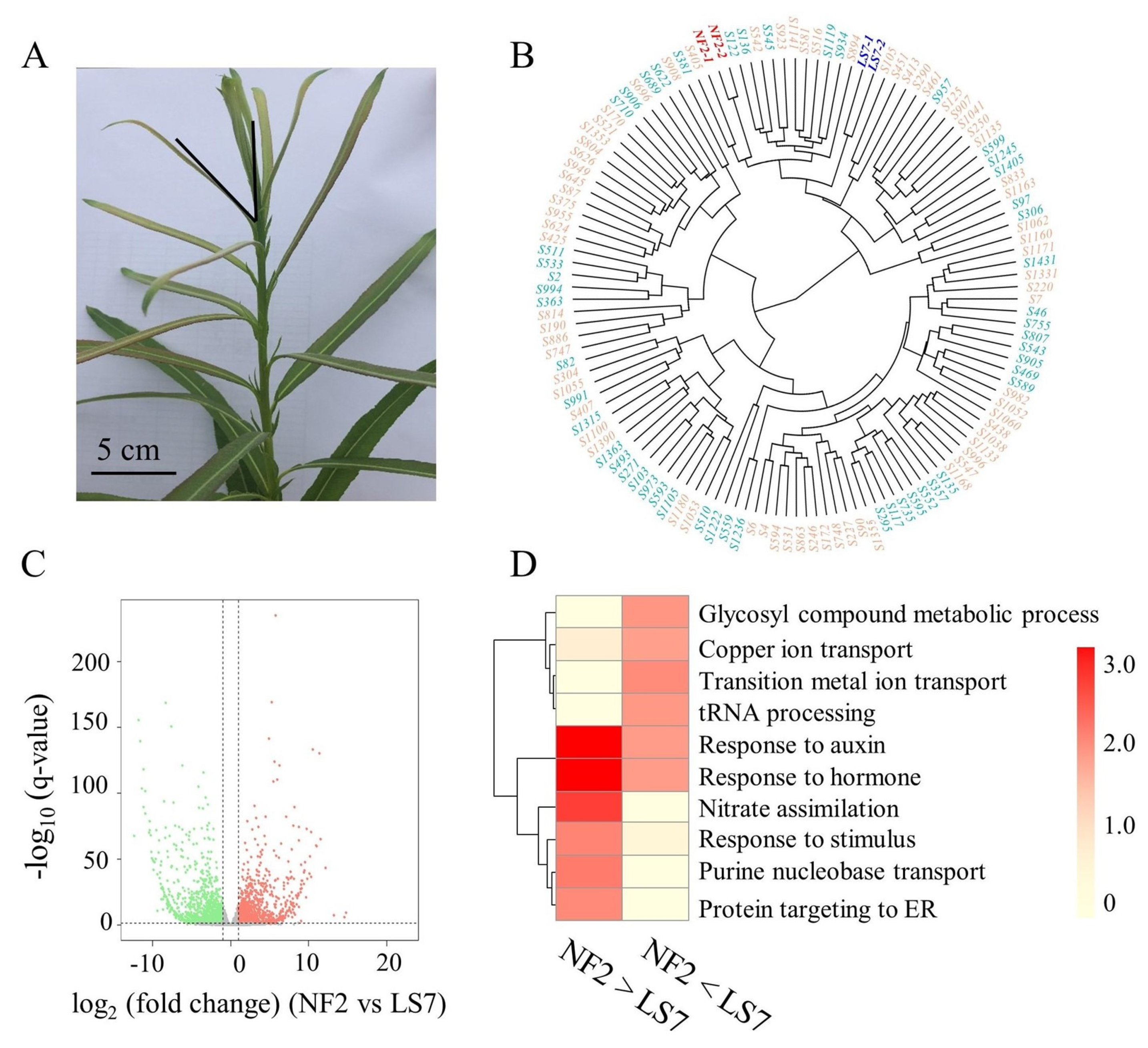
2.1. Gene Expression Variability in the Transcriptome Dataset
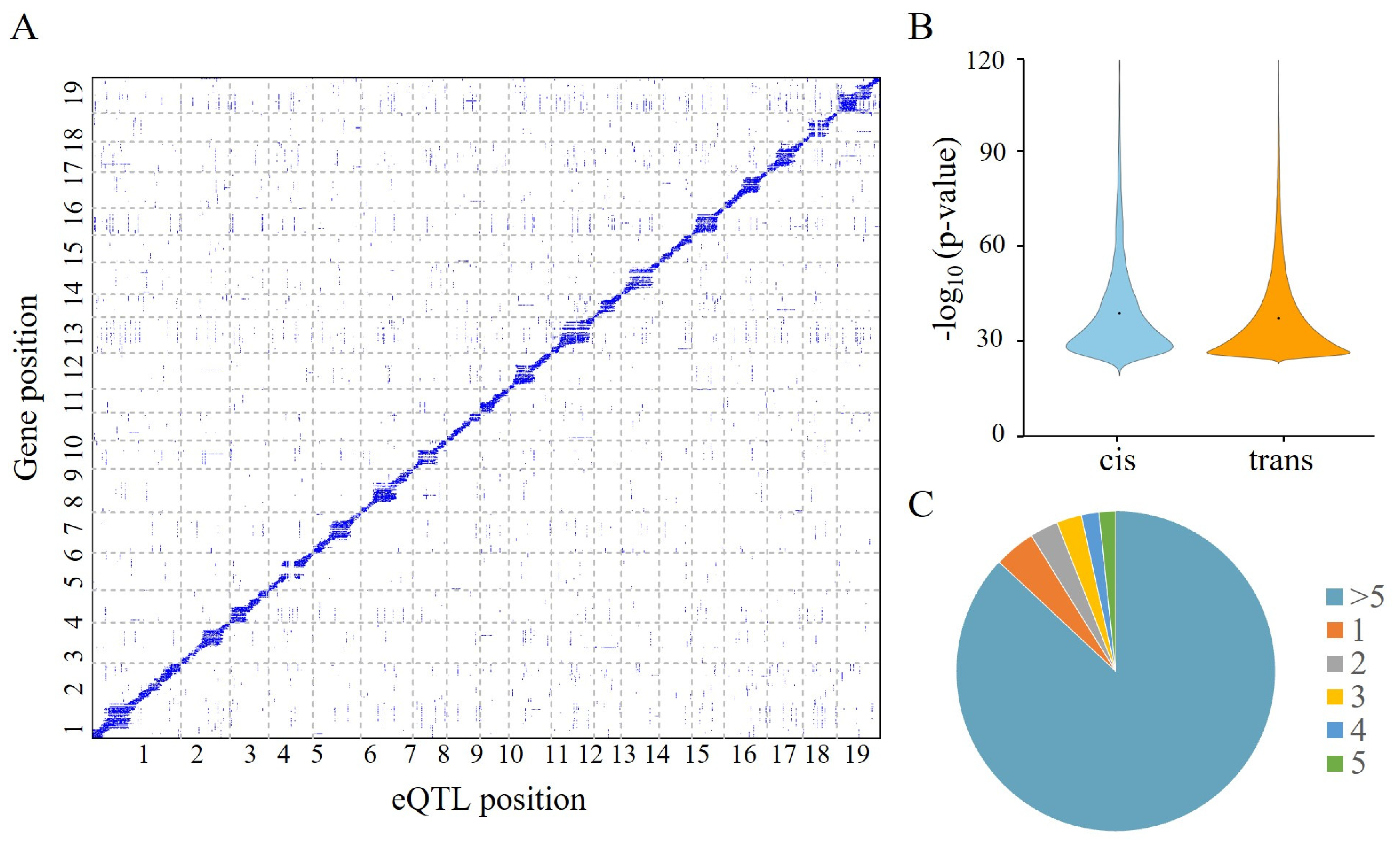
2.2. Identification of SNPs and the Genomic Location of Variants
2.3. Global eQTL Mapping-Linked Variations and Gene Expression
2.4. Expression Characteristics of WFRGs
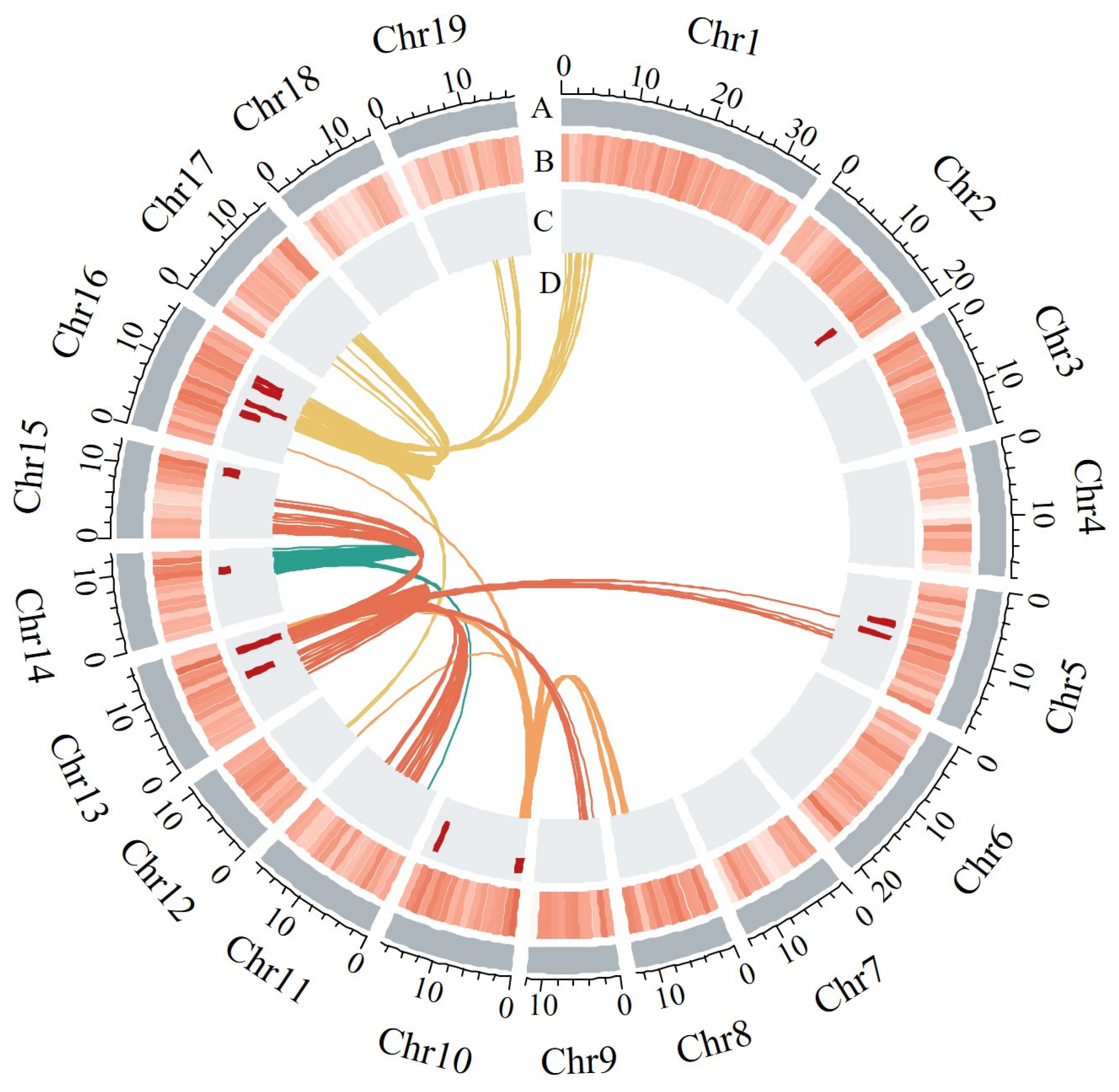
2.5. Identification of eQTL Hotspots, Their Functional Annotation and Effect on S. suchowensis Wood Traits
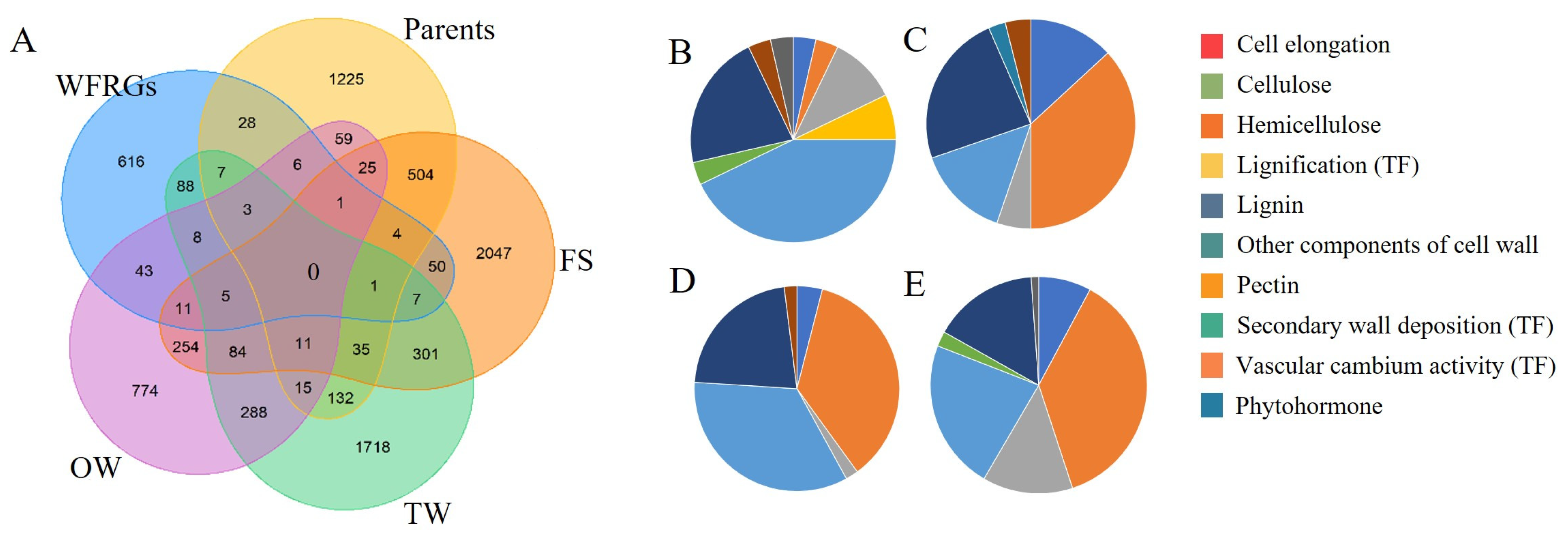
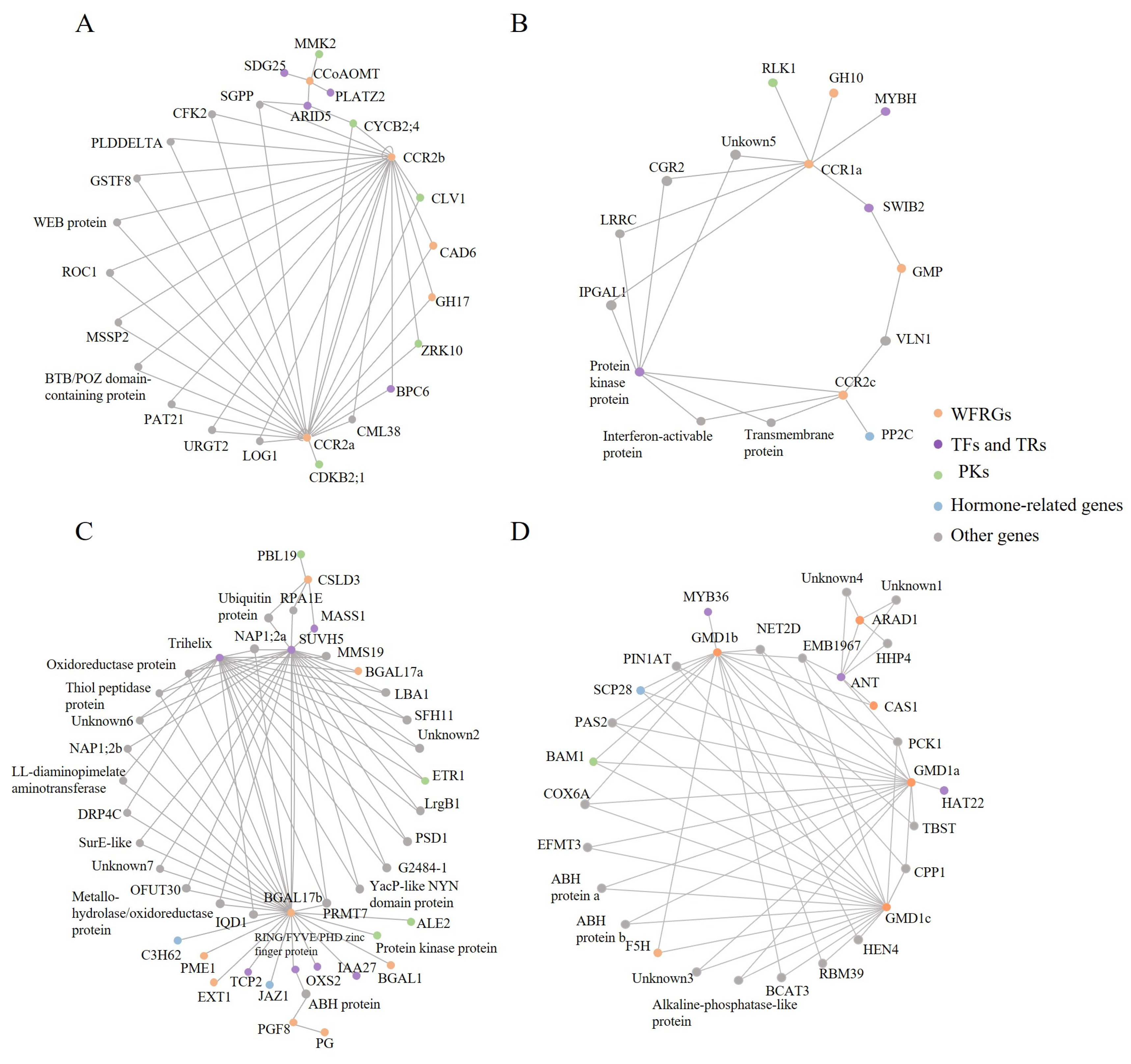
2.6. Regulatory Networks Provide a Comprehensive View of Wood Formation
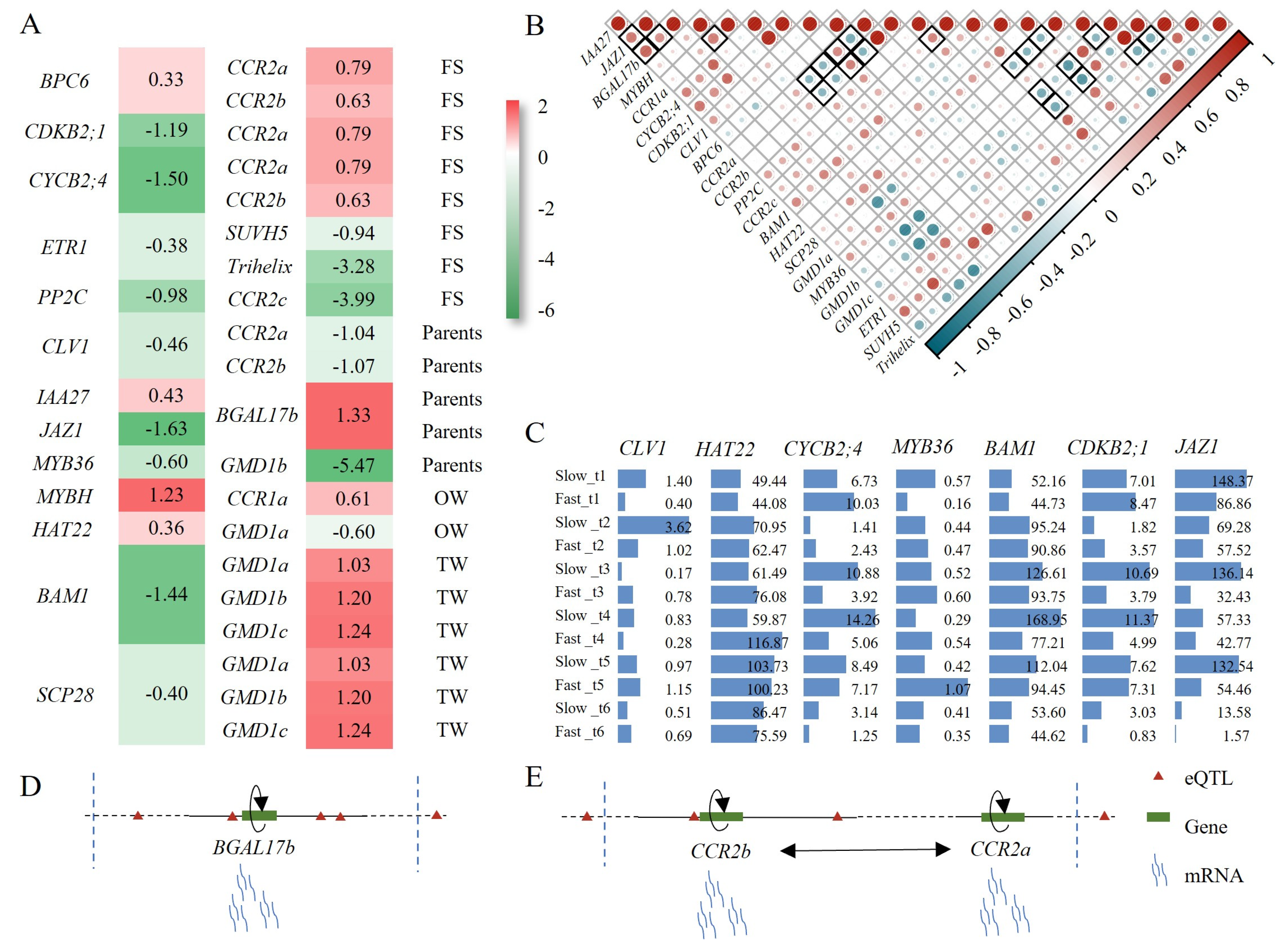
2.7. Potential Regulators and Their Properties in Wood Formation
3. Discussion
3.1. A Large Population Helps to Interpret the Rapid Growth of S. suchowensis
3.2. Potential Regulators at Distant eQTL Hotspots
3.3. eQTLs Linked Gene Expression with Genotype, Which Was Helpful for Mining Candidate Genes
4. Materials and Methods
4.1. Plant Material
4.2. RNA Sequencing and Expression Profiling
4.3. SNP Calling of Population
4.4. Construction of Homologous Gene Sets
4.5. Identification and Analysis of eQTL
4.6. Construction of Regulatory Networks for Wood Formation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Volk, T.A.; Abrahamson, L.P.; Nowak, C.A.; Smart, L.B.; Tharakan, P.J.; White, E.H. The development of short-rotation willow in the northeastern United States for bioenergy and bioproducts, agroforestry and phytoremediation. Biomass Bioenergy 2006, 30, 715–727. [Google Scholar] [CrossRef]
- Karp, A.; Shield, L. Bioenergy from plants and the sustainable yield challenge. New Phytol. 2008, 179, 15–32. [Google Scholar] [CrossRef]
- Karp, A.; Steve, J.H.; Trybush, S.O.; Macalpine, W.; Pei, M.; Shield, I. Genetic improvement of willow for bioenergy and biofuels. J. Integr. Plant Biol. 2011, 53, 151–165. [Google Scholar] [CrossRef]
- Meers, E.; Vandecasteele, B.; Ruttens, A.; Vangronsveld, J.; Tack, F.M.G. Potential of five willow species (Salix spp.) for phytoextraction of heavy metals. Plant Physiol. 2007, 60, 57–68. [Google Scholar] [CrossRef]
- Brereton, N.J.B.; Gonzalez, E.; Marleau, J.; Nissim, W.G.; Labrecque, M.; Joly, S.; Pitre, F.E. Comparative transcriptomic approaches exploring contamination stress tolerance in Salix sp. reveal the importance for a metaorganismal de novo assembly approach for nonmodel plants. Plant Physiol. 2016, 171, 3–24. [Google Scholar] [CrossRef]
- Li, Y.; Xie, T.; Zha, Y.; Du, W.; Ying, Y.; Guo, H. Urea-enhanced phytoremediation of cadmium with willow in pyrene and cadmium contaminated soil. J. Hazard. Mater. 2021, 405, 124257. [Google Scholar] [CrossRef]
- Guo, N.; Fan, L.; Cao, Y.; Xu, G.; Zhou, J.; Chen, Q.; Tao, J. Comparison of two willow genotypes reveals potential roles of iron-regulated transporter 9 and heavy-metal ATPase 1 in cadmium accumulation and resistance in Salix suchowensis. Ecotox. Environ. Safe 2022, 244, 114065. [Google Scholar] [CrossRef]
- Demura, T.; Fukuda, H. Visualization by comprehensive microarray analysis of gene expression programs during transdifferentiation of mesophyll cells into xylem cells. Trends Plant Sci. 2007, 12, 64–70. [Google Scholar] [CrossRef]
- Déjardin, A.; Laurans, F.; Arnaud, D.; Breton, C.; Pilate, G.; Leplé, J. Wood formation in Angiosperms. Comptes Rendus Biol. 2010, 333, 325–334. [Google Scholar] [CrossRef]
- Ye, Z.; Zhong, R. Molecular control of wood formation in trees. J. Exp. Bot. 2015, 66, 4119–4131. [Google Scholar] [CrossRef]
- Turner, S.R.; Somerville, C.R. Collapsed xylem phenotype of arabidopsis identifies mutants deficient in cellulose deposition in the secondary cell wall. Plant Cell 1997, 9, 689–701. [Google Scholar]
- Taylor, G. Populus: Arabidopsis for forestry. Do we need a model tree? Ann. Bot. 2002, 90, 681–689. [Google Scholar] [CrossRef]
- Yokoyama, R.; Nishitani, K. Identification and characterization of Arabidopsis thaliana genes involved in xylem secondary cell walls. J. Plant Res. 2006, 119, 189–194. [Google Scholar] [CrossRef]
- Yadun-Lev, S. Induction of sclereid differentiation in the pith of Arabidopsis thaliana (L) Heynh. J. Exp. Bot. 1994, 45, 1845–1849. [Google Scholar] [CrossRef]
- Oh, S.; Park, S.; Han, K. Transcriptional regulation of secondary growth in Arabidopsis thaliana. J. Exp. Bot. 2003, 54, 2709–2722. [Google Scholar] [CrossRef]
- Ko, J.; Han, K.; Park, S.; Yang, J. Plant body weight-induced secondary growth in arabidopsis and its transcription phenotype revealed by whole-transcriptome profiling. Plant Physiol. 2004, 135, 1069–1083. [Google Scholar] [CrossRef]
- Zhao, C.; Craig, J.C.; Petzold, H.E.; Dickerman, A.W.; Beers, E.P. The xylem and phloem transcriptomes from secondary tissues of the arabidopsis root-hypocotyl. Plant Physiol. 2005, 138, 803–818. [Google Scholar] [CrossRef]
- Schrader, J.; Moyle, R.; Bhalerao, R.; Hertzberg, M.; Lundeberg, J.; Nilsson, P.; Bhalerao, R.P. Cambial meristem dormancy in trees involves extensive remodelling of the transcriptome. Plant J. 2004, 40, 173–187. [Google Scholar] [CrossRef]
- Shi, R.; Sun, Y.; Li, Q.; Heber, S.; Sederoff, R.; Chiang, V.L. Towards a systems approach for lignin biosynthesis in Populus trichocarpa: Transcript abundance and specificity of the monolignol biosynthetic genes. Plant Cell Physiol. 2010, 51, 144–163. [Google Scholar] [CrossRef]
- Lu, S.; Li, Q.; Wei, H.; Chang, M.; Tunlaya-Anukit, S.; Kim, J.; Song, J.; Sun, Y.; Yuan, L.; Yeh, T.; et al. Ptr-miR397a is a negative regulator of laccase genes affecting lignin content in Populus trichocarpa. Proc. Natl. Acad. Sci. USA 2013, 110, 10848–10853. [Google Scholar] [CrossRef]
- Lin, Y.; Li, W.; Sun, Y.; Kumari, S.; Wei, H.; Li, Q.; Tunlaya-Anukit, S.; Sederoff, R.R.; Chiang, V.L. SND1 transcription factor-directed quantitative functional hierarchical genetic regulatory network in wood formation in Populus trichocarpa. Plant Cell 2013, 25, 4324–4341. [Google Scholar] [CrossRef]
- Taylor-Teeples, M.; Lin, L.; Lucas, M.D.; Turco, G.; Toal, T.W.; Young, N.F.; Trabucco, G.M. An arabidopsis gene regulatory network for secondary cell wall synthesis. Nature 2015, 517, 517–575. [Google Scholar] [CrossRef]
- Kubo, M.; Udagawa, M.; Nishikubo, N.; Horiguchi, G.; Yamaguchi, M.; Ito, J.; Mimura, T.; Fukuda, H.; Demura, T. Transcription switches for protoxylem and metaxylem vessel formation. Genes Dev. 2005, 19, 1860–1885. [Google Scholar] [CrossRef]
- Zhong, R.; Demura, T.; Ye, Z. SND1, a NAC domain transcription factor, is a key regulator of secondary wall synthesis in fibers of arabidopsis. Plant Cell 2006, 18, 3158–3170. [Google Scholar] [CrossRef]
- Zhong, R.; Ye, Z. Complexity of the transcriptional network controlling secondary wall biosynthesis. Plant Sci. 2014, 229, 193–207. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, L. Multi-layered regulation of plant cell wall thickening. Plant Cell Physiol. 2021, 62, 1867–1873. [Google Scholar] [CrossRef]
- Mitsuda, N.; Seki, M.; Shinozaki, K.; Ohme-Takagi, M. The NAC transcription factors NST1 and NST2 of arabidopsis regulate secondary wall thickenings and are required for anther dehiscence. Plant Cell 2005, 17, 2993–3006. [Google Scholar] [CrossRef]
- Ohashi-Ito, K.; Oda, Y.; Fukuda, H. Arabidopsis vascular-related NACDOMAIN6 directly regulates the genes that govern programmed cell death and secondary wall formation during xylem differentiation. Plant Cell 2010, 22, 3461–3473. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Mitsuda, N.; Ohtani, M.; Ohme-Takagi, M.; Kato, K. vascular-related NAC-DOMAIN7 directly regulates the expression of a broad range of genes for xylem vessel formation. Plant J. 2011, 66, 579–590. [Google Scholar] [CrossRef]
- Zhong, R.; Lee, C.; Ye, Z. Functional characterization of poplar wood-associated NAC domain transcription factors. Plant Physiol. 2010, 152, 1044–1055. [Google Scholar] [CrossRef]
- Ohtani, M.; Nishikubo, N.; Xu, B.; Yamaguchi, M.; Mitsuda, N.; Goué, N.; Shi, F.; Ohme-Takagi, M.; Demura, T. A NAC domain protein family contributing to the regulation of wood formation in poplar. Plant J. 2011, 67, 499–512. [Google Scholar] [CrossRef]
- Bomal, C.; Duval, I.; Giguère, I.; Fortin, É.; Caron, S.; Stewart, D.; Boyle, B.; Séguin, A.; MacKay, J.J. Opposite action of R2R3-MYBs from different subgroups on key genes of the shikimate and monolignol pathways in spruce. J. Exp. Bot. 2014, 65, 495–508. [Google Scholar] [CrossRef]
- Zhong, R.; McCarthy, R.L.; Lee, C.; Ye, Z. Dissection of the transcriptional program regulating secondary wall biosynthesis during wood formation in poplar. Plant Physiol. 2011, 157, 1452–1468. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, J.; Xu, P.; Zhang, R.; Li, L. Intron-mediated alternative splicing of wood-associated NAC transcription FACTOR1B regulates cell wall thickening during fiber development in Populus species. Plant Physiol. 2014, 164, 765–776. [Google Scholar] [CrossRef]
- Lin, Y.J.; Chen, H.; Li, Q.; Li, W.; Wang, J.P.; Shi, R.; Wang, Z.; Shuai, P.; Demura, T.; Wang, J.P.; et al. Reciprocal cross-regulation of VND and SND multigene TF families for wood formation in Populus trichocarpa. Proc. Natl. Acad. Sci. USA 2017, 114, E9722–E9729. [Google Scholar] [CrossRef]
- McCarthy, R.L.; Zhong, R.; Fowler, S.; Lyskowski, D.; Piyasena, H.; Carleton, K.; Spicer, C.; Ye, Z.; Demura, T. The poplar MYB transcription factors, PtrMYB3 and PtrMYB20, are involved in the regulation of secondary wall biosynthesis. Plant Cell Physiol. 2010, 51, 1084–1090. [Google Scholar] [CrossRef]
- Zhong, R.; McCarthy, R.L.; Haghighat, M.; Ye, Z. The poplar MYB master switches bind to the SMRE site and activate the secondary wall biosynthetic program during wood formation. PLoS ONE 2013, 8, e69219. [Google Scholar] [CrossRef]
- Patzlaff, A.; McInnis, S.; Courtenay, A.; Surman, C.; Newman, L.J.; Smith, C.; Bevan, M.W.; Mansfield, S.; Whetten, R.W.; Sederoff, R.R.; et al. Characterisation of a pine MYB that regulates lignification. Plant J. 2003, 36, 743–754. [Google Scholar] [CrossRef]
- Goicoechea, M.; Lacombe, E.; Legay, S.; Mihaljevic, S.; Rech, P.; Jauneau, A.; Lapierre, C.; Pollet, B.; Verhaegen, D.; Chaubet-Gigot, N.; et al. EgMYB2, a new transcriptional activator from Eucalyptus xylem, regulates secondary cell wall formation and lignin biosynthesis. Plant J. 2005, 43, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Gui, J.; Luo, L.; Zhong, Y.; Sun, J.; Umezawa, T.; Li, L. Phosphorylation of LTF1, an myb transcription factor in populus, acts as a sensory switch regulating lignin biosynthesis in wood cells. Mol. Plant. 2019, 12, 1325–1337. [Google Scholar] [CrossRef] [PubMed]
- King, M.; Wilson, A.C. Evolution at two levels in humans and chimpanzees. Science 1975, 188, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Konishi, S.; Izawa, T.; Lin, S.Y.; Ebana, K.; Fukuta, Y.; Sasaki, T.; Yano, M. An SNP caused loss of seed shattering during rice domestication. Science 2006, 312, 1392–1396. [Google Scholar] [CrossRef] [PubMed]
- Hung, H.; Shannon, L.M.; Tian, F.; Bradbury, P.J.; Chen, C.; Flint-Garcia, S.A.; McMullen, M.D.; Ware, D.; Buckler, E.S.; Doebley, J.F.; et al. ZmCCT and the genetic basis of day-length adaptation underlying the postdomestication spread of maize. Proc. Natl. Acad. Sci. USA 2012, 109, E1913–E1921. [Google Scholar] [CrossRef] [PubMed]
- Si, L.; Chen, J.; Huang, X.; Gong, H.; Luo, J.; Hou, Q.; Zhou, T.; Lu, T.; Zhu, J.; Shangguan, Y.; et al. OsSPL13 controls grain size in cultivated rice. Nat. Genet. 2016, 48, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Luo, X.; Niu, L.; Xiao, Y.; Chen, L.; Wang, X.; Jin, M.; Li, W.; Zhang, Q.; Yan, J. Distant eQTLs and non-coding sequences play critical roles in regulating gene expression and quantitative trait variation in maize. Mol. Plant. 2016, 10, 414–426. [Google Scholar] [CrossRef]
- Wang, J.; Yu, H.; Weng, X.; Xie, W.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. An expression quantitative trait loci-guided co-expression analysis for constructing regulatory network using a rice recombinant inbred line population. J. Exp. Bot. 2014, 65, 1069–1079. [Google Scholar] [CrossRef]
- Zhang, L.; Su, W.; Tao, R.; Zhang, W.; Chen, J.; Wu, P.; Yan, C.; Jia, Y.; Larkin, R.M.; Lavelle, D.; et al. RNA sequencing provides insights into the evolution of lettuce and the regulation of flavonoid biosynthesis. Nat. Commun. 2017, 8, 2264. [Google Scholar] [CrossRef]
- Wei, S.; Yang, Y.; Yin, T. The chromosome-scale assembly of the willow genome provides insight into Salicaceae genome evolution. Hortic. Res. 2020, 7, 45. [Google Scholar] [CrossRef]
- Yang, G.; Xu, Q.; Li, W.; Ling, J.; Li, X.; Yin, T. Sex-related differences in growth, herbivory, and defense of two salix species. Forests 2020, 11, 450. [Google Scholar] [CrossRef]
- Li, Z.; Wang, P.; You, C.; Yu, J.; Zhang, X.; Yan, F.; Ye, Z.; Shen, C.; Li, B.; Guo, K.; et al. Combined GWAS and eQTL analysis uncovers a genetic regulatory network orchestrating the initiation of secondary cell wall development in cotton. New Phytol. 2020, 226, 1738–1752. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, Y.; Shi, T.; Kou, M.; Sun, J.; Xu, T.; Li, Q.; Wu, S.; Cao, Q.; Hou, W.; et al. Genome-wide analysis of expression quantitative trait loci (eQTLs) reveals the regulatory architecture of gene expression variation in the storage roots of sweet potato. Hort. Res. 2020, 7, 90. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, D.; Batoux, M.; Fulton, L.; Pfister, K.; Yadav, R.K.; Schellenberg, M.; Schneitz, K. STRUBBELIG defines a receptor kinase-mediated signaling pathway regulating organ development in arabidopsis. Proc. Natl. Acad. Sci. USA 2005, 102, 9074–9079. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jin, Y.; Wang, C.; Li, B.; Jiang, C.; Sun, Z.; Zhang, Z.; Kong, F.; Demura, H. Fasciclin-like arabinogalactan proteins, PtFLAs, play important roles in GA-mediated tension wood formation in Populus. Sci. Rep. 2017, 7, 6182. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, A.; Chen, X.; Gao, J.; Leśniewska, B.; Hammerl, R.; Dawid, C.; Schneitz, K. The arabidopsis receptor kinase STRUBBELIG regulates the response to cellulose deficiency. PLoS Genet. 2020, 16, e1008433. [Google Scholar] [CrossRef] [PubMed]
- Vries, L.D.; Brouckaert, M.; Chanoca, A.; Kim, H.; Regner, M.R.; Timokhin, V.I.; Sun, Y.; Meester, B.D.; Doorsselaere, J.V.; Goeminne, G.; et al. CRISPR-Cas9 editing of CAFFEOYL SHIKIMATE ESTERASE 1 and 2 shows their importance and partial redundancy in lignification in Populus tremula × P. alba. Plant Biotechnol. J. 2021, 19, 2221–2234. [Google Scholar] [CrossRef]
- Nibbering, P.; Castilleux, R.; Wingsle, G.; Niittylä, T. CAGEs are Golgi-localized GT31 enzymes involved in cellulose biosynthesis in arabidopsis. Plant J. 2022, 110, 1271–1285. [Google Scholar] [CrossRef]
- Suzuki, S.; Li, L.; Sun, Y.; Chiang, V.L. The cellulose synthase gene superfamily and biochemical functions of xylem-specific cellulose synthase-like genes in Populus trichocarpa. Plant Physiol. 2006, 142, 1233–1245. [Google Scholar] [CrossRef]
- Dwevedi, A.; Kayastha, A.M. Plant β-Galactosidases: Physiological significance and recent advances in technological applications. J. Plant Biochem. Biotechnol. 2010, 32, 9–20. [Google Scholar] [CrossRef]
- Du, J.; Anderson, C.T.; Xiao, C. Dynamics of pectic homogalacturonan in cellular morphogenesis and adhesion, wall integrity sensing and plant development. Nat. Plants 2022, 8, 332–340. [Google Scholar] [CrossRef]
- Su, Y.H.; Zhou, C.; Li, Y.J.; Yu, Y.; Tang, L.P.; Zhang, W.J.; Yao, W.J.; Huang, R.; Laux, T.; Zhang, X.S. Integration of pluripotency pathways regulates stem cell maintenance in the arabidopsis shoot meristem. Proc. Natl. Acad. Sci. USA 2020, 117, 22561–22571. [Google Scholar] [CrossRef]
- Reyt, G.; Ramakrishna, P.; Salas-González, I.; Fujita, S.; Love, A.; Tiemessen, D.; Lapierre, C.; Morreel, K.; Calvo-Polanco, M.; Flis, P.; et al. Two chemically distinct root lignin barriers control solute and water balance. Nat. Commun. 2019, 19, 2320. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Zhang, Y.; Wang, R.; Liu, Y.; Li, M.; Wang, X.; Chen, X.; Luan, X.; Zhang, H.; Wei, H.; et al. PtrHAT22, as a higher hierarchy regulator, coordinately regulates secondary cell wall component biosynthesis in Populus trichocarpa. Plant Sci. 2022, 316, 111170. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Liu, P.; Wang, C.; Wu, S.; Dong, C.; Lin, Q.; Sun, W.; Huang, B.; Xu, M.; Tauqeer, A.; et al. Transcriptional networks regulating suberin and lignin in endodermis link development and ABA response. Plant Physiol. 2022, 190, 1165–1181. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ding, Q.; Chen, J.; Cui, K.; He, X. Induction of PtoCDKB and PtoCYCB transcription by temperature during cambium reactivation in Populus tomentosa Carr. J. Exp. Bot. 2009, 60, 2621–2630. [Google Scholar] [CrossRef] [PubMed]
- Nimchuk, Z.L. CLAVATA1 controls distinct signaling outputs that buffer shoot stem cell proliferation through a two-step transcriptional compensation loop. BMC Plant Biol. 2017, 13, e1006681. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Aguilar, E.; Bradai, M.; Xue, H.; Wang, H.; Rosas-Diaz, T.; Tang, W.; Wolf, S.; Zhang, H.; Xu, L.; et al. The receptor-like kinases BAM1 and BAM2 are required for root xylem patterning. Proc. Natl. Acad. Sci. USA 2021, 118, e2022547118. [Google Scholar] [CrossRef]
- Grunewald, W.; Vanholme, B.; Pauwels, L.; Plovie, E.; Inzé, D.; Gheysen, G.; Goossens, A. Expression of the arabidopsis jasmonate signalling repressor JAZ1/TIFY10A is stimulated by auxin. EMBO Rep. 2009, 10, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Fischer, U.; Kucukoglu, M.; Helariutta, Y.; Bhalerao, R.P. The dynamics of cambial stem cell activity. Annu. Rev. Plant Biol. 2019, 70, 293–319. [Google Scholar] [CrossRef]
- Du, J.; Gerttula, S.; Li, Z.; Zhao, S.; Liu, Y.; Liu, Y.; Lu, M.; Groover, A.T. Brassinosteroid regulation of wood formation in poplar. New Phytol. 2020, 225, 1516–1530. [Google Scholar] [CrossRef]
- Liu, C.; Yu, H.; Rao, X.; Li, L.; Dixon, R.A. Abscisic acid regulates secondary cell-wall formation and lignin deposition in Arabidopsis thaliana through phosphorylation of NST1. Proc. Natl. Acad. Sci. USA 2021, 118, e2010911118. [Google Scholar] [CrossRef]
- Hendelman, A.; Zebell, S.; Rodriguez-Leal, D.; Dukler, N.; Robitaille, G.; Wu, X.; Kostyun, J.; Tal, L.; Wang, P.; Bartlett, M.E.; et al. Conserved pleiotropy of an ancient plant homeobox gene uncovered by cis-regulatory dissection. Cell 2021, 184, 1724–1739. [Google Scholar] [CrossRef] [PubMed]
- Marand, A.P.; Eveland, A.L.; Kaufmann, K.; Springer, N.M. cis-Regulatory elements in plant development, adaptation, and evolution. Annu. Rev. Plant Biol. 2023, 74, 111–137. [Google Scholar] [CrossRef]
- Pavlides, J.M.W.; Zhu, Z.; Gratten, J.; McRae, A.F.; Wray, N.R.; Yang, J. Predicting gene targets from integrative analyses of summary data from GWAS and eQTL studies for 28 human complex traits. Genome Med. 2016, 8, 84. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.C.; Nap, J. Genetical genomics: The added value from segregation. BMC Plant Biol. 2001, 17, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, B.A.; Mezey, J. Gene expression network reconstruction by convex feature selection when incorporating genetic perturbations. PLoS Comput. Biol. 2010, 6, e1001014. [Google Scholar] [CrossRef] [PubMed]
- Wittkopp, P.J.; Haerum, B.K.; Clark, A.G. Evolutionary changes in cis and trans gene regulation. Nature 2004, 430, 85–88. [Google Scholar] [CrossRef]
- Zhu, G.; Wang, S.; Huang, Z.; Zhang, S.; Liao, Q.; Zhang, C.; Lin, T.; Qin, M.; Peng, M.; Yang, C.; et al. Rewiring of the fruit metabolome in tomato breeding. Cell 2018, 172, 249–261. [Google Scholar] [CrossRef]
- Keurentjes, J.J.B.; Fu, J.; Terpstra, I.R.; Garcia, J.M.; van den Ackerveken, G.; Snoek, L.B.; Peeters, A.J.M.; Vreugdenhil, D.; Koornneef, M.; Jansen, R.C. Regulatory network construction in arabidopsis by using genome-wide gene expression quantitative trait loci. Proc. Natl. Acad. Sci. USA 2007, 104, 1708–1713. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, Y.; Zheng, K.; Xie, M.; Feng, K.; Jawdy, S.S.; Gunter, L.E.; Ranjan, P.; Singan, V.R.; Engle, N.L.; et al. Genome-wide association studies and expression-based quantitative trait loci analyses reveal roles of HCT2 in caffeoylquinic acid biosynthesis and its regulation by defenseresponsive transcription factors in Populus. New Phytol. 2018, 220, 502–516. [Google Scholar] [CrossRef]
- Wang, X.; Gao, L.; Jiao, C.; Stravoravdis, S.; Hosmani, P.S.; Saha, S.; Zhang, J.; Mainiero, S.; Strickler, S.R.; Catala, C.; et al. Genome of Solanum pimpinellifolium provides insights into structural variants during tomato breeding. Nat. Commun. 2020, 11, 5817. [Google Scholar] [CrossRef]
- Rhee, S.Y.; Osborne, E.; Poindexter, P.D.; Somerville, C.R. Microspore Separation in the quartet 3 mutants of arabidopsis is impaired by a defect in a developmentally regulated polygalacturonase required for pollen mother cell wall degradation. Plant Physiol. 2019, 133, 1170–1180. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Somerville, C.; Anderson, C.T. Polygalacturonase involved in expansion1 functions in cell elongation and flower development in arabidopsis. Plant Cell 2014, 26, 1018–1035. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Branes, W.J.; Zamil, M.S.; Yi, H.; Puri, V.M.; Anderson, C.T. Activation tagging of arabidopsis POLYGALACTURONASE INVOLVED IN EXPANSION2 promotes hypocotyl elongation, leaf expansion, stem lignification, mechanical stiffening, and lodging. Plant J. 2017, 89, 1159–1173. [Google Scholar] [CrossRef]
- Hu, J.; Su, H.; Cao, H.; Wei, H.; Fu, X.; Jiang, X.; Song, Q.; He, X.; Xu, C.; Luo, K. AUXIN RESPONSE FACTOR7 integrates gibberellin and auxin signaling via interactions between DELLA and AUX/IAA proteins to regulate cambial activity in poplar. Plant Cell 2022, 34, 2688–2707. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lu, S.; Chiang, V. A genomic and molecular view of wood formation. Plant Sci. 2006, 25, 215–233. [Google Scholar] [CrossRef]
- Schrader, J.; Nilsson, J.; Mellerowicz, E.; Berglund, A.; Nilsson, P.; Hertzberg, M.; Sandberg, G. A high-resolution transcript profile across the wood-forming meristem of poplar identifies potential regulators of cambial stem cell identity. Plant Cell 2004, 16, 2278–2292. [Google Scholar] [CrossRef]
- Liberman, L.M.; Sparks, E.E.; Moreno-Risueno, M.A.; Petricka, J.J.; Benfey, P.N. The wild sweetpotato (Ipomoea trifida) genome provides insights into storage root development. Proc. Natl. Acad. Sci. USA 2015, 112, 12099–12104. [Google Scholar] [CrossRef]
- Kamiya, T.; Borghi, M.; Wang, P.; Danku, J.M.C.; Kalmbach, L.; Hosmani, P.S.; Naseer, S.; Fujiwara, T.; Geldner, N.; Salt, D.E. The MYB36 transcription factor orchestrates Casparian strip formation. Proc. Natl. Acad. Sci. USA 2015, 112, 10533–10538. [Google Scholar] [CrossRef]
- Sun, X.; Jiao, C.; Schwaninger, H.; Chao, C.; Ma, Y.; Duan, N.; Khan, A.; Ban, S.; Xu, K.; Cheng, L.; et al. Phased diploid genome assemblies and pan-genomes provide insights into the genetic history of apple domestication. Nat Genet. 2020, 52, 1423–1432. [Google Scholar] [CrossRef]
- Lee, S.; Karp, G.; Kim, K.; Chu, S.; Shin, J.; Lee, Y.; Park, Y.; Park, S. Dissection of expression-quantitative trait locus and allele specificity using a haploid/diploid plant system-insights into compensatory evolution of transcriptional regulation within populations. Rice 2019, 12, 84. [Google Scholar] [CrossRef]
- Ma, Q.; Chen, Y.; Ju, J.; Dai, X.G. Modeling and analyzing the dynamic growth for progeny in a full-sib family of Salix xuzhouenesis. J. Nanjing For. Univ. (Nat. Sci. Ed.) 2013, 37, 13–16. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Calvo-Polanco, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Zinkgraf, M.; Zhao, S.T.; Canning, C.; Gerttula, S.; Lu, M.Z.; Filkov, V.; Groover, A. Evolutionary network genomics of wood formation in a phylogenetic survey of angiosperm forest trees. New Phytol. 2020, 228, 1811–1823. [Google Scholar] [CrossRef]
- Wei, S.; Chen, Y.; Hou, J.; Yang, Y.; Yin, T. Aux/IAA and ARF gene families in Salix suchowensis: Identification, evolution, and dynamic transcriptome profiling during the plant growth process. Front. Plant Sci. 2021, 12, 666310. [Google Scholar] [CrossRef]
- Li, J. High-Density Genetic Linkage Map Construction and Detection of Quantitative Trait Loci for Ground Diameter and Number of Branches in Salix suchowensis; Nanjing Forestry University: Nanjing, China, 2008. [Google Scholar]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303-3997. [Google Scholar]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207-3907. [Google Scholar]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Zhong, R.; Himmelsbach, D.S.; McPhail, B.T.; Ye, Z. Molecular characterization of PoGT8D and PoGT43B, two secondary wall-associated glycosyltransferases in poplar. Plant Cell Physiol. 2007, 48, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Shabalin, A.A. Matrix eQTL: Ultra fast eQTL analysis via large matrix operations. Bioinformatics 2012, 28, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Li Zhan, L.; et al. Transcription switches for protoxylem and metaxylem vessel formation. Innovation 2021, 2, 100141. [Google Scholar] [PubMed]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef]
- Demchak, B.; Hull, T.; Reich, M.; Liefeld, T.; Smoot, M.; Fei, Z. Cytoscape: The network visualization tool for GenomeSpace workflows. F1000Research 2014, 3, 151. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiao, C.; Sun, H.; Rosil, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hotspot ID | Chr | Hotspot Start | Hotspot End | Enriched Special Pathway | Target(s) Involved in Special Pathway | Candidate Regulators |
|---|---|---|---|---|---|---|
| Hot4 | Chr10 | 0 | 1,410,000 | hormone-mediated signaling pathway | EVM0000387 (GH3.10) | - |
| Hot7 | Chr13 | 11,810,000 | 13,040,000 | hormone-mediated signaling pathway | EVM0000387 (GH3.10) | EVM0029643 (FLA13) |
| Hot8 | Chr14 | 8,740,000 | 10,000,000 | hormone-mediated signaling pathway | EVM0002776 (GH3.6), EVM0041547 (ARF2) | EVM0029357 (CAGE1) |
| Hot12 | Chr16 | 11,110,000 | 12,390,000 | plant-type cell wall assembly | EVM0036007 (PME3), EVM0034292 (PME3), EVM0005816 (COBL2) | EVM0011519 (SRF6), EVM0008575 (PGF11), EVM0008454 (CSE) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Liu, L.; Yang, G.; Li, X.; Dai, X.; Xue, L.; Yin, T. Expression Quantitative Trait Locus of Wood Formation-Related Genes in Salix suchowensis. Int. J. Mol. Sci. 2024, 25, 247. https://doi.org/10.3390/ijms25010247
Chen L, Liu L, Yang G, Li X, Dai X, Xue L, Yin T. Expression Quantitative Trait Locus of Wood Formation-Related Genes in Salix suchowensis. International Journal of Molecular Sciences. 2024; 25(1):247. https://doi.org/10.3390/ijms25010247
Chicago/Turabian StyleChen, Li, Liyan Liu, Guo Yang, Xiaoping Li, Xiaogang Dai, Liangjiao Xue, and Tongming Yin. 2024. "Expression Quantitative Trait Locus of Wood Formation-Related Genes in Salix suchowensis" International Journal of Molecular Sciences 25, no. 1: 247. https://doi.org/10.3390/ijms25010247