
Interdependence of Molecular Lesions That Drive Uveal Melanoma Metastasis

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
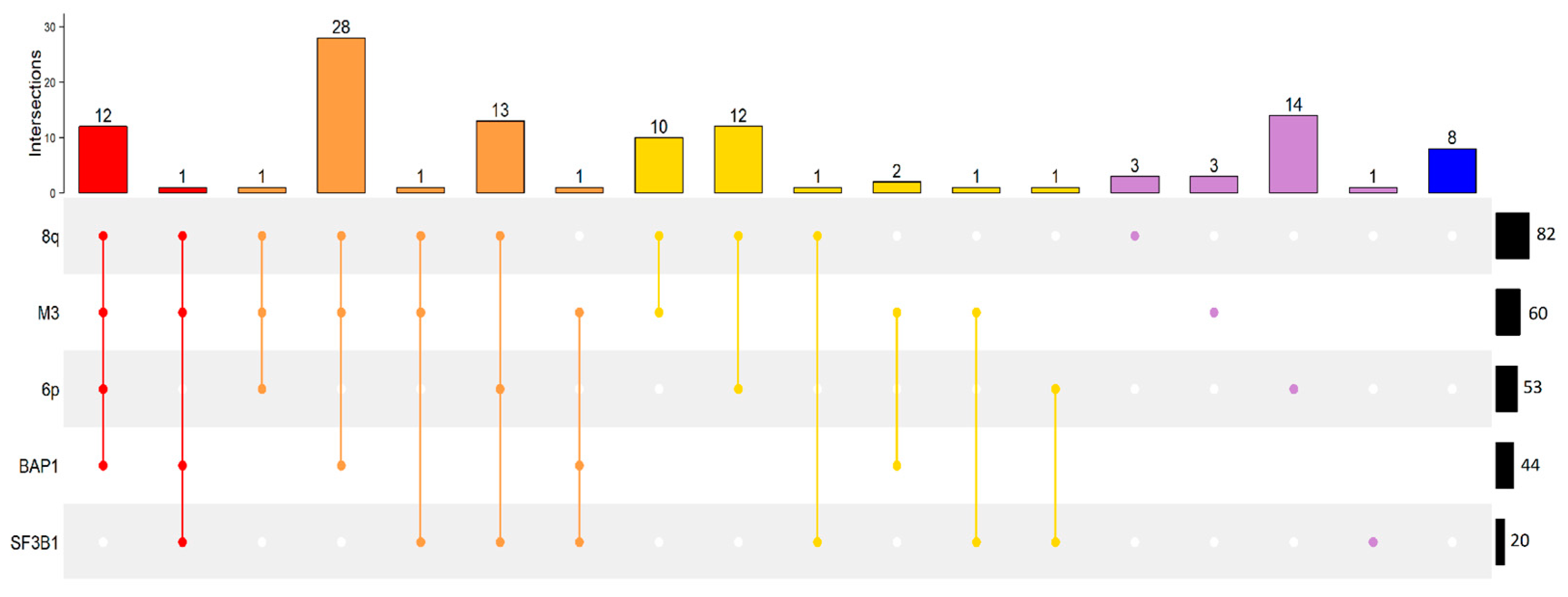
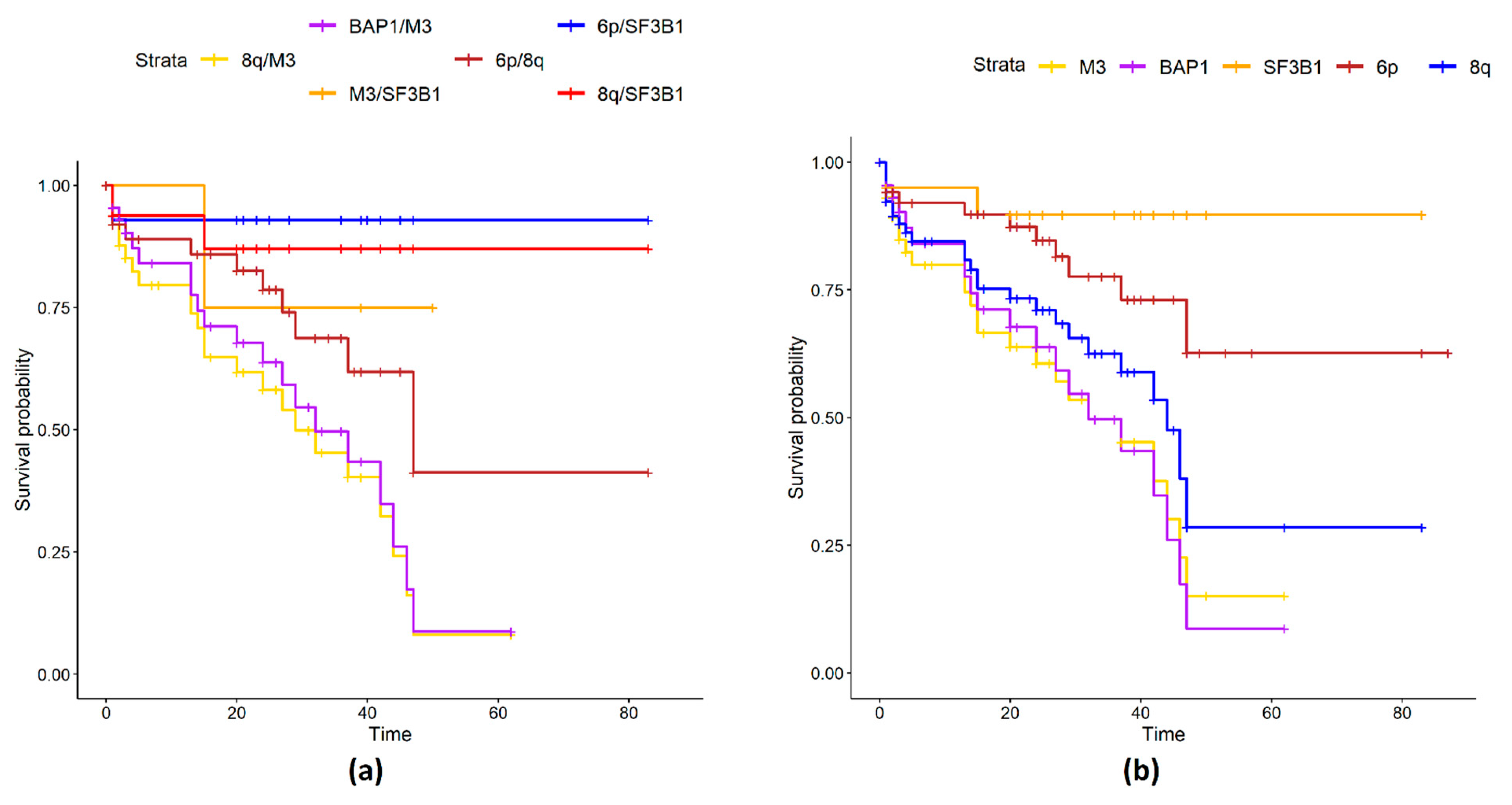
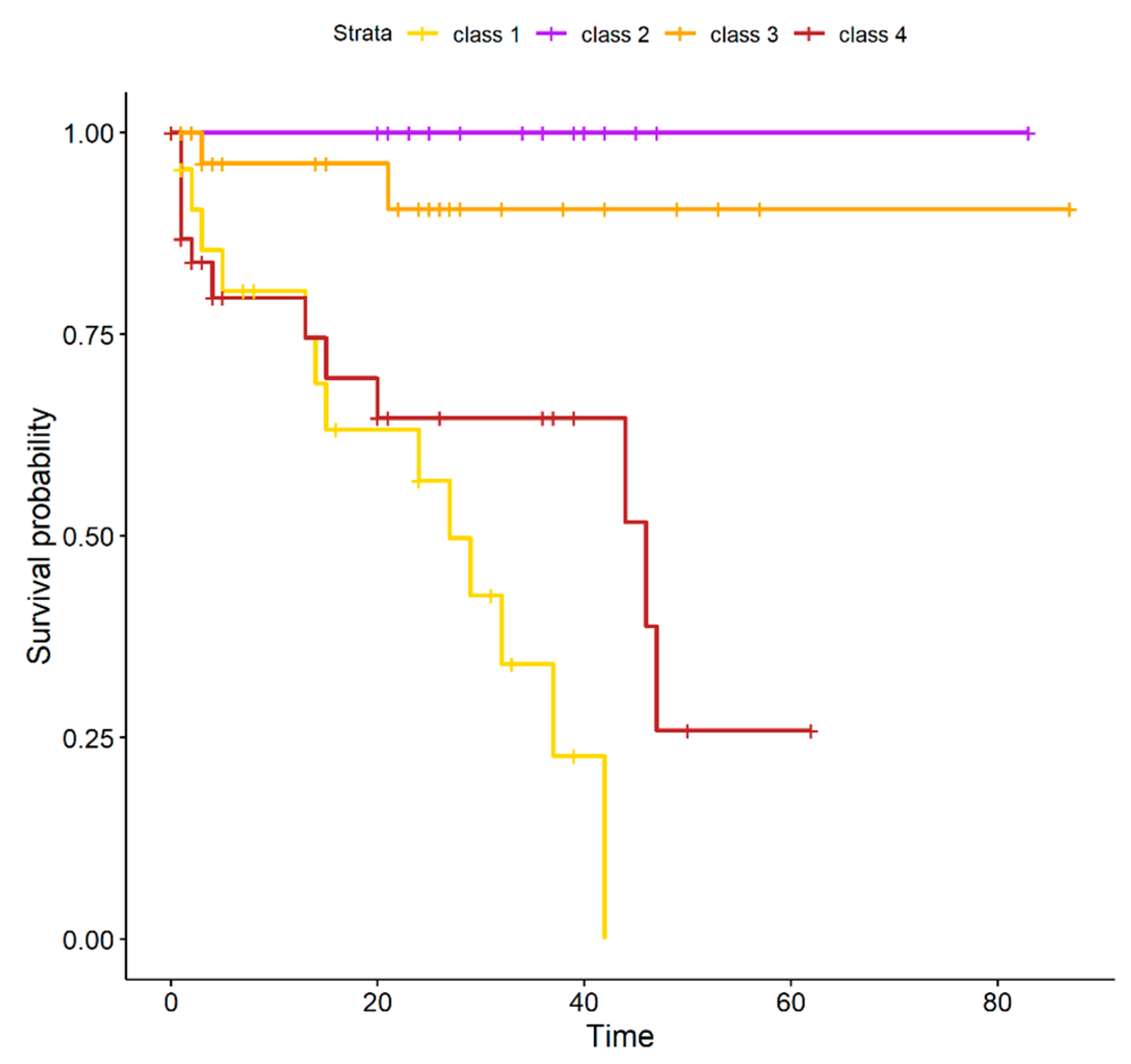
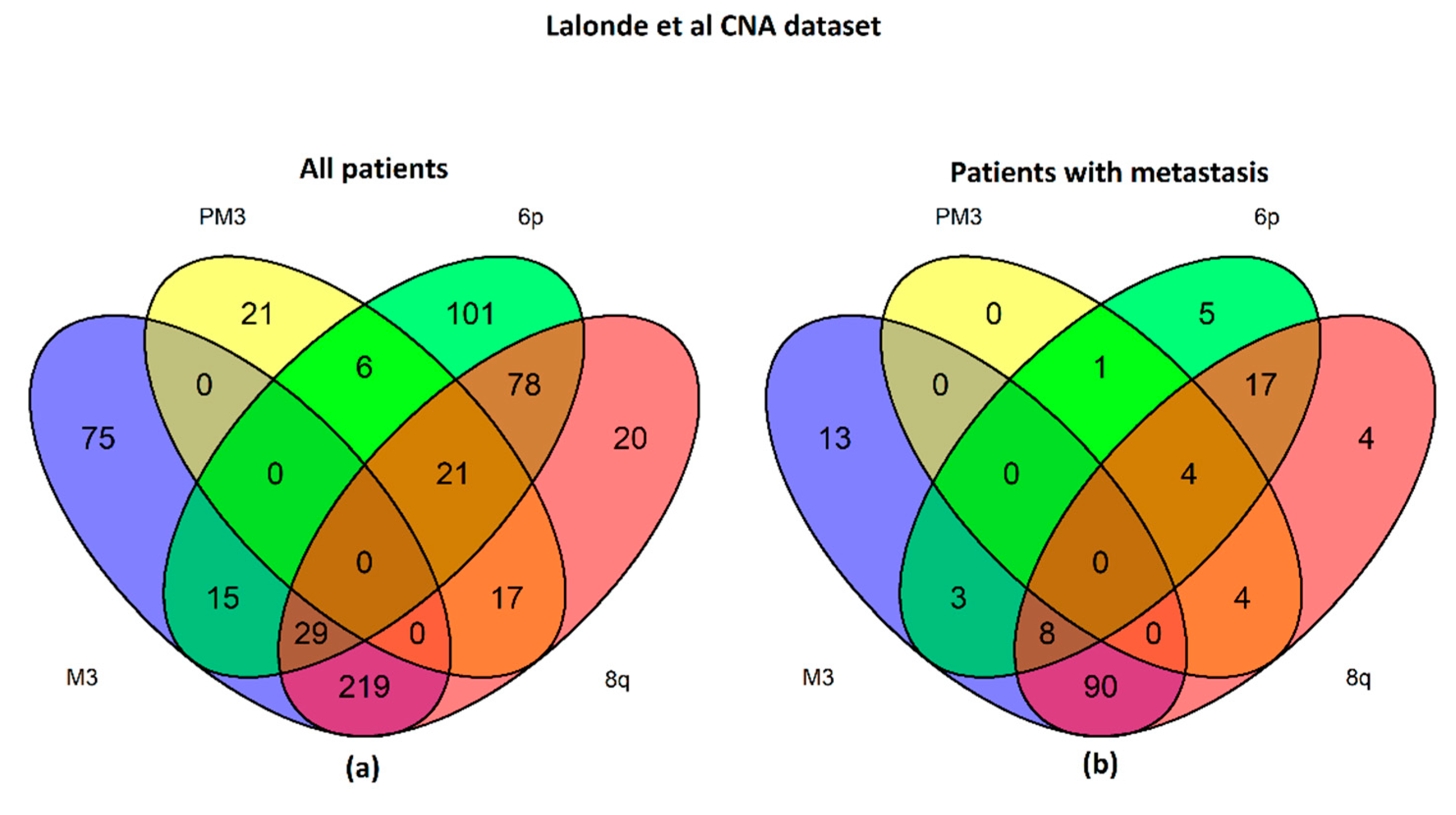
2.1. UM Genomic Features and Metastases
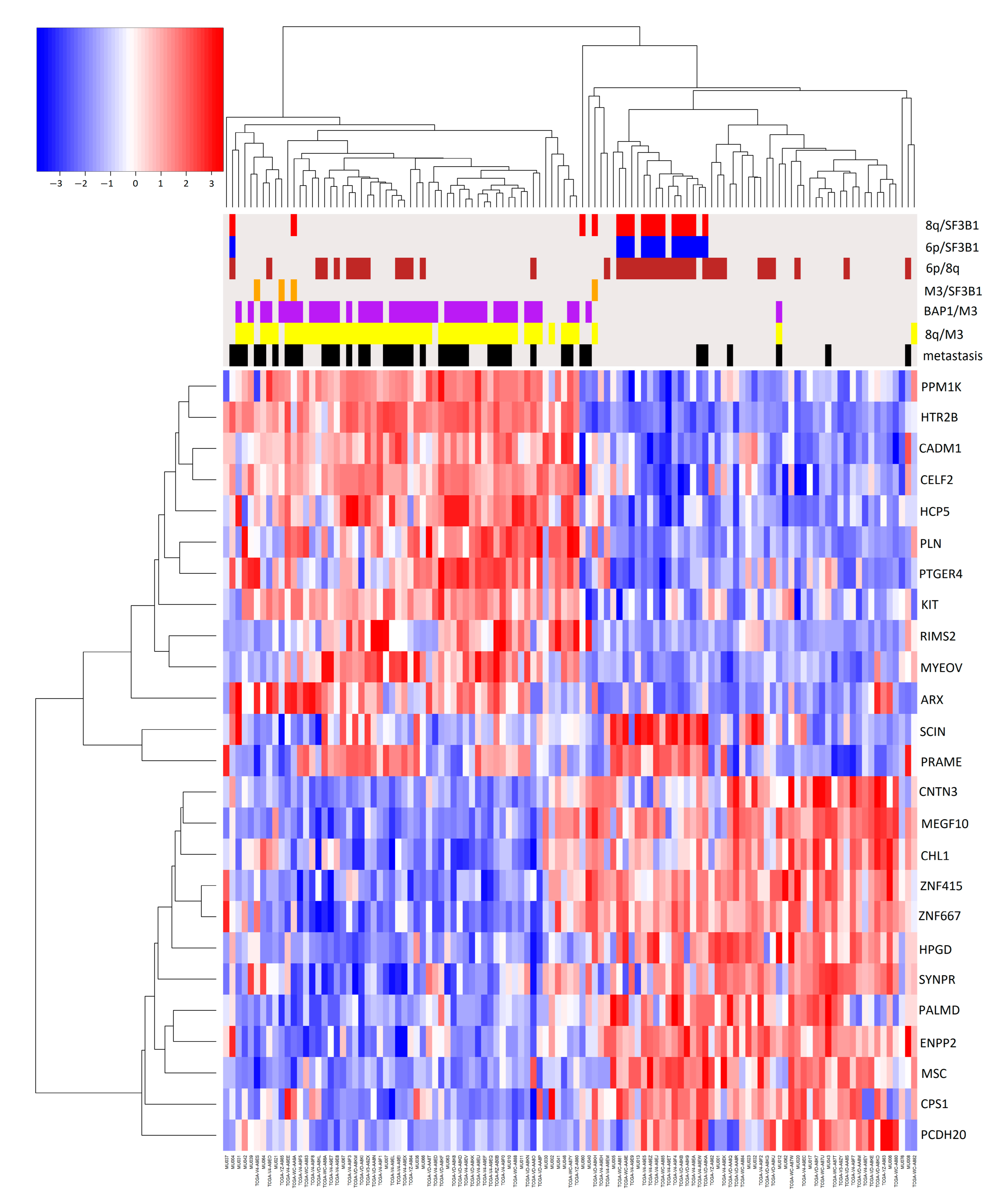
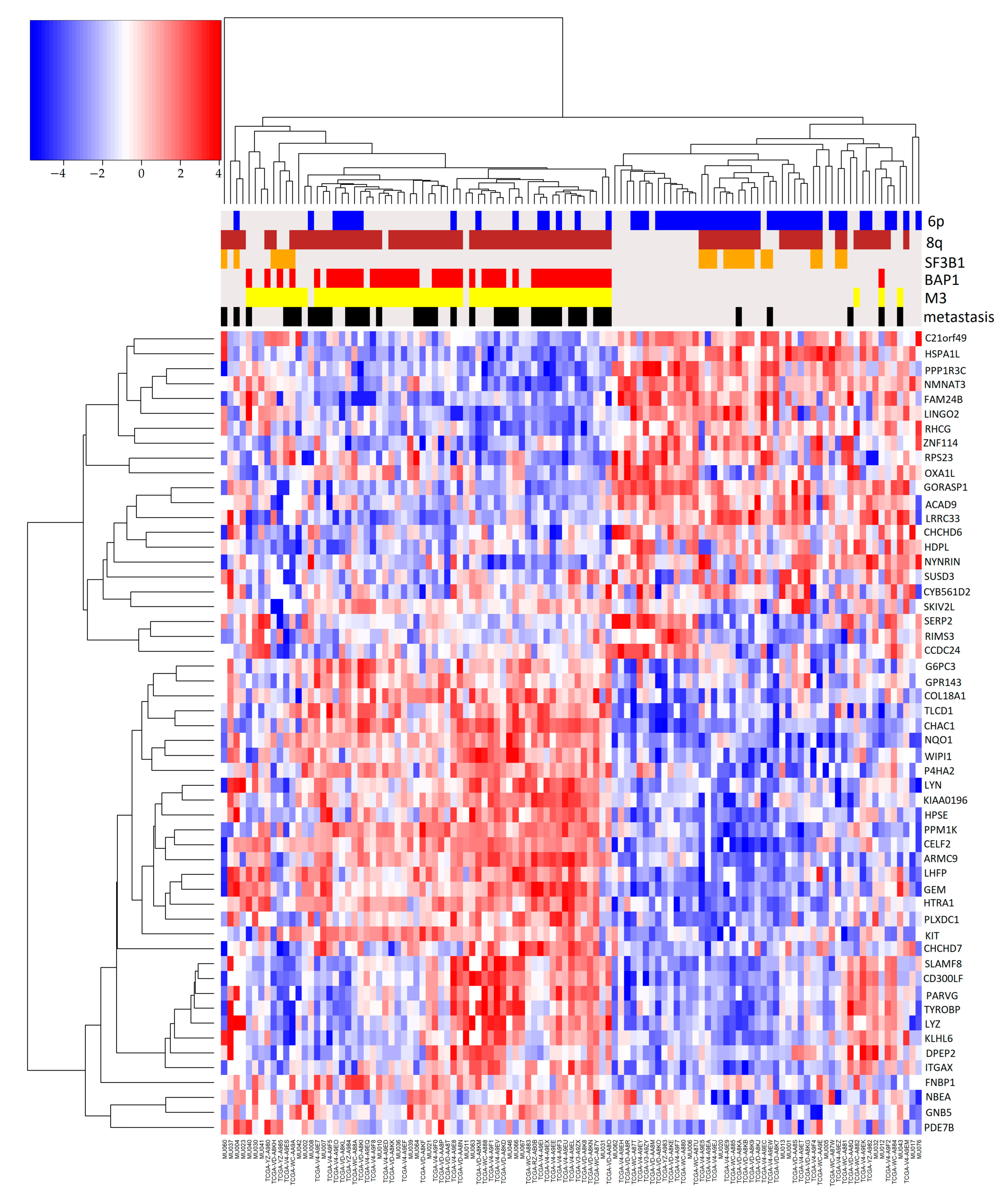
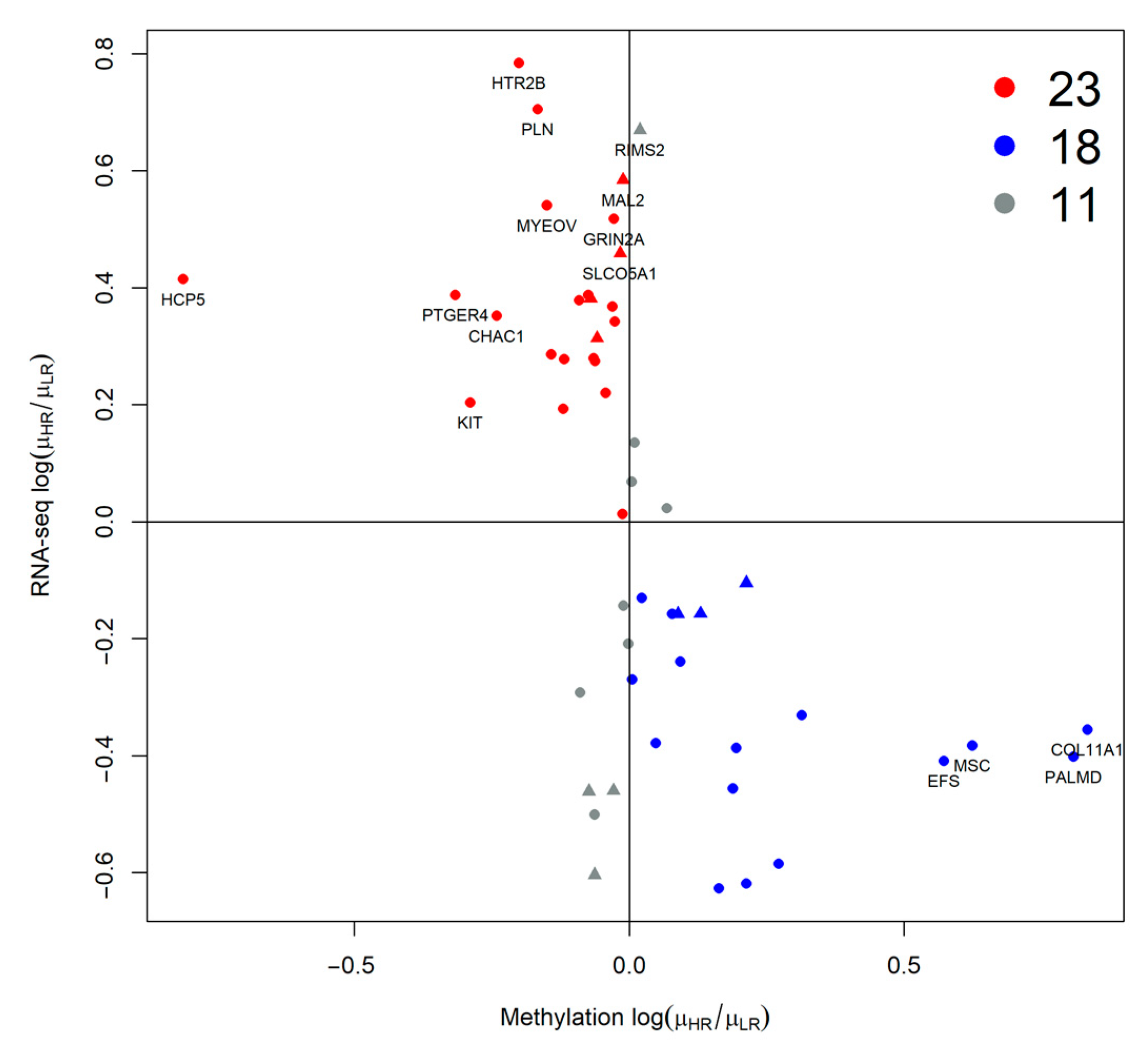
2.2. Association of Pairs of Risk Factors with Gene Expression
3. Discussion
4. Materials and Methods
4.1. Data Source
4.2. UM Gene Expression Data Collection
4.3. UM Expression Data Analysis
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jager, M.J.; Shields, C.L.; Cebulla, C.M.; Abdel-Rahman, M.H.; Grossniklaus, H.E.; Stern, M.-H.; Carvajal, R.D.; Belfort, R.N.; Jia, R.; Shields, J.A.; et al. Uveal Melanoma. Nat. Rev. Dis. Prim. 2020, 6, 24. [Google Scholar] [CrossRef]
- Amaro, A.; Gangemi, R.; Piaggio, F.; Angelini, G.; Barisione, G.; Ferrini, S.; Pfeffer, U. The Biology of Uveal Melanoma. Cancer Metastasis Rev. 2017, 36, 109–140. [Google Scholar] [CrossRef]
- Lalonde, E.; Ewens, K.; Richards-Yutz, J.; Ebrahimzedeh, J.; Terai, M.; Gonsalves, C.F.; Sato, T.; Shields, C.L.; Ganguly, A. Improved Uveal Melanoma Copy Number Subtypes Including an Ultra-High-Risk Group. Ophthalmol. Sci. 2022, 2, 100121. [Google Scholar] [CrossRef]
- Montazeri, K.; Pattanayak, V.; Sullivan, R.J. Tebentafusp in the Treatment of Metastatic Uveal Melanoma: Patient Selection and Special Considerations. Drug Des. Devel. Ther. 2023, 17, 333–339. [Google Scholar] [CrossRef]
- Piaggio, F.; Tozzo, V.; Bernardi, C.; Croce, M.; Puzone, R.; Viaggi, S.; Patrone, S.; Barla, A.; Coviello, D.; Jager, M.J.; et al. Secondary Somatic Mutations in G-Protein-Related Pathways and Mutation Signatures in Uveal Melanoma. Cancers 2019, 11, 1688. [Google Scholar] [CrossRef]
- Davis, E.J.; Johnson, D.B.; Sosman, J.A.; Chandra, S. Melanoma: What Do All the Mutations Mean? Cancer 2018, 124, 3490–3499. [Google Scholar] [CrossRef]
- Johansson, P.; Aoude, L.G.; Wadt, K.; Glasson, W.J.; Warrier, S.K.; Hewitt, A.W.; Kiilgaard, J.F.; Heegaard, S.; Isaacs, T.; Franchina, M.; et al. Deep Sequencing of Uveal Melanoma Identifies a Recurrent Mutation in PLCB4. Oncotarget 2016, 7, 4624–4631. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Heppt, M.V.; Amaral, T.; Kähler, K.C.; Heinzerling, L.; Hassel, J.C.; Meissner, M.; Kreuzberg, N.; Loquai, C.; Reinhardt, L.; Utikal, J.; et al. Combined Immune Checkpoint Blockade for Metastatic Uveal Melanoma: A Retrospective, Multi-Center Study. J. Immunother. Cancer 2019, 7, 299. [Google Scholar] [CrossRef] [PubMed]
- Newell, F.; Johansson, P.A.; Wilmott, J.S.; Nones, K.; Lakis, V.; Pritchard, A.L.; Lo, S.N.; Rawson, R.V.; Kazakoff, S.H.; Colebatch, A.J.; et al. Comparative Genomics Provides Etiologic and Biological Insight into Melanoma Subtypes. Cancer Discov. 2022, 12, 2856–2879. [Google Scholar] [CrossRef]
- Onken, M.D.; Worley, L.A.; Ehlers, J.P.; Harbour, J.W. Gene Expression Profiling in Uveal Melanoma Reveals Two Molecular Classes and Predicts Metastatic Death. Cancer Res. 2004, 64, 7205–7209. [Google Scholar] [CrossRef]
- Piaggio, F.; Croce, M.; Reggiani, F.; Monti, P.; Bernardi, C.; Ambrosio, M.; Banelli, B.; Dogrusöz, M.; Jockers, R.; Bordo, D.; et al. In Uveal Melanoma Gα-Protein GNA11 Mutations Convey a Shorter Disease-Specific Survival and Are More Strongly Associated with Loss of BAP1 and Chromosomal Alterations than Gα-Protein GNAQ Mutations. Eur. J. Cancer 2022, 170, 27–41. [Google Scholar] [CrossRef]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2017, 32, 204–220.e15. [Google Scholar] [CrossRef]
- Bakhoum, M.F.; Francis, J.H.; Agustinus, A.; Earlie, E.M.; Di Bona, M.; Abramson, D.H.; Duran, M.; Masilionis, I.; Molina, E.; Shoushtari, A.N.; et al. Loss of Polycomb Repressive Complex 1 Activity and Chromosomal Instability Drive Uveal Melanoma Progression. Nat. Commun. 2021, 12, 5402. [Google Scholar] [CrossRef]
- Kaler, C.J.; Dollar, J.J.; Cruz, A.M.; Kuznetsoff, J.N.; Sanchez, M.I.; Decatur, C.L.; Licht, J.D.; Smalley, K.S.M.; Correa, Z.M.; Kurtenbach, S.; et al. BAP1 Loss Promotes Suppressive Tumor Immune Microenvironment via Upregulation of PROS1 in Class 2 Uveal Melanomas. Cancers 2022, 14, 3678. [Google Scholar] [CrossRef]
- Smit, K.N.; Jager, M.J.; De Klein, A.; Kiliҫ, E. Uveal Melanoma: Towards a Molecular Understanding. Prog. Retin. Eye Res. 2020, 75, 100800. [Google Scholar] [CrossRef] [PubMed]
- Shields, C.L.; Mayro, E.L.; Bas, Z.; Dockery, P.W.; Yaghy, A.; Lally, S.E.; Ganguly, A.; Shields, J.A. Ten-Year Outcomes of Uveal Melanoma Based on The Cancer Genome Atlas (TCGA) Classification in 1001 Cases. Indian J. Ophthalmol. 2021, 69, 1839–1845. [Google Scholar] [CrossRef] [PubMed]
- Coupland, S.; Kalirai, H.; Taktak, A.; Eleuteri, A.; Damato, B. Re: Gelmi et al.: Adding The Cancer Genome Atlas Chromosome Classes to American Joint Committee on Cancer System Offers More Precise Prognostication in Uveal Melanoma (Ophthalmology. 2022:129:431-437). Ophthalmology 2022, 129, e78–e79. [Google Scholar] [CrossRef] [PubMed]
- Damato, B.; Eleuteri, A.; Hussain, R.; Kalirai, H.; Thornton, S.; Taktak, A.; Heimann, H.; Coupland, S.E. Parsimonious Models for Predicting Mortality from Choroidal Melanoma. Investig. Ophthalmol. Vis. Sci. 2020, 61, 35. [Google Scholar] [CrossRef]
- Trolet, J.; Hupé, P.; Huon, I.; Lebigot, I.; Decraene, C.; Delattre, O.; Sastre-Garau, X.; Saule, S.; Thiéry, J.-P.; Plancher, C.; et al. Genomic Profiling and Identification of High-Risk Uveal Melanoma by Array CGH Analysis of Primary Tumors and Liver Metastases. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2572–2580. [Google Scholar] [CrossRef]
- Davoli, T.; Xu, A.W.; Mengwasser, K.E.; Sack, L.M.; Yoon, J.C.; Park, P.J.; Elledge, S.J. Cumulative Haploinsufficiency and Triplosensitivity Drive Aneuploidy Patterns and Shape the Cancer Genome. Cell 2013, 155, 948–962. [Google Scholar] [CrossRef]
- Royer-Bertrand, B.; Torsello, M.; Rimoldi, D.; El Zaoui, I.; Cisarova, K.; Pescini-Gobert, R.; Raynaud, F.; Zografos, L.; Schalenbourg, A.; Speiser, D.; et al. Comprehensive Genetic Landscape of Uveal Melanoma by Whole-Genome Sequencing. Am. J. Hum. Genet. 2016, 99, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef]
- Kutcher, R.; Sprayregen, S.; Rosenblatt, R.; Goldman, M. The Sonographic Appearance of Segmental Polycystic Kidney. J. Ultrasound Med. 1983, 2, 425–427. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.; Croce, M.; Reggiani, F.; Schinzari, G.; Ambrosio, M.; Gangemi, R.; Tortora, G.; Pfeffer, U.; Amaro, A. Uveal Melanoma Metastasis. Cancers 2021, 13, 5684. [Google Scholar] [CrossRef]
- Souri, Z.; Wierenga, A.P.A.; Mulder, A.; Jochemsen, A.G.; Jager, M.J. HLA Expression in Uveal Melanoma: An Indicator of Malignancy and a Modifiable Immunological Target. Cancers 2019, 11, 1132. [Google Scholar] [CrossRef]
- Liu-Smith, F.; Lu, Y. Opposite Roles of BAP1 in Overall Survival of Uveal Melanoma and Cutaneous Melanoma. J. Clin. Med. 2020, 9, 411. [Google Scholar] [CrossRef] [PubMed]
- van Essen, T.H.; van Pelt, S.I.; Versluis, M.; Bronkhorst, I.H.G.; van Duinen, S.G.; Marinkovic, M.; Kroes, W.G.M.; Ruivenkamp, C.A.L.; Shukla, S.; de Klein, A.; et al. Prognostic Parameters in Uveal Melanoma and Their Association with BAP1 Expression. Br. J. Ophthalmol. 2014, 98, 1738–1743. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.; Mobuchon, L.; Houy, A.; Alsafadi, S.; Baulande, S.; Mariani, O.; Marande, B.; Ait Rais, K.; Van der Kooij, M.K.; Kapiteijn, E.; et al. Evolutionary Routes in Metastatic Uveal Melanomas Depend on MBD4 Alterations. Clin. Cancer Res. 2019, 25, 5513–5524. [Google Scholar] [CrossRef]
- Uner, O.E.; See, T.R.O.; Szalai, E.; Grossniklaus, H.E.; Stålhammar, G. Author Correction: Estimation of the Timing of BAP1 Mutation in Uveal Melanoma Progression. Sci. Rep. 2021, 11, 17036. [Google Scholar] [CrossRef]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang Model of Human Colorectal Tumor Growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Ceresa, D.; Alessandrini, F.; Lucchini, S.; Marubbi, D.; Piaggio, F.; Mena Vera, J.M.; Ceccherini, I.; Reverberi, D.; Appolloni, I.; Malatesta, P. Early Clonal Extinction in Glioblastoma Progression Revealed by Genetic Barcoding. Cancer Cell 2023, 41, 1466–1479.e9. [Google Scholar] [CrossRef] [PubMed]
- Yavuzyigitoglu, S.; Koopmans, A.E.; Verdijk, R.M.; Vaarwater, J.; Eussen, B.; van Bodegom, A.; Paridaens, D.; Kiliç, E.; de Klein, A.; Rotterdam Ocular Melanoma Study Group. Uveal Melanomas with SF3B1 Mutations: A Distinct Subclass Associated with Late-Onset Metastases. Ophthalmology 2016, 123, 1118–1128. [Google Scholar] [CrossRef]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent Somatic Mutations of GNAQ in Uveal Melanoma and Blue Naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in Uveal Melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Amaro, A.; Pfeffer, M.; Pfeffer, U.; Reggiani, F. Evaluation and Comparison of Multi-Omics Data Integration Methods for Subtyping of Cutaneous Melanoma. Biomedicines 2022, 10, 3240. [Google Scholar] [CrossRef]
- Kulski, J.K. Long Noncoding RNA HCP5, a Hybrid HLA Class I Endogenous Retroviral Gene: Structure, Expression, and Disease Associations. Cells 2019, 8, 480. [Google Scholar] [CrossRef]
- Hu, S.; Ge, M.; Gao, L.; Jiang, M.; Hu, K. LncRNA HCP5 as a Potential Therapeutic Target and Prognostic Biomarker for Various Cancers: A Meta-analysis and Bioinformatics Analysis. Cancer Cell Int. 2021, 21, 686. [Google Scholar] [CrossRef]
- Lei, S.; Zhang, Y. Integrative Analysis Identifies Key Genes Related to Metastasis and a Robust Gene-Based Prognostic Signature in Uveal Melanoma. BMC Med. Genom. 2022, 15, 61. [Google Scholar] [CrossRef]
- Amaro, A.; Mirisola, V.; Angelini, G.; Musso, A.; Tosetti, F.; Esposito, A.I.; Perri, P.; Lanza, F.; Nasciuti, F.; Mosci, C.; et al. Evidence of Epidermal Growth Factor Receptor Expression in Uveal Melanoma: Inhibition of Epidermal Growth Factor-Mediated Signalling by Gefitinib and Cetuximab Triggered Antibody-Dependent Cellular Cytotoxicity. Eur. J. Cancer 2013, 49, 3353–3365. [Google Scholar] [CrossRef]
- Gangemi, R.; Mirisola, V.; Barisione, G.; Fabbi, M.; Brizzolara, A.; Lanza, F.; Mosci, C.; Salvi, S.; Gualco, M.; Truini, M.; et al. Mda-9/Syntenin Is Expressed in Uveal Melanoma and Correlates with Metastatic Progression. PLoS ONE 2012, 7, e29989. [Google Scholar] [CrossRef] [PubMed]
- Rappoport, N.; Shamir, R. NEMO: Cancer Subtyping by Integration of Partial Multi-Omic Data. Bioinformatics 2019, 35, 3348–3356. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.; Uschmajew, A.; Amaro, A.; Pfeffer, U. Data Fusion Techniques for the Integration of Multi-Domain Genomic Data from Uveal Melanoma. Cancers 2019, 11, 1434. [Google Scholar] [CrossRef]
- Vanharanta, S.; Massagué, J. Origins of Metastatic Traits. Cancer Cell 2013, 24, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, D.A.; Yang, J.; Durante, M.A.; Shoushtari, A.N.; Moschos, S.J.; Wrzeszczynski, K.O.; Harbour, J.W.; Carvajal, R.D. Multiregional Genetic Evolution of Metastatic Uveal Melanoma. NPJ Genom. Med. 2021, 6, 70. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Mirisola, V.; Pfeffer, U. Metastasis Signatures: Genes Regulating Tumor-Microenvironment Interactions Predict Metastatic Behavior. Cancer Metastasis Rev. 2008, 27, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, Y.; Chen, L.; Zhang, J. Expression Analysis of Genes and Pathways Associated with Liver Metastases of the Uveal Melanoma. BMC Med. Genet. 2014, 15, 29. [Google Scholar] [CrossRef]
- Amaro, A.; Chiara, S.; Pfeffer, U. Molecular Evolution of Colorectal Cancer: From Multistep Carcinogenesis to the Big Bang. Cancer Metastasis Rev. 2016, 35, 63–74. [Google Scholar] [CrossRef]
- Field, M.G.; Durante, M.A.; Anbunathan, H.; Cai, L.Z.; Decatur, C.L.; Bowcock, A.M.; Kurtenbach, S.; Harbour, J.W. Punctuated Evolution of Canonical Genomic Aberrations in Uveal Melanoma. Nat. Commun. 2018, 9, 116. [Google Scholar] [CrossRef]
- Stålhammar, G.; Gill, V.T. Digital Morphometry and Cluster Analysis Identifies Four Types of Melanocyte during Uveal Melanoma Progression. Commun. Med. 2023, 3, 60. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Taminau, J.; Meganck, S.; Lazar, C.; Steenhoff, D.; Coletta, A.; Molter, C.; Duque, R.; de Schaetzen, V.; Weiss Solís, D.Y.; Bersini, H.; et al. Unlocking the Potential of Publicly Available Microarray Data Using inSilicoDb and inSilicoMerging R/Bioconductor Packages. BMC Bioinform. 2012, 13, 335. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Therneau, T.M.; Grambsch, P.M. Modeling Survival Data: Extending the Cox Model; Statistics for Biology and Health; Springer: New York, NY, USA, 2000; ISBN 978-0-387-98784-2. [Google Scholar]
- Theraneau, T.M. R Package, Version 3.2-11; A Package for Survival Analysis in R; R Foundation for Statistical Computing: Vienna, Austria, 2021.
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Yan, L. Ggvenn: Draw Venn Diagram by “Ggplot2”; R Foundation for Statistical Computing: Vienna, Austria, 2023. [Google Scholar]
- Wilke, C.O. Cowplot: Streamlined Plot Theme and Plot Annotations for “Ggplot2”; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Zhao, S.; Guo, Y.; Sheng, Q.; Shyr, Y. Heatmap3: An Improved Heatmap Package with More Powerful and Convenient Features. BMC Bioinform. 2014, 15, P16. [Google Scholar] [CrossRef]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | M3/BAP1 | Metastasis | All |
|---|---|---|---|
| class 1 | 24 | 16 | 25 |
| class 2 | 0 | 2 | 18 |
| class 3 | 2 | 3 | 31 |
| class 4 | 34 | 20 | 39 |
| Patients_ID | Metastasis | M3 | 6p | 8q | BAP1 | SF3B1 | Cluster |
|---|---|---|---|---|---|---|---|
| TCGA-V4-A9EW | 1 | 0 | 1 | 0 | 0 | 1 | 2 |
| TCGA-VD-A8KA | 1 | 0 | 1 | 1 | 0 | 1 | 2 |
| MU012 | 1 | 1 | 0 | 1 | 1 | 0 | 3 |
| MU043 | 1 | 1 | 0 | 0 | 0 | 0 | 3 |
| TCGA-VD-AA8Q | 1 | 0 | 0 | 0 | 0 | 0 | 3 |
| Parameter | Category | Genoa Dataset (33) | TCGA Dataset (80) |
|---|---|---|---|
| Gender | Male/Female | 19, 14 | 45, 35 |
| Age | Years | 65 (42–83) | 62 (22–86) |
| Metastases | Yes | 15 | 26 |
| No | 18 | 54 | |
| Alive | 21 | 56 | |
| State | Dead (metastatic disease) | 8 | 20 |
| Dead (other) | 4 | 4 | |
| Follow up | Months | 2 (0–5) | 27 (0–87) |
| Chromosome 3p | loss | 18 | 42 |
| Chromosome 8q | gain | 23 | 59 |
| Chromosome 6p | gain | 8 | 45 |
| BAP1 | mutation | 9 | 35 |
| SF3B1 | mutation | 2 | 18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reggiani, F.; Ambrosio, M.; Croce, M.; Tanda, E.T.; Spagnolo, F.; Raposio, E.; Petito, M.; El Rashed, Z.; Forlani, A.; Pfeffer, U.; et al. Interdependence of Molecular Lesions That Drive Uveal Melanoma Metastasis. Int. J. Mol. Sci. 2023, 24, 15602. https://doi.org/10.3390/ijms242115602
Reggiani F, Ambrosio M, Croce M, Tanda ET, Spagnolo F, Raposio E, Petito M, El Rashed Z, Forlani A, Pfeffer U, et al. Interdependence of Molecular Lesions That Drive Uveal Melanoma Metastasis. International Journal of Molecular Sciences. 2023; 24(21):15602. https://doi.org/10.3390/ijms242115602
Chicago/Turabian StyleReggiani, Francesco, Marianna Ambrosio, Michela Croce, Enrica Teresa Tanda, Francesco Spagnolo, Edoardo Raposio, Mariangela Petito, Zeinab El Rashed, Alessandra Forlani, Ulrich Pfeffer, and et al. 2023. "Interdependence of Molecular Lesions That Drive Uveal Melanoma Metastasis" International Journal of Molecular Sciences 24, no. 21: 15602. https://doi.org/10.3390/ijms242115602