
Structural and Electromagnetic Signatures of Anatase and Rutile NTs and Sheets in Three Different Water Models under Different Temperature Conditions
 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
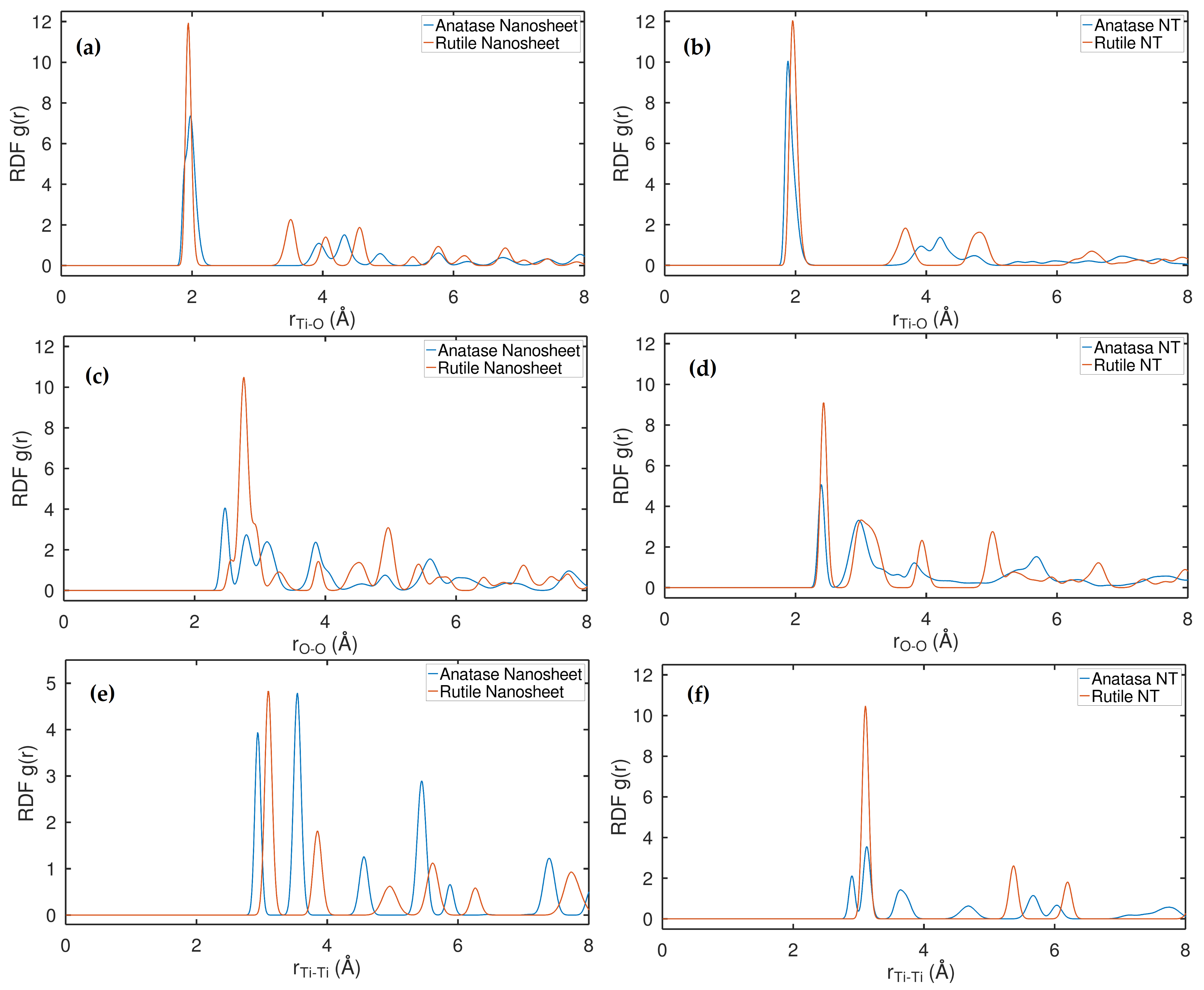
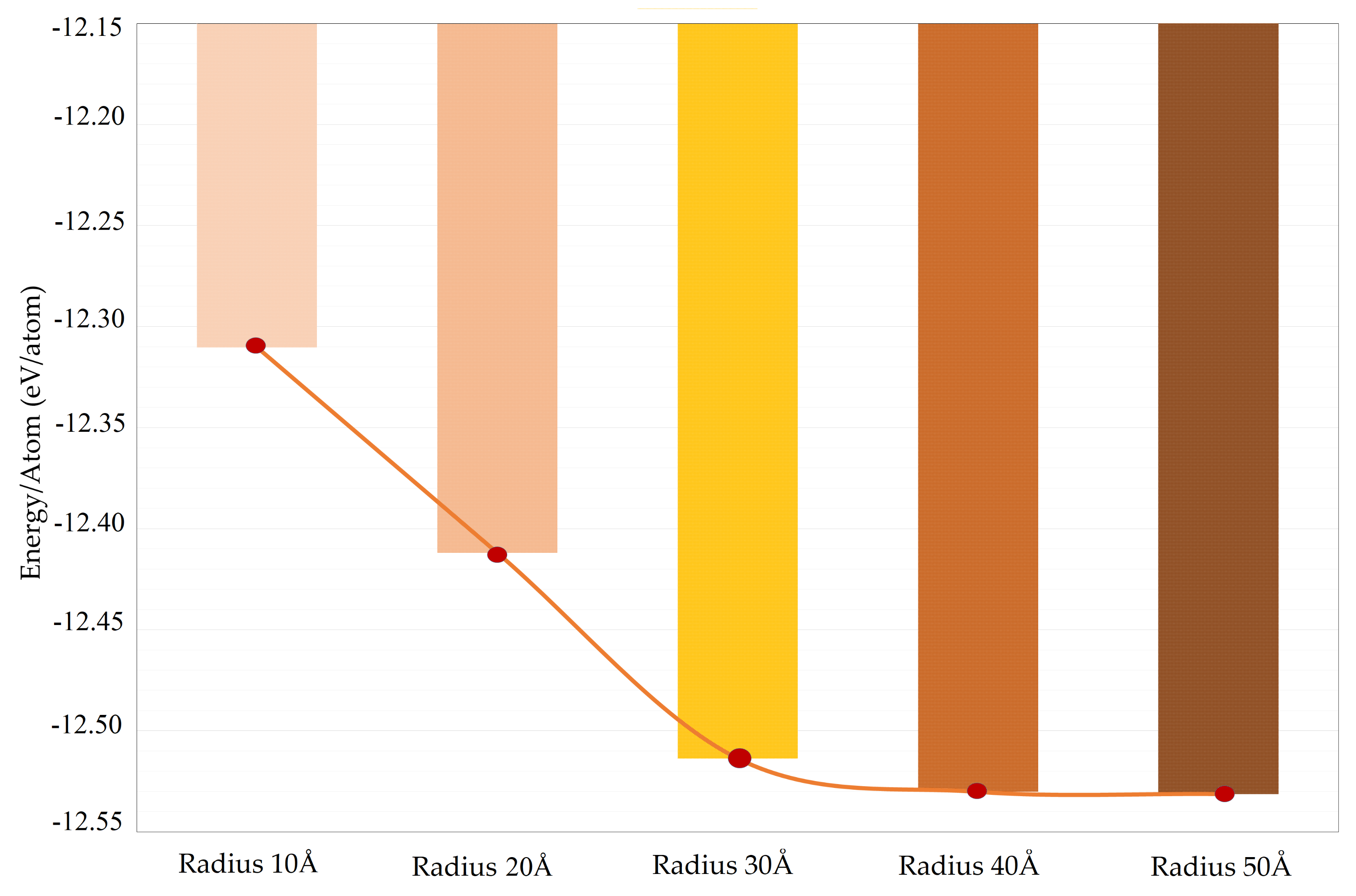
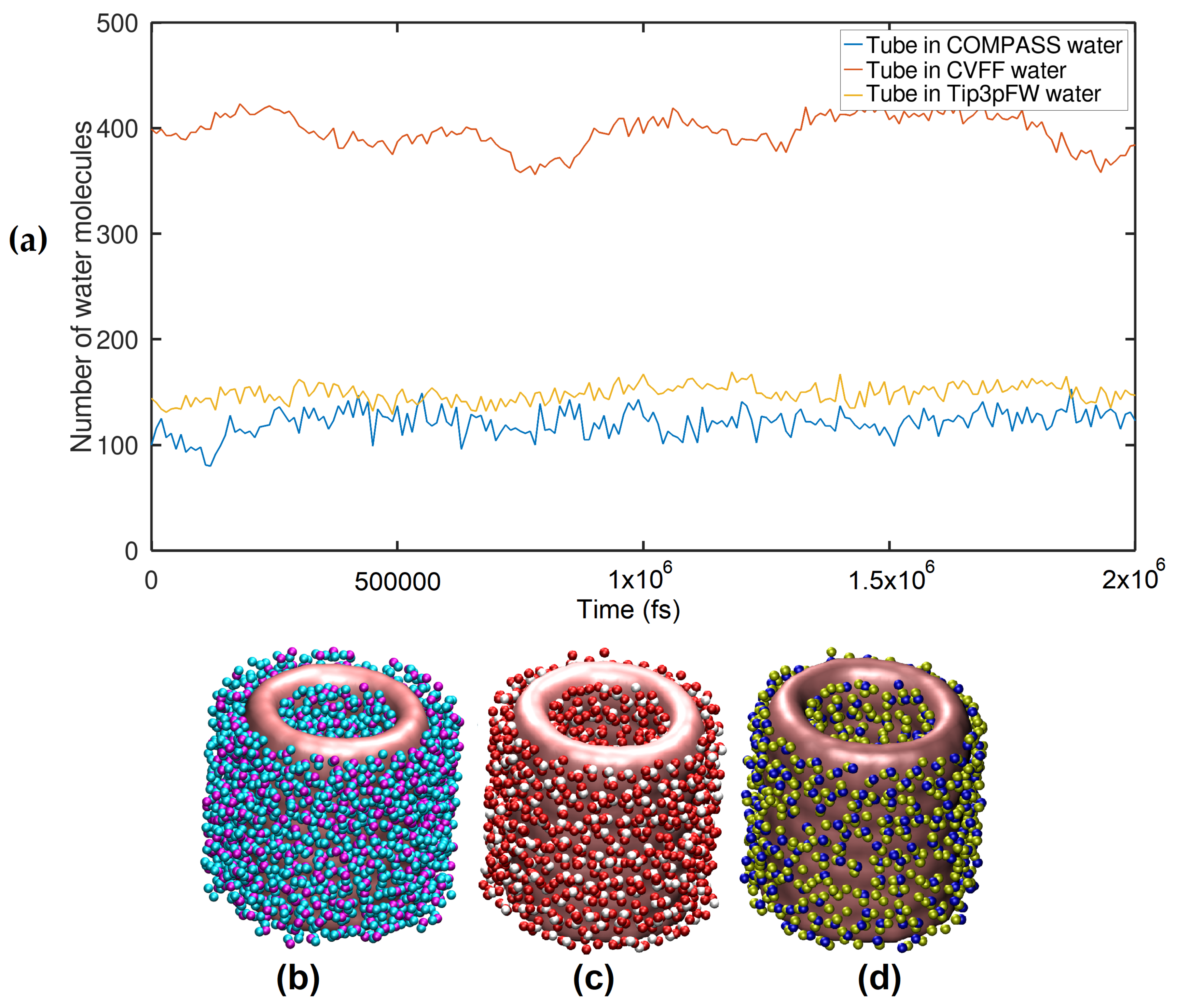
2.1. Strutural Analysis in the Aspect of RDF
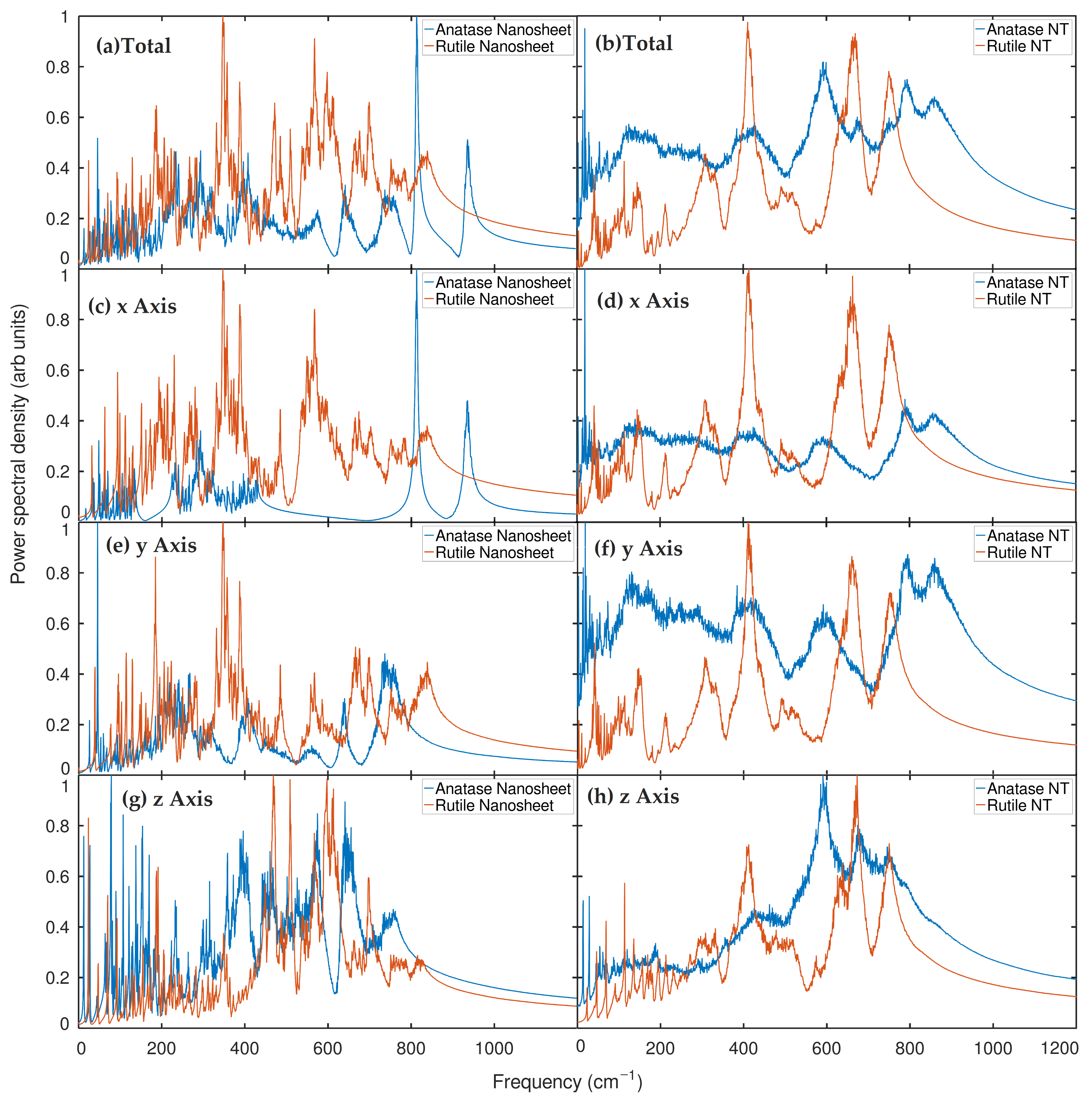
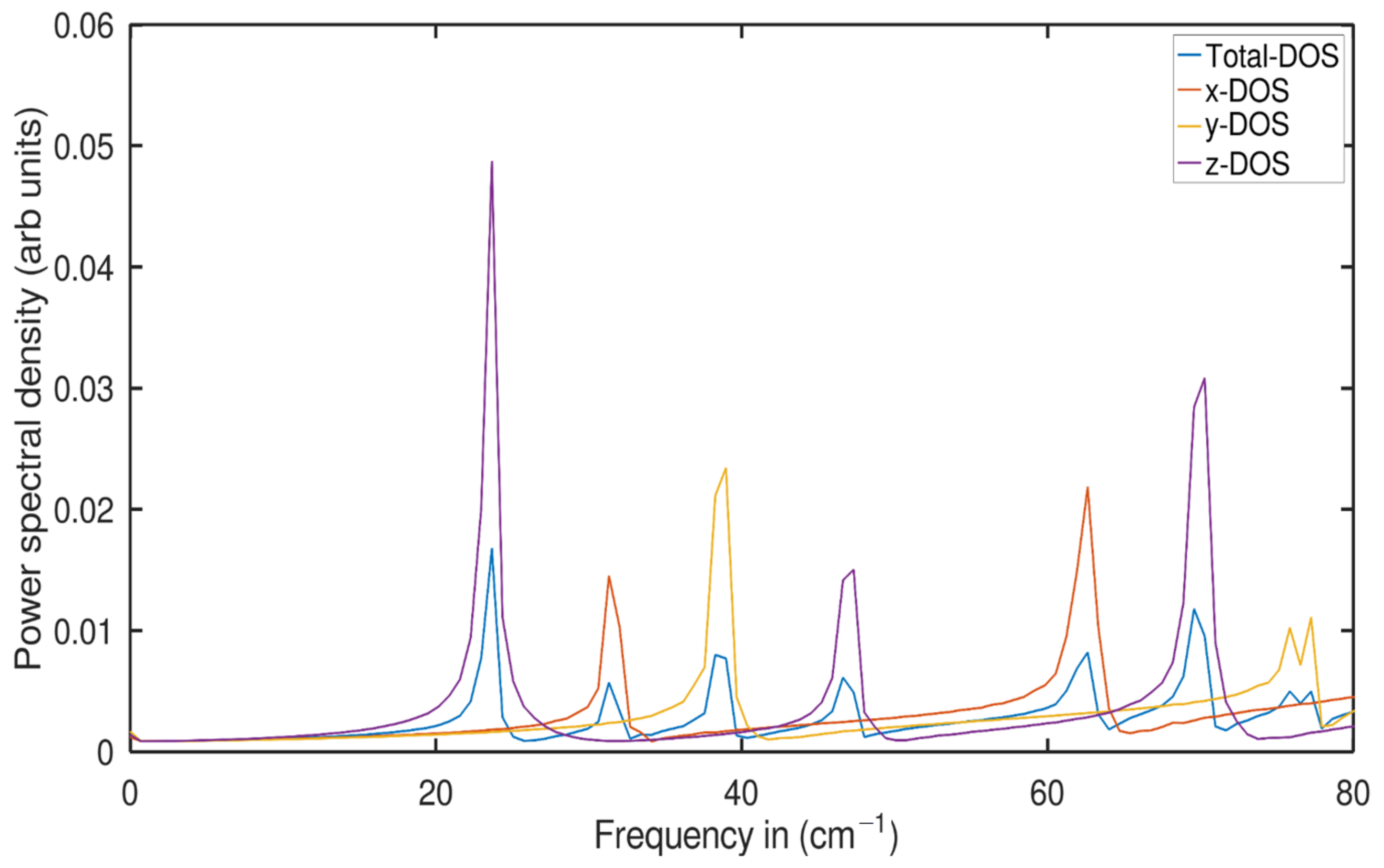
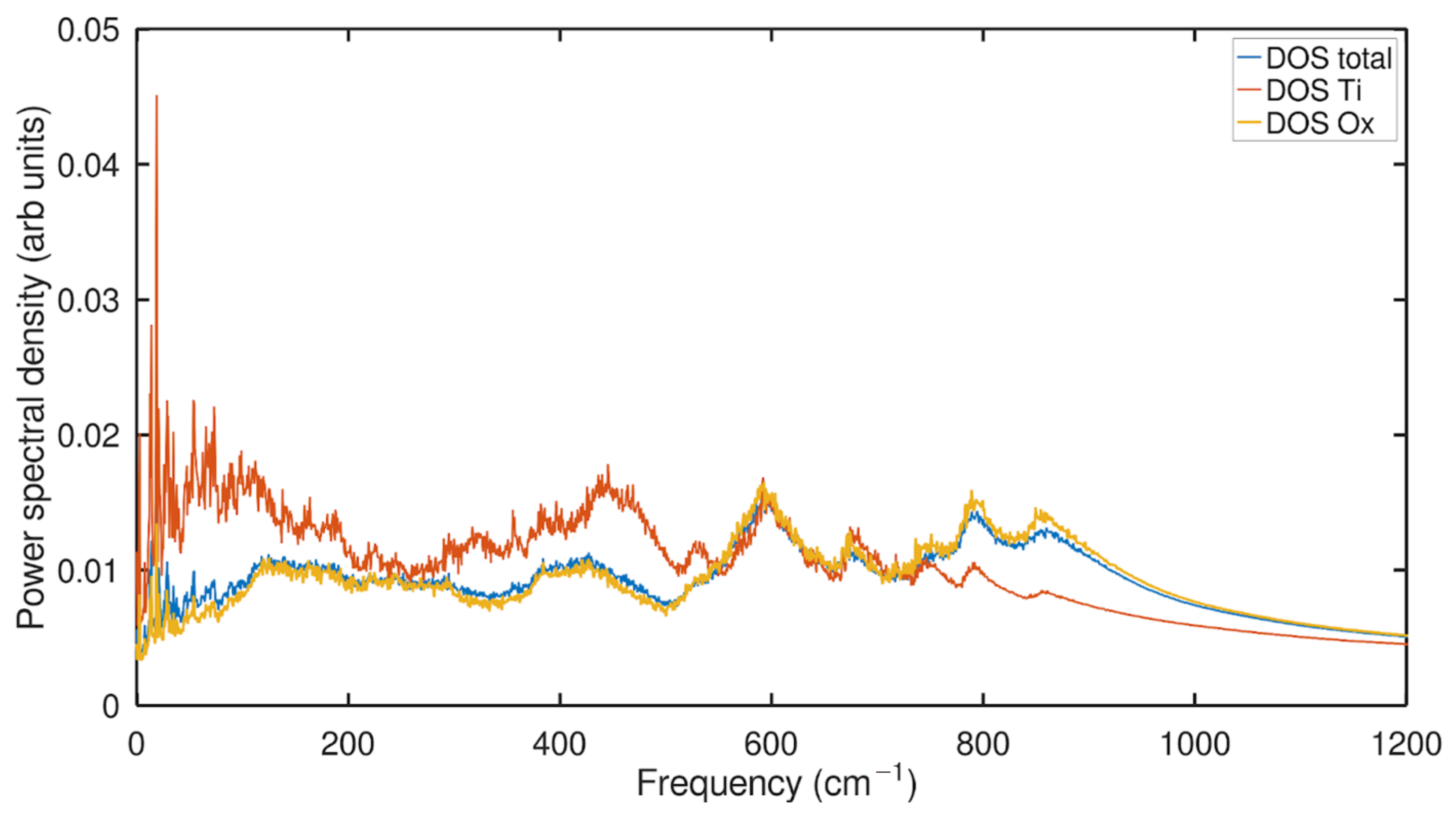
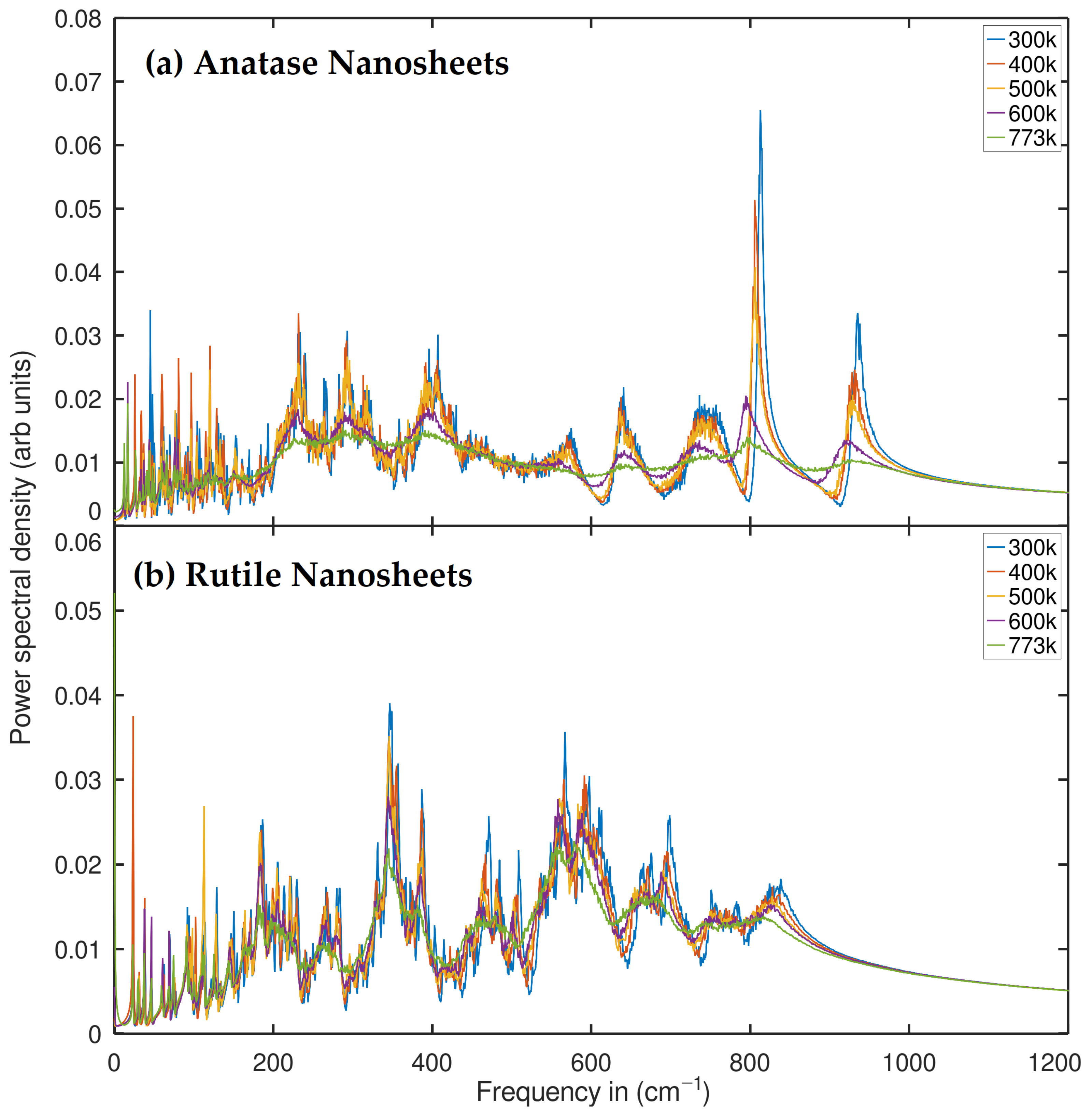
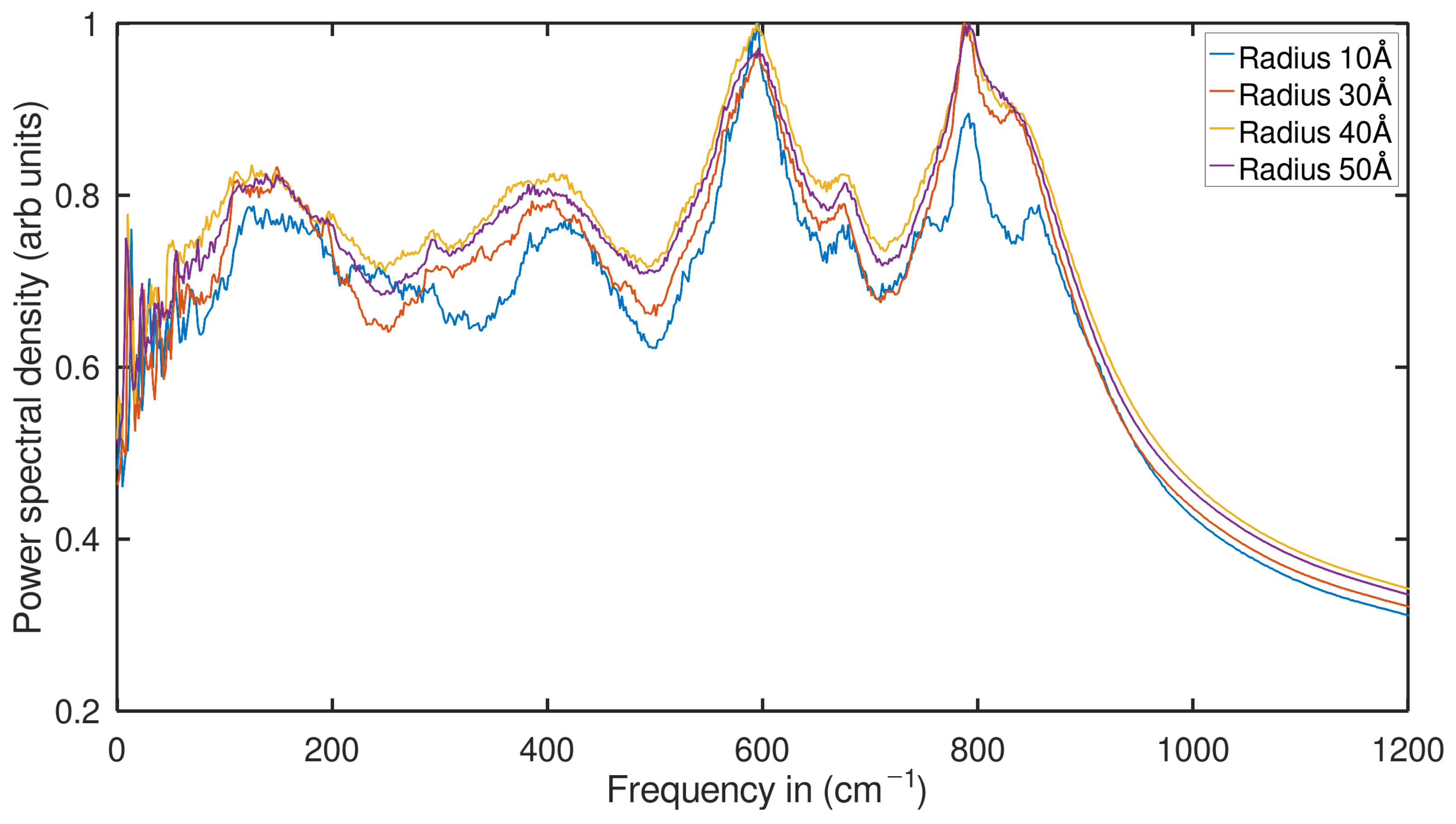
2.2. Phonon Density of States (PDOS) Analysis
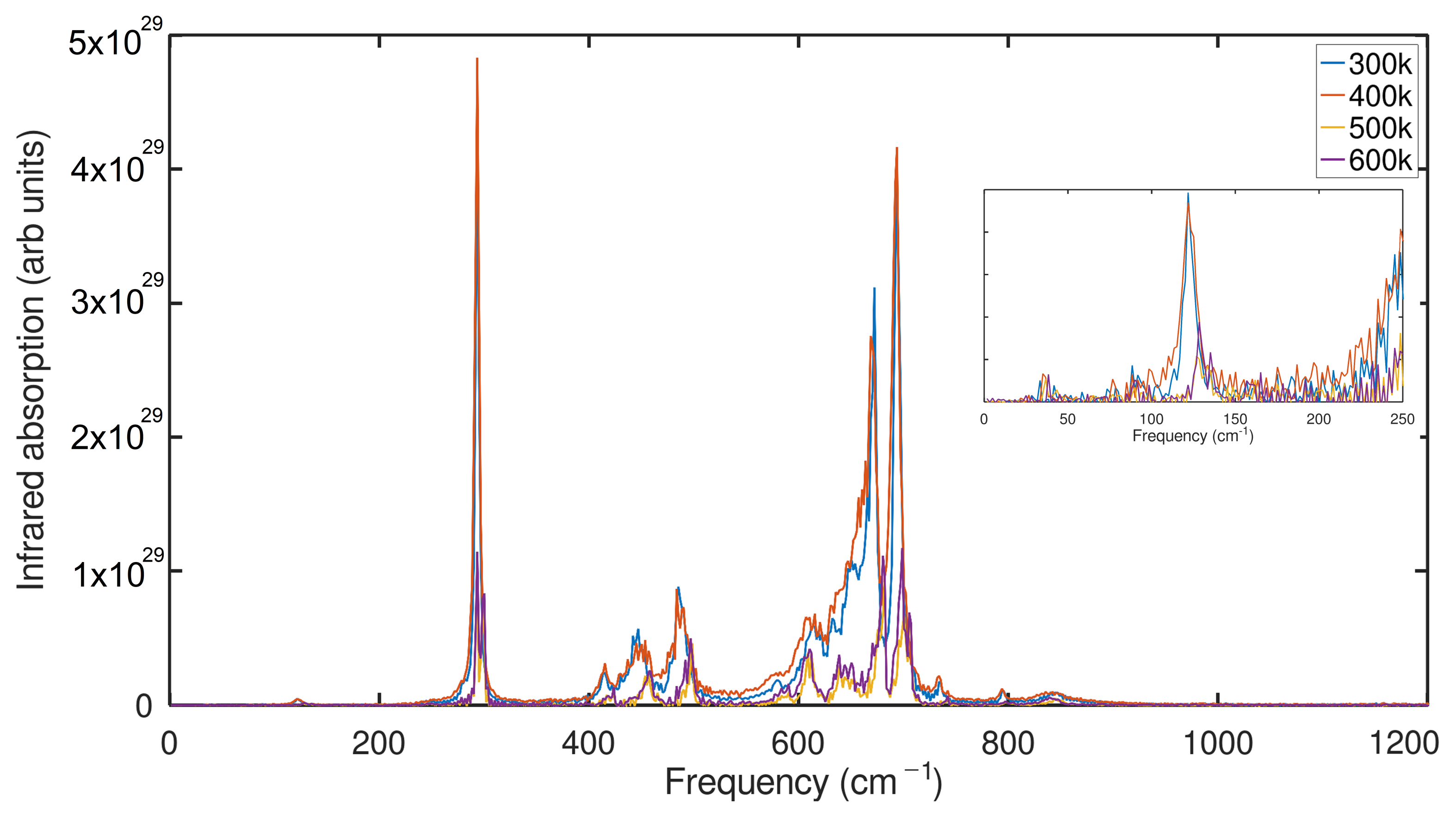
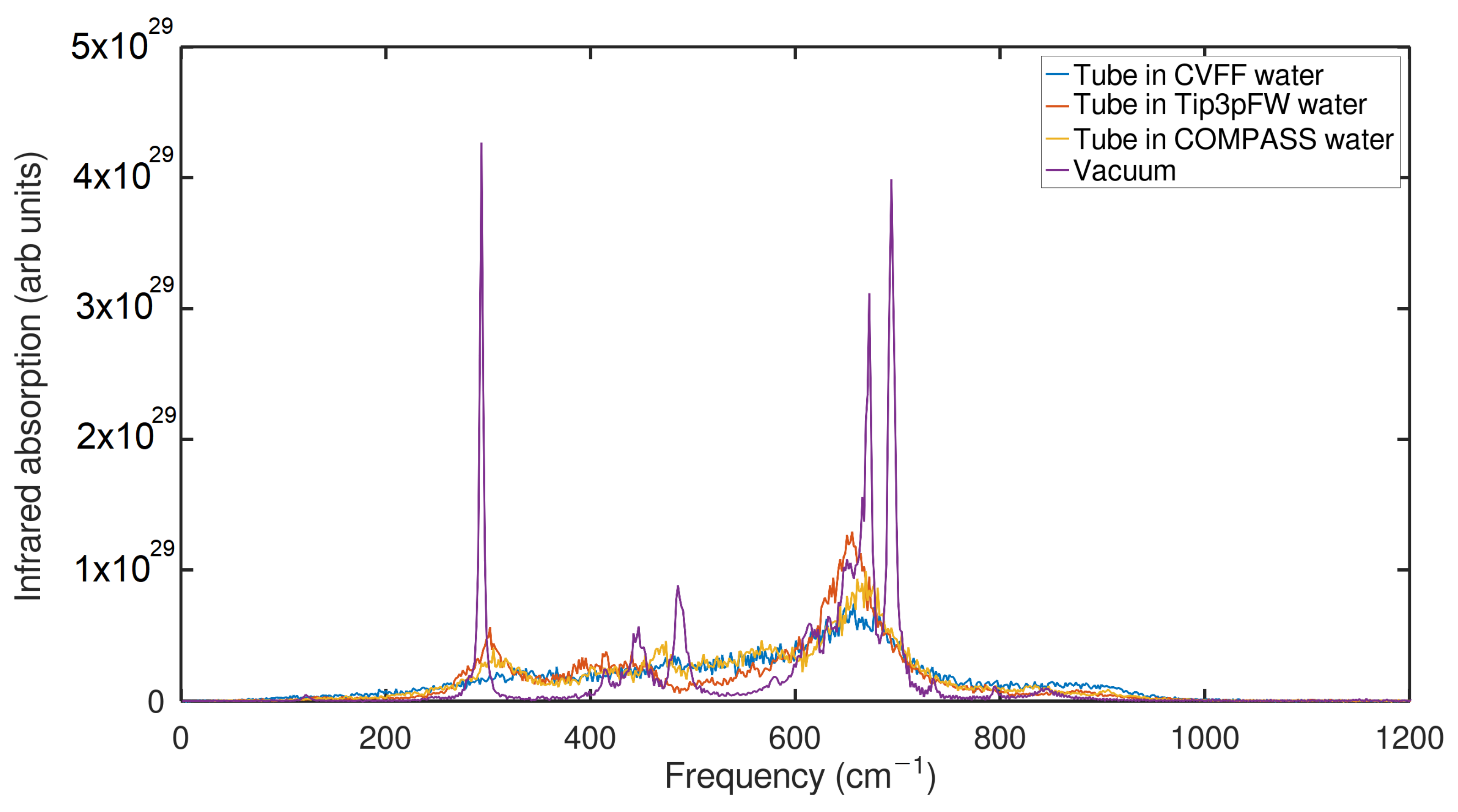
2.3. Infrared Spectrum (IR) Analysis
3. Materials and Methods
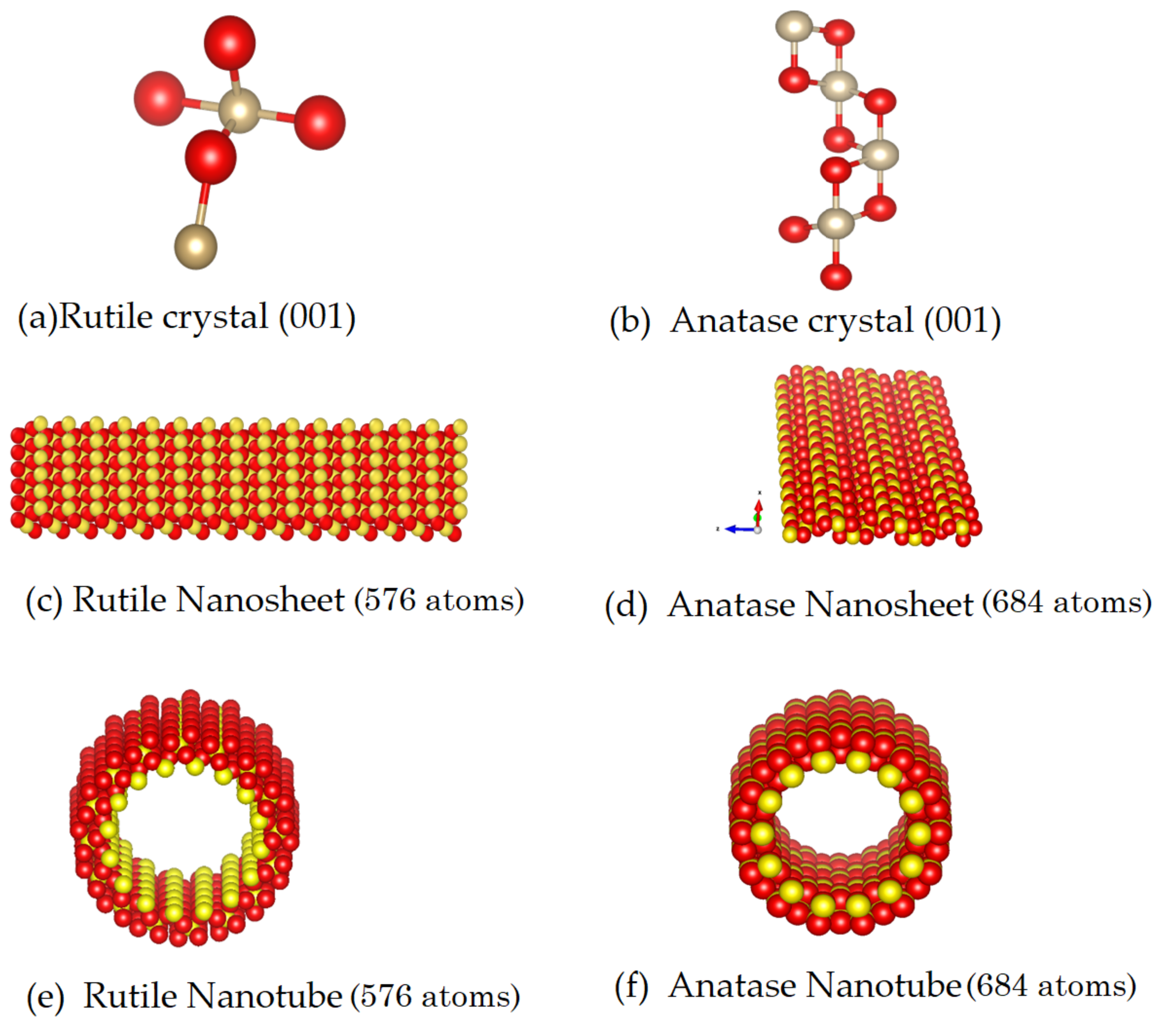
3.1. Creating the Nanotubes
3.2. Radial Distribution Function (RDF)
3.3. Phonon Density of States (PDOS)
3.4. Infrared Spectrum (IR)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, X.; Mao, S. Titanium dioxide nanomaterials: Synthesis, properties, modifications, and applications. Chem. Rev. 2007, 107, 2891–2959. [Google Scholar] [CrossRef] [PubMed]
- Almomani, M.; Ahmed, N.; Rashid, M.; Ibnaouf, K.; Aldaghri, O.; Madkhali, N.; Cabrera, H. Performance improvement of graded bandgap solar cell via optimization of energy levels alignment in Si quantum dot, TiO2 nanoparticles, and porous Si. Photonics 2022, 9, 843. [Google Scholar] [CrossRef]
- Moma, J.; Baloyi, J. Modified titanium dioxide for photocatalytic applications. In Photocatalysts: Applications and Attributes; BoD—Books on Demand: Norderstedt, Germany, 2019; Volume 18. [Google Scholar]
- Nunzi, F.; De Angelis, F. Modeling titanium dioxide nanostructures for photocatalysis and photovoltaics. Chem. Sci. 2022, 13, 9485–9497. [Google Scholar] [CrossRef] [PubMed]
- Kılınç, N.; Şennik, E.; Işık, M.; Ahsen, A.; Öztürk, O.; Öztürk, Z. Fabrication and gas sensing properties of C-doped and un-doped TiO2 nanotubes. Ceram. Int. 2014, 40, 109–115. [Google Scholar] [CrossRef]
- Zhu, X.; Zheng, Y.; Chen, L.; Wu, J.; Li, S.; Xin, Y.; Su, M.; Cui, Y. Degradation of chemical warfare agents by nickel doped titanium dioxide powders: Enhanced surface activity. Arab. J. Chem. 2022, 15, 103678. [Google Scholar] [CrossRef]
- Cheng, F.; Chen, J. Storage of hydrogen and lithium in inorganic nanotubes and nanowires. J. Mater. Res. 2006, 21, 2744–2757. [Google Scholar] [CrossRef]
- Paul, S.; Rahman, M.; Sharif, S.; Kim, J.; Siddiqui, S.; Hossain, M. TiO2 as an Anode of high-performance lithium-ion batteries: A Comprehensive Review towards Practical Application. Nanomaterials 2022, 12, 2034. [Google Scholar] [CrossRef]
- Madian, M.; Eychmüller, A.; Giebeler, L. Current advances in TiO2-based nanostructure electrodes for high performance lithium ion batteries. Batteries 2018, 4, 7. [Google Scholar] [CrossRef]
- Cheng, Y.; Yang, H.; Yang, Y.; Huang, J.; Wu, K.; Chen, Z.; Wang, X.; Lin, C.; Lai, Y. Progress in TiO2 nanotube coatings for biomedical applications: A review. J. Mater. Chem. B 2018, 6, 1862–1886. [Google Scholar] [CrossRef]
- Weir, A.; Westerhoff, P.; Fabricius, L.; Hristovski, K.; Von Goetz, N. Titanium dioxide nanoparticles in food and personal care products. Environ. Sci. Technol. 2012, 46, 2242–2250. [Google Scholar] [CrossRef]
- Ziental, D.; Czarczynska-Goslinska, B.; Mlynarczyk, D.; Glowacka-Sobotta, A.; Stanisz, B.; Goslinski, T.; Sobotta, L. Titanium dioxide nanoparticles: Prospects and applications in medicine. Nanomaterials 2020, 10, 387. [Google Scholar] [CrossRef] [PubMed]
- Llave, E. Modelado computacional del comportamiento molecular en interfases y entornos nanoestructurados; Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires: Aires, Argentina, 2012. [Google Scholar]
- Born, M.; Huang, K.; Lax, M. Dynamical theory of crystal lattices. Am. J. Phys. 1955, 23, 474. [Google Scholar] [CrossRef]
- Dove, M. Introduction to Lattice Dynamics; Cambridge University Press: Cambridge, UK, 1993. [Google Scholar]
- Wei, S.; Chou, M. Ab initio calculation of force constants and full phonon dispersions. Phys. Rev. Lett. 1992, 69, 2799. [Google Scholar] [CrossRef] [PubMed]
- Kunc, K.; Dacosta, P. Real-space convergence of the force series in the lattice dynamics of germanium. Phys. Rev. B 1985, 32, 2010. [Google Scholar] [CrossRef]
- Srivastava, G.; Kunc, K. Phonon dispersion in the (110) direction: A testing ground for phenomenological models of germanium. J. Phys. C Solid State Phys. 1988, 21, 5087. [Google Scholar] [CrossRef]
- Lewis, A.; Grisafi, A.; Ceriotti, M.; Rossi, M. Learning electron densities in the condensed phase. J. Chem. Theory Comput. 2021, 17, 7203–7214. [Google Scholar] [CrossRef]
- Baroni, S.; Giannozzi, P.; Testa, A. Green’s-function approach to linear esponse in solids. Phys. Rev. Lett. 1987, 58, 1861. [Google Scholar] [CrossRef]
- Gonçalves, S.; Bonadeo, H. Vibrational densities of states from molecular-dynamics calculations. Phys. Rev. B 1992, 46, 12019. [Google Scholar] [CrossRef]
- Baroni, S.; De Gironcoli, S.; Dal Corso, A.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515. [Google Scholar] [CrossRef]
- Kong, L.; Lewis, L. Transition state theory of the preexponential factors for self-diffusion on Cu, Ag, and Ni surfaces. Phys. Rev. B 2006, 74, 073412. [Google Scholar] [CrossRef]
- Kong, L.; Lewis, L. Surface diffusion coefficients: Substrate dynamics matters. Phys. Rev. B. 2008, 77, 165422. [Google Scholar] [CrossRef]
- Mortier, W.; Van Genechten, K.; Gasteiger, J. Electronegativity equalization: Application and parametrization. J. Am. Chem. Soc. 1985, 107, 829–835. [Google Scholar] [CrossRef]
- Nistor, R.; Müser, M. Dielectric properties of solids in the regular and split-charge equilibration formalisms. Phys. Rev. B 2009, 79, 104303. [Google Scholar] [CrossRef]
- Rutledge, G.; Lacks, D.; Martoňák, R.; Binder, K. A comparison of quasi-harmonic lattice dynamics and Monte Carlo simulation of polymeric crystals using orthorhombic polyethylene. J. Chem. Phys. 1998, 108, 10274–10280. [Google Scholar] [CrossRef]
- Estévez Ruiz, E.P.; Lago, J.L.; Thirumuruganandham, S.P. Experimental Studies on TiO2. NT with Metal Dopants through Co-Precipitation, Sol–Gel, Hydrothermal Scheme and Corresponding Computational Molecular Evaluations. Materials 2023, 16, 3076. [Google Scholar] [CrossRef]
- Ogata, S.; Iyetomi, H.; Tsuruta, K.; Shimojo, F.; Nakano, A.; Vashishta, P.; Kalia, R.; Loong, C. Initial stages of sintering of TiO2 nanoparticles: Variable-charge molecular dynamics simulations. MRS Online Proc. Libr. (OPL) 2000, 634, B7-6. [Google Scholar] [CrossRef]
- Ghuman, K.; Goyal, N.; Prakash, S. Vibrational density of states of TiO2 nanoparticles. J. Non-Cryst. Solids 2013, 373, 28–33. [Google Scholar] [CrossRef]
- Fuertes, V.; Negre, C.; Oviedo, M.; Bonafé, F.; Oliva, F.; Sánchez, C. A theoretical study of the optical properties of nanostructured TiO2. J. Phys. Condens. Matter 2013, 25, 115304. [Google Scholar] [CrossRef]
- Soussi, A.; Ait Hssi, A.; Boujnah, M.; Boulkadat, L.; Abouabassi, K.; Asbayou, A.; Elfanaoui, A.; Markazi, R.; Ihlal, A.; Bouabid, K. Electronic and optical properties of TiO2 thin films: Combined experimental and theoretical study. J. Electron. Mater. 2021, 50, 4497–4510. [Google Scholar] [CrossRef]
- Qin, H.; Qin, Q.; Luo, H.; Wei, W.; Liu, L.; Li, L. Theoretical study on adsorption characteristics and environmental effects of dimetridazole on TiO2 surface. Comput. Theor. Chem. 2019, 1150, 10–17. [Google Scholar] [CrossRef]
- Rosiński, W.S. Development of the Method for Analysis of Contact Angle between Water Drolet and Surface of Materials Used in Aerospace Industry. Ph.D. Thesis, Zakład Projektowania Materiałów, Warsaw, Poland, 2017. [Google Scholar]
- Wang, D.; Luo, H.; Liu, L.; Wei, W.; Li, L. Adsorption characteristics and degradation mechanism of metronidazole on the surface of photocatalyst TiO2: A theoretical study. Appl. Surf. Sci. 2019, 478, 896–905. [Google Scholar] [CrossRef]
- Yang, C.; Olsen, T.; Lau, M.L.; Smith, K.A.; Hattar, K.; Sen, A.; Wu, Y.; Hou, D.; Narayanan, B.; Long, M.; et al. In situ ion irradiation of amorphous TiO2. nanotubes. J. Mater. Res. 2022, 37, 1144–1155. [Google Scholar] [CrossRef]
- Cromer, D.; Herrington, K. The structures of anatase and rutile. J. Am. Chem. Soc. 1955, 77, 4708–4709. [Google Scholar] [CrossRef]
- Demkowicz, M.; Argon, A. Liquidlike atomic environments act as plasticity carriers in amorphous silicon. Phys. Rev. B 2005, 72, 245205. [Google Scholar] [CrossRef]
- Kerrache, A.; Mousseau, N.; Lewis, L. Crystallization of amorphous silicon induced by mechanical shear deformations. Phys. Rev. B 2011, 84, 014110. [Google Scholar] [CrossRef]
- Hoang, V.; Zung, H.; Trong, N. Structural properties of amorphous TiO2 nanoparticles. Eur. Phys. J. D 2007, 44, 515–524. [Google Scholar] [CrossRef]
- Van Hoang, V. Structural properties of simulated liquid and amorphous TiO2. Phys. Status Solidi B 2007, 244, 1280–1287. [Google Scholar] [CrossRef]
- Treacy, M.; Borisenko, K. The local structure of amorphous silicon. Science 2012, 335, 950–953. [Google Scholar] [CrossRef]
- Oba, Y.; Tadano, T.; Akashi, R.; Tsuneyuki, S. First-principles study of phonon anharmonicity and negative thermal expansion in ScF 3. Phys. Rev. Mater. 2019, 3, 033601. [Google Scholar] [CrossRef]
- Pool, A.; Jain, S.; Barkema, G. Structural characterization of carbon nanotubes via the vibrational density of states. Carbon 2017, 118, 58–65. [Google Scholar] [CrossRef]
- Svishchev, I.; Kusalik, P.; Murashov, V. Orthorhombic quartzlike polymorph of silica: A molecular-dynamics simulation study. Phys. Rev. B 1997, 55, 721. [Google Scholar] [CrossRef]
- Cui, Q.; Ren, K.; Zheng, R.; Zhang, Q.; Yu, L.; Li, J. Tunable thermal properties of the biphenylene and the lateral heterostructure formed with graphene: A molecular dynamics investigation. Front. Phys. 2022, 10, 1222. [Google Scholar] [CrossRef]
- Ni, Y.; Chalopin, Y.; Volz, S. Calculation of inter-plane thermal resistance of few-layer graphene from equilibrium molecular dynamics simulations. J. Phys. Conf. Ser. 2012, 395, 012106. [Google Scholar] [CrossRef]
- Chakraborty, H.; Mogurampelly, S.; Yadav, V.; Waghmare, U.; Klein, M. Phonons and thermal conducting properties of borocarbonitride (BCN) nanosheets. Nanoscale 2018, 10, 22148–22154. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, P.; Guo, L.; Chen, Z.; Wu, Q.; Ding, Y.; Zheng, W.; Cao, Y. The design of TiO2 nanostructures (nanoparticle, nanotube, and nanosheet) and their photocatalytic activity. J. Phys. Chem. C 2014, 118, 12727–12733. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Q.; Chen, X.; Jena, P. Stable three-dimensional metallic carbon with interlocking hexagons. Proc. Natl. Acad. Sci. USA 2013, 110, 18809–18813. [Google Scholar] [CrossRef]
- Hermet, P.; Lignie, A.; Fraysse, G.; Armand, P.; Papet, P. Thermodynamic properties of the α-quartz-type and rutile-type GeO2 from first-principles calculations. Phys. Chem. Chem. Phys. 2013, 15, 15943–15948. [Google Scholar] [CrossRef]
- Zhang, Y.; Harris, C.; Wallenmeyer, P.; Murowchick, J.; Chen, X. Asymmetric lattice vibrational characteristics of rutile TiO2 as revealed by laser power dependent Raman spectroscopy. J. Phys. Chem. C 2013, 117, 24015–24022. [Google Scholar] [CrossRef]
- Hamden, Z.; Conceição, D.; Boufi, S.; Vieira Ferreira, L.; Bouattour, S. Structural, morphological, optical and photocatalytic properties of Y, N-doped and codoped TiO2 thin films. Materials 2017, 10, 600. [Google Scholar] [CrossRef]
- Nunzi, F.; De Angelis, F. DFT investigations of formic acid adsorption on single-wall TiO2 nanotubes: Effect of the surface curvature. J. Phys. Chem. C 2011, 115, 2179–2186. [Google Scholar] [CrossRef]
- Enyashin, A.; Seifert, G. Structure, stability and electronic properties of TiO2 nanostructures. Phys. Status Solidi B 2005, 242, 1361–1370. [Google Scholar] [CrossRef]
- Wang, J.; Wang, L.; Ma, L.; Zhao, J.; Wang, B.; Wang, G. Structures, electronic properties, and hydrogen-storage capacity of single-walled TiO2 nanotubes. Phys. E Low-Dimens. Syst. Nanostructures 2009, 41, 838–842. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, Q.; Qin, L. Reduction in the electronic band gap of titanium oxide nanotubes. Solid State Commun. 2007, 141, 168–171. [Google Scholar] [CrossRef]
- Bandura, A.; Evarestov, R. From anatase (1 0 1) surface to TiO2 nanotubes: Rolling procedure and first principles LCAO calculations. Surf. Sci. 2009, 603, L117–L120. [Google Scholar] [CrossRef]
- Hossain, F.; Evteev, A.; Belova, I.; Nowotny, J.; Murch, G. Electronic and optical properties of anatase TiO2 nanotubes. Comput. Mater. Sci. 2010, 48, 854–858. [Google Scholar] [CrossRef]
- Lin, F.; Zhou, G.; Li, Z.; Li, J.; Wu, J.; Duan, W. Molecular and atomic adsorption of hydrogen on TiO2 nanotubes: An ab initio study. Chem. Phys. Lett. 2009, 475, 82–85. [Google Scholar] [CrossRef]
- Zhao, H.; Dong, Y.; Jiang, P.; Wang, G.; Zhang, J. Highly dispersed CeO2 on TiO2 nanotube: A synergistic nanocomposite with superior peroxidase-like activity. ACS Appl. Mater. Interfaces 2015, 7, 6451–6461. [Google Scholar] [CrossRef]
- Ambroziak, R.; Hołdyński, M.; Płociński, T.; Pisarek, M.; Kudelski, A. Cubic silver nanoparticles fixed on TiO2 nanotubes as simple and efficient substrates for surface enhanced Raman scattering. Materials 2019, 12, 3373. [Google Scholar] [CrossRef]
- Cao, W.; Lu, L.; Huang, L.; Dong, Y.; Lu, X. Molecular behavior of water on titanium dioxide nanotubes: A molecular dynamics simulation study. J. Chem. Eng. Data 2016, 61, 4131–4138. [Google Scholar] [CrossRef]
- Wang, S.; Hou, K.; Heinz, H. Accurate and compatible force fields for molecular oxygen, nitrogen, and hydrogen to simulate gases, electrolytes, and heterogeneous interfaces. J. Chem. Theory Comput. 2021, 17, 5198–5213. [Google Scholar] [CrossRef]
- Matsui, M.; Akaogi, M. Molecular dynamics simulation of the structural and physical properties of the four polymorphs of TiO2. Mol. Simul. 1991, 6, 239–244. [Google Scholar] [CrossRef]
- Luan, B.; Huynh, T.; Zhou, R. Simplified TiO2 force fields for studies of its interaction with biomolecules. J. Chem. Phys. 2015, 142, 234102. [Google Scholar] [CrossRef] [PubMed]
- Zeydabadi-Nejad, I.; Zolfaghari, N.; Mosavi-Mashhadi, M.; Baniassadi, M. Exceptional behavior of anatase TiO2 nanotubes in axial loading: A molecular dynamics study of the effect of surface wrinkles. Comput. Mater. Sci. 2019, 158, 307–314. [Google Scholar] [CrossRef]
- Lim, T. Alignment of Buckingham parameters to generalized Lennard-Jones potential functions. Z. Naturforschung A 2009, 64, 200–204. [Google Scholar] [CrossRef]
- Laguna, M.; Jagla, E. Classical isotropic two-body potentials generating martensitic transformations. J. Stat. Mech. Theory Exp. 2009, 2009, P09002. [Google Scholar] [CrossRef]
- Laguna, M. Testing conditions for reversibility of martensitic transformations with an isotropic potential for identical particles. Phys. Status Solidi B 2015, 252, 538–544. [Google Scholar] [CrossRef]
- Zhang, Y.; Farsinezhad, S.; Wiltshire, B.; Kisslinger, R.; Kar, P.; Shankar, K. Optical anisotropy in vertically oriented TiO2 nanotube arrays. Nanotechnology 2017, 28, 374001. [Google Scholar] [CrossRef]
- Reinhardt, A. Phase behavior of empirical potentials of titanium dioxide. J. Chem. Phys. 2019, 151, 064505. [Google Scholar] [CrossRef]
- Yang, L.; Wang, C.; Lin, S.; Chen, T.; Cao, Y.; Zhang, P.; Liu, X. Thermal conductivity of TiO2 nanotube: A molecular dynamics study. J. Phys. Condens. Matter 2018, 31, 055302. [Google Scholar] [CrossRef]
- Yang, K.; Yang, L.; Ai, C.; Wang, Z.; Lin, S. Elastic properties of anatase titanium dioxide nanotubes: A molecular dynamics study. Chin. Phys. B 2019, 28, 103102. [Google Scholar] [CrossRef]
- Mitev, P.; Hermansson, K. Surface properties of rutile TiO2 (1 1 0) from molecular dynamics and lattice dynamics at 300 K: Variable-charge model results. Surf. Sci. 2007, 601, 5359–5367. [Google Scholar] [CrossRef]
- Allen, M.; Tildesley, D. Computer Simulation of Liquids; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Thompson, A.; Aktulga, H.; Berger, R.; Bolintineanu, D.; Brown, W.; Crozier, P.; Veld, P.; Kohlmeyer, A.; Moore, S.; Nguyen, T.; et al. LAMMPS—A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comp. Phys. Comm. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Brostow, W. Radial distribution function peaks and coordination numbers in liquids and in amorphous solids. Chem. Phys. Lett. 1977, 49, 285–288. [Google Scholar] [CrossRef]
- Hansen, J.; McDonald, I. Theory of Simple Liquids: With Applications to Soft Matter; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Thirumuruganandham, S.; Urbassek, H. Low-frequency vibrational modes and infrared absorbance of red, blue and green opsin. J. Mol. Model. 2009, 15, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Leach, A.; Leach, A. Molecular Modelling: Principles and Applications; Pearson Education: Hoboken, NJ, USA, 2001. [Google Scholar]
- Guillot, B. A molecular dynamics study of the far infrared spectrum of liquid water. J. Chem. Phys. 1991, 95, 1543–1551. [Google Scholar] [CrossRef]
- Boulard, B.; Kieffer, J.; Phifer, C.; Angell, C. Vibrational spectra in fluoride crystals and glasses at normal and high pressures by computer simulation. J. Non-Cryst. Solids 1992, 140, 350–358. [Google Scholar] [CrossRef]
- Martinez-Gonzalez, J.; Nandi, P.; English, N.; Gowen, A. Infrared spectra and density of states at the interface between water and protein: Insights from classical molecular dynamics. Chem. Phys. Lett. 2020, 757, 137867. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interaction | (Kcal/mol) | (Å) | (Kcal/mol)/Å |
|---|---|---|---|
| Ti-Ti | 717,653.9571 | 0.154 | 120.9967 |
| Ti-O | 391,052.7442 | 0.194 | 290.3920 |
| O-O | 271,718.8311 | 0.234 | 696.9407 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz, E.P.E.; Thirumuruganandham, S.P.; Lago, J.C.L. Structural and Electromagnetic Signatures of Anatase and Rutile NTs and Sheets in Three Different Water Models under Different Temperature Conditions. Int. J. Mol. Sci. 2023, 24, 14878. https://doi.org/10.3390/ijms241914878
Ruiz EPE, Thirumuruganandham SP, Lago JCL. Structural and Electromagnetic Signatures of Anatase and Rutile NTs and Sheets in Three Different Water Models under Different Temperature Conditions. International Journal of Molecular Sciences. 2023; 24(19):14878. https://doi.org/10.3390/ijms241914878
Chicago/Turabian StyleRuiz, Eduardo Patricio Estévez, Saravana Prakash Thirumuruganandham, and Joaquín Cayetano López Lago. 2023. "Structural and Electromagnetic Signatures of Anatase and Rutile NTs and Sheets in Three Different Water Models under Different Temperature Conditions" International Journal of Molecular Sciences 24, no. 19: 14878. https://doi.org/10.3390/ijms241914878