
Post-Ischemic Permeability of the Blood–Brain Barrier to Amyloid and Platelets as a Factor in the Maturation of Alzheimer’s Disease-Type Brain Neurodegeneration

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Search Strategy and Selection Criteria
3. Structure and Functions of the BBB
4. BBB Permeability Post-Ischemia
4.1. Permeability of Non-Cellular Blood Elements through the Ischemic BBB in the Gray Matter
4.2. Permeability of Non-Cellular Blood Elements through the Ischemic BBB in the White Matter
4.3. Permeability of Cellular Blood Elements through the Ischemic BBB in the Gray Matter
4.4. Permeability of Cellular Blood Elements through the Ischemic BBB in the White Matter
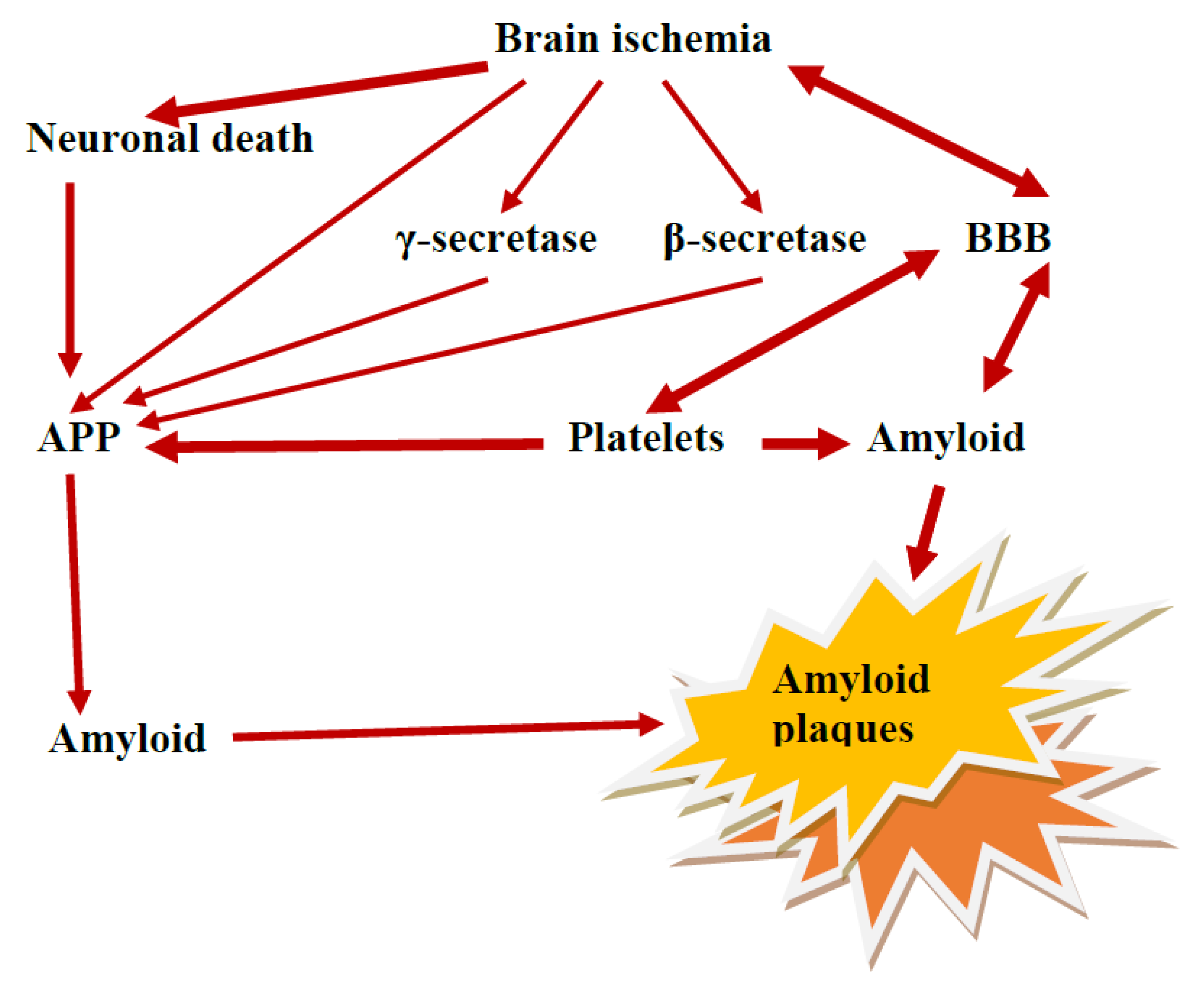
5. Platelets as Source of Amyloid Protein Precursor and Amyloid
6. Amyloid and BBB Vascular Angiogenesis
7. Amyloid, Platelets, and Post-Ischemic Vasoconstriction
8. Cerebral Amyloid Angiopathy Post-Ischemia
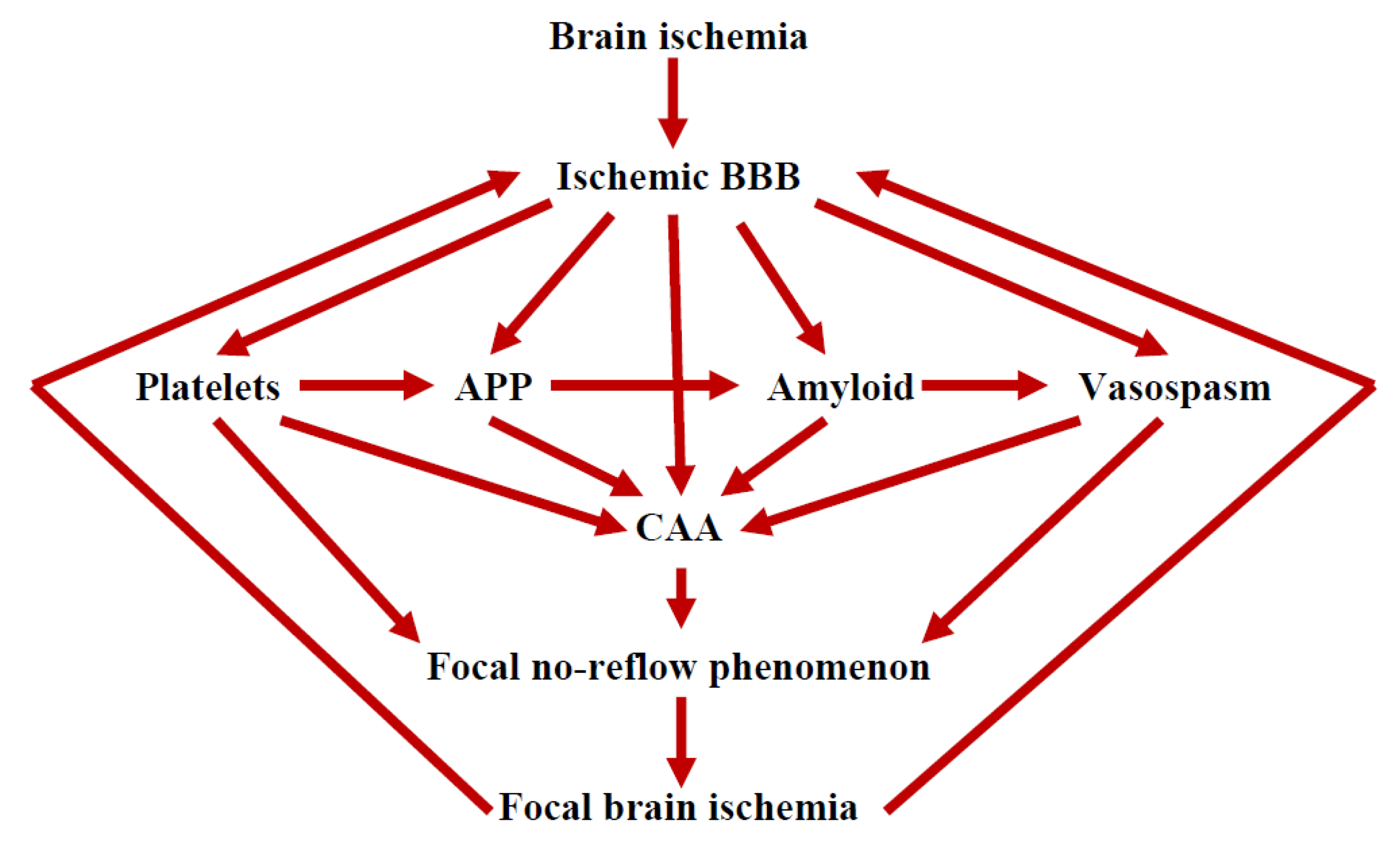
9. The Role of the Ischemic BBB in the Maturation of AD
10. Conclusions
11. Current Message from the Lab for the Clinic
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Biron, K.E.; Dickstein, D.L.; Gopaul, R.; Jefferies, W.A. Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in Alzheimer’s disease. PLoS ONE 2011, 6, e23789. [Google Scholar] [CrossRef] [Green Version]
- Pluta, R.; Lossinsky, A.S.; Wisniewski, H.M.; Mossakowski, M.J. Early blood-brain barrier changes in the rat following transient complete cerebral ischemia induced by cardiac arrest. Brain Res. 1994, 633, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Kida, E.; Lossinsky, A.S.; Golabek, A.A.; Mossakowski, M.J.; Wisniewski, H.M. Complete cerebral ischemia with short-term survival in rats induced by cardiac arrest. I. Extracellular accumulation of Alzheimer’s β-amyloid protein precursor in the brain. Brain Res. 1994, 649, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Lossinsky, A.S.; Walski, M.; Wisniewski, H.M.; Mossakowski, M.J. Platelet occlusion phenomenon after short- and long-term survival following complete cerebral ischemia in rats produced by cardiac arrest. J. Brain Res. 1994, 35, 463–471. [Google Scholar]
- Wardlow, J.M.; Sandercock, P.A.; Dennis, M.S.; Starr, J. Is breakdown of the blood-brain barrier responsible for lacunar stroke; leukoaraiosis; and dementia? Stroke 2003, 34, 806–812. [Google Scholar] [CrossRef]
- Goulay, R.; Mena Romo, L.; Hol, E.M.; Dijkhuizen, R.M. From stroke to dementia: A Comprehensive review exposing tight interactions between stroke and amyloid-β formation. Transl. Stroke Res. 2020, 11, 601–614. [Google Scholar] [CrossRef] [Green Version]
- Gürler, G.; Soylu, K.O.; Yemisci, M. Importance of pericytes in the pathophysiology of cerebral ischemia. Noro Psikiyatr. Ars. 2022, 59 (Suppl. 1), S29–S35. [Google Scholar]
- Han, W.; Song, Y.; Rocha, M.; Shi, Y. Ischemic brain edema: Emerging cellular mechanisms and therapeutic approaches. Neurobiol. Dis. 2023, 178, 106029. [Google Scholar] [CrossRef]
- Blennow, K.; Wallin, A.; Fredman, P.; Karlsson, I.; Gottfries, C.G.; Svennerholm, L. Blood-brain barrier disturbance in patients with Alzheimer’s disease is related to vascular factors. Acta Neurol. Scand. 1990, 81, 323–326. [Google Scholar] [CrossRef]
- Zipser, B.D.; Johanson, C.E.; Gonzalez, L.; Berzin, T.M.; Tavares, R.; Hultte, C.M.; Vitek, M.P.; Hovanesian, V.; Stopa, E.G. Microvascular injury and blood-brain barrier leakage in Alzheimer’s disease. Neurobiol. Aging 2007, 28, 977–986. [Google Scholar] [CrossRef]
- Liesz, A. The vascular side of Alzheimer’s disease. Science 2019, 365, 223–224. [Google Scholar] [CrossRef]
- Fisher, R.A.; Miners, J.S.; Love, S. Pathological changes within the cerebral vasculature in Alzheimer’s disease: New perspectives. Brain Pathol. 2022, 32, e13061. [Google Scholar] [CrossRef] [PubMed]
- Kurz, C.; Walker, L.; Rauchmann, B.S.; Perneczky, R. Dysfunction of the blood-brain barrier in Alzheimer’s disease: Evidence from human studies. Neuropathol. Appl. Neurobiol. 2022, 48, e12782. [Google Scholar] [CrossRef] [PubMed]
- Eisenmenger, L.B.; Peret, A.; Famakin, B.M.; Spahic, A.; Roberts, G.S.; Bockholt, J.H.; Johnson, K.M.; Paulsen, J.S. Vascular contributions to Alzheimer’s disease. Transl. Res. 2023, 254, 41–53. [Google Scholar] [CrossRef]
- Sousa, J.A.; Bernardes, C.; Bernardo-Castro, S.; Lino, M.; Albino, I.; Ferreira, L.; Brás, J.; Guerreiro, R.; Tábuas-Pereira, M.; Baldeiras, I.; et al. Reconsidering the role of blood-brain barrier in Alzheimer’s disease: From delivery to target. Front. Aging Neurosci. 2023, 15, 1102809. [Google Scholar] [CrossRef] [PubMed]
- Ujiie, M.; Dickstein, D.L.; Carlow, D.A.; Jefferies, W.A. Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation 2003, 10, 463–470. [Google Scholar] [PubMed]
- Ferrer, I.; Boada Rovira, M.; Sanchez Guerra, M.L.; Rey, M.J.; Costa-Jussa, F. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer’s disease. Brain Pathol. 2004, 14, 11–20. [Google Scholar] [CrossRef]
- Ramos-Cejudo, J.; Wisniewski, T.; Marmar, C.; Zetterberg, H.; Blennow, K.; de Leon, M.J.; Fossati, S. Traumatic brain injury and Alzheimer’s disease: The cerebrovascular link. EBioMedicine 2018, 28, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Michalicova, A.; Majerova, P.; Kovac, A. Tau protein and its role in blood-brain barrier dysfunction. Front. Mol. Neurosci. 2020, 13, 570045. [Google Scholar] [CrossRef]
- Pluta, R.; Czuczwar, S.J.; Januszewski, S.; Jabłoński, M. The many faces of post-ischemic tau protein in brain neurodegeneration of the Alzheimer’s disease type. Cells 2021, 10, 2213. [Google Scholar] [CrossRef]
- Pluta, R.; Barcikowska, M.; Januszewski, S.; Misicka, A.; Lipkowski, A.W. Evidence of blood-brain barrier permeability/leakage for circulating human Alzheimer’s β-amyloid- (1-42)-peptide. NeuroReport 1996, 7, 1261–1265. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Misicka, A.; Januszewski, J.; Barcikowska, M.; Lipkowski, A.W. Transport of human β-amyloid peptide through the rat blood-brain barrier after global cerebral ischemia. Acta Neurochir. Suppl. 1997, 70, 247–249. [Google Scholar] [PubMed]
- Pluta, R.; Barcikowska, M.; Misicka, A.; Januszewski, S.; Lipkowski, A.W. Disappearing diffuse amyloid plaques. Neurobiol. Aging 1998, 19, S131. [Google Scholar]
- Pluta, R.; Barcikowska, M.; Misicka, A.; Lipkowski, A.W.; Spisacka, S.; Januszewski, S. Ischemic rats as a model in the study of the neurobiological role of human β-amyloid peptide. Time-dependent disappearing diffuse amyloid plaques in brain. NeuroReport 1999, 10, 3615–3619. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R. The role of apolipoprotein E in the deposition of β-amyloid peptide during ischemia-reperfusion brain injury. A model of early Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2000, 903, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R. Blood-brain barrier dysfunction and amyloid precursor protein accumulation in microvascular compartment following ischemia-reperfusion brain injury with 1-year survival. Acta Neurochir. Suppl. 2003, 86, 117–122. [Google Scholar]
- Pluta, R.; Ułamek, M.; Łuczyk, W.; Hodun, R.; Niczyporuk, P.; Smyrgała, B.; Januszewski, S. Chronic blood-brain barrier opening following ischemia-reperfusion brain injury with 1-year survival. J. Cereb. Blood Flow Metab. 2003, 23 (Suppl. 1), 165. [Google Scholar]
- Pluta, R. Pathological opening of the blood-brain barrier to horseradish peroxidase and amyloid precursor protein following ischemia-reperfusion brain injury. Chemotherapy 2005, 51, 223–226. [Google Scholar] [CrossRef]
- Pluta, R.; Januszewski, S.; Ułamek, M. Chronic blood-brain barrier insufficiency and cytotoxic fragment of amyloid precursor protein activity in white matter following ischemia-reperfusion brain injury in long-lived animals. J. Cereb. Blood Flow Metab. 2005, 25, S251. [Google Scholar] [CrossRef]
- Pluta, R.; Ułamek, M.; Januszewski, S. Micro-blood-brain barrier openings and cytotoxic fragments of amyloid precursor protein accumulation in white matter after ischemic brain injury in long-lived rats. Acta Neurochir. Suppl. 2006, 96, 267–271. [Google Scholar]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Hypoxia/ischemia activate processing of amyloid precursor protein: Impact of vascular dysfunction in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2017, 140, 536–549. [Google Scholar] [CrossRef] [Green Version]
- Pluta, R.; Januszewski, S.; Czuczwar, S.J. Brain ischemia as a prelude to Alzheimer’s disease. Front Aging Neurosci. 2021, 13, 636653. [Google Scholar] [CrossRef]
- Elman-Shina, K.; Efrati, S. Ischemia as a common trigger for Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 1012779. [Google Scholar] [CrossRef]
- Pluta, R. Alzheimer’s disease connected genes in the post-ischemic hippocampus and temporal cortex. Genes 2022, 13, 1059. [Google Scholar] [CrossRef]
- Pluta, R. Brain ischemia as a bridge to Alzheimer’s disease. Neural. Regen. Res. 2022, 17, 791–792. [Google Scholar] [CrossRef]
- Dickstein, D.L.; Biron, K.E.; Ujiie, M.; Pfeifer, C.G.; Jeffries, A.R.; Jefferies, W.A. Aβ peptide immunization restores blood-brain barrier integrity in Alzheimer disease. FASEB J. 2006, 20, 426–433. [Google Scholar] [CrossRef]
- Kawai, M.; Kalaria, R.N.; Harik, S.I.; Perry, G. The relationship of amyloid plaques to cerebral capillaries in Alzheimer’s disease. Am. J. Pathol. 1990, 137, 1435–1446. [Google Scholar]
- Leybaert, L. Neurobarrier coupling in the brain: A partner of neurovascular and neurometabolic coupling? J. Cereb. Blood Flow Metab. 2005, 25, 2–16. [Google Scholar] [CrossRef] [Green Version]
- Reese, T.S.; Karnovsky, M.J. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J. Cell Biol. 1967, 34, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Grieb, P.; Forster, R.E.; Strome, D.; Goodwin, C.W.; Pape, P.C. O2 exchange between blood and brain tissues studied with 18O2 indicator-dilution technique. J. Appl. Physiol. 1985, 58, 1929–1941. [Google Scholar] [CrossRef]
- Zhang, Y.; Pardridge, W.M. Rapid transferrin efflux from brain to blood across the blood-brain barrier. J. Neurochem. 2001, 76, 1597–1600. [Google Scholar] [CrossRef]
- Pluta, R.; Salinska, E.; Puka, M.; Stafiej, A.; Lazarewicz, J.W. Early changes in extracellular amino acids and calcium concentrations in rabbit hippocampus following complete 15-min cerebral ischemia. Resuscitation 1988, 16, 193–210. [Google Scholar] [CrossRef]
- Abbott, N.J. Astrocyte-endothelial interactions and blood-brain barrier permeability. J. Anat. 2002, 200, 629–638. [Google Scholar] [CrossRef]
- Begley, D.J.; Brightman, M.W. Structural and functional aspects of the blood-brain barrier. Prog. Drug Res. 2003, 61, 39–78. [Google Scholar]
- Atwood, C.S.; Bowen, R.L.; Smith, M.A.; Perry, G. Cerebrovascular requirement for sealant; anti-coagulant and remodeling molecules that allow for the maintenance of vascular integrity and blood supply. Brain Res. Brain Res. Rev. 2003, 43, 164–178. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 2005, 28, 202–208. [Google Scholar] [CrossRef]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef] [Green Version]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef] [Green Version]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Gao, C.; Gao, D.; Sun, R.; Li, W.; Wang, F.; Wang, Y.; Cao, H.; Zhou, G.; Zhang, J.; et al. Reduction in pericyte coverage leads to blood-brain barrier dysfunction via endothelial transcytosis following chronic cerebral hypoperfusion. Fluids Barriers CNS 2021, 18, 21. [Google Scholar] [CrossRef]
- Pluta, R.; Misicka, A.; Barcikowska, M.; Spisacka, S.; Lipkowski, A.W.; Januszewski, S. Possible reverse transport of β-amyloid peptide across the blood-brain barrier. Acta Neurochir. Suppl. 2000, 76, 73–77. [Google Scholar]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood-brain barrier: An overview: Structure; regulation; and clinical implications. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef]
- Michinaga, S.; Koyama, Y. Dual roles of astrocyte-derived factors in regulation of blood-brain barrier function after brain damage. Int. J. Mol. Sci. 2019, 20, 571. [Google Scholar] [CrossRef] [Green Version]
- Lien, C.F.; Mohanta, S.K.; Frontczak-Baniewicz, M.; Swinny, J.D.; Zablocka, B.; Gorecki, D.C. Absence of glial alpha-dystrobrevin causes abnormalities of the blood–brain barrier and progressive brain edema. J. Biol. Chem. 2012, 287, 41374–41385. [Google Scholar] [CrossRef] [Green Version]
- Ramsauer, M.; Krause, D.; Dermietzel, R. Angiogenesis of the blood-brain barrier in vitro and the function of cerebral pericytes. FASEB J. 2002, 16, 1274–1276. [Google Scholar] [CrossRef]
- Mossakowski, M.J.; Lossinsky, A.S.; Pluta, R.; Wisniewski, H.M. Changes in cerebral microcirculation system following experimentally induced cardiac arrest: A SEM and TEM study. In Microcirculatory Stasis in the Brain; Tomita, M., Ed.; Elsevier Science Publishers B.V.: Amsterdam, The Netherlands, 1993; pp. 99–106. [Google Scholar]
- Mossakowski, M.J.; Lossinsky, A.S.; Pluta, R.; Wisniewski, H.M. Abnormalities of the blood-brain barrier in global cerebral ischemia in rats due to experimental cardiac arrest. Acta Neurochir. Suppl. 1994, 60, 274–276. [Google Scholar]
- Wisniewski, H.M.; Pluta, R.; Lossinsky, A.S.; Mossakowski, M.J. Ultrastructural studies of cerebral vascular spasm after cardiac arrest-related global cerebral ischemia in rats. Acta Neuropathol. 1995, 90, 432–440. [Google Scholar] [CrossRef]
- Ueno, M.; Akiguchi, I.; Hosokawa, M.; Shinnou, M.; Sakamoto, H.; Takemura, M.; Higuchi, K. Age-related changes in the brain transfer of blood-borne horseradish peroxidase in the hippocampus of senescence-accelerated mouse. Acta Neuropathol. 1997, 93, 233–240. [Google Scholar] [CrossRef]
- Shinnou, M.; Ueno, M.; Sakamoto, H.; Ide, M. Blood-brain barrier damage in reperfusion following ischemia in the hippocampus of the Mongolian gerbil brain. Acta Neurol. Scand. 1998, 98, 406–411. [Google Scholar] [CrossRef]
- Lippoldt, A.; Kniesel, U.; Liebner, S.; Kalbacher, H.; Kirsch, T.; Wolburg, H.; Haller, H. Structural alterations of tight junctions are associated with loss of polarity in stroke-prone spontaneously hypertensive rat blood-brain barrier endothelial cells. Brain Res. 2000, 885, 251–261. [Google Scholar] [CrossRef]
- Ueno, M.; Tomimoto, H.; Akiguchi, I.; Wakita, H.; Sakamoto, H. Blood-brain barrier disruption in white matter lesions in a rat model of chronic cerebral hypoperfusion. J. Cereb. Blood Flow Metab. 2002, 22, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Sakamoto, H.; Liao, Y.J.; Onodera, M.; Huang, C.L.; Miyanaka, H.; Nakagawa, T. Blood-brain barrier disruption in the hypothalamus of young adult spontaneously hypertensive rats. Histochem. Cell Biol. 2004, 122, 131–137. [Google Scholar] [CrossRef]
- Hallenbeck, J.M.; Dutka, A.J.; Tanishima, T.; Kochanek, P.M.; Kumaroo, K.K.; Thompson, C.B.; Obrenovitch, T.P.; Contreras, T.J. Polymorphonuclear leukocyte accumulation in brain regions with low blood flow during the early postischemic period. Stroke 1986, 17, 246–253. [Google Scholar] [CrossRef] [Green Version]
- Pluta, R.; Januszewski, S.; Czuczwar, S.J. Neuroinflammation in post-ischemic neurodegeneration of the brain: Friend; foe; or both? Int. J. Mol. Sci. 2021, 22, 4405. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.D.; Prittila, T. Biological markers of Alzheimer’s disease. Drug Dev. Res. 2002, 56, 74–84. [Google Scholar] [CrossRef]
- Jendroska, K.; Poewe, W.; Daniel, S.E.; Pluess, J.; Iwerssen-Schmidt, H.; Paulsen, J.; Barthel, S.; Schelosky, L.; Cervos-Navarr, J.; DeArmond, S.J. Ischemic stress induces deposition of amyloid beta immunoreactivity in human brain. Acta Neuropathol. 1995, 90, 461–466. [Google Scholar] [CrossRef]
- Wisniewski, H.M.; Maslinska, D. Beta-protein immunoreactivity in the human brain after cardiac arrest. Folia Neuropathol. 1996, 34, 65–71. [Google Scholar]
- Jendroska, K.; Hoffmann, O.M.; Patt, S. Amyloid β peptide and precursor protein (APP) in mild and severe brain ischemia. Ann. N. Y. Acad. Sci. 1997, 826, 401–405. [Google Scholar] [CrossRef]
- Lee, P.H.; Bang, O.Y.; Hwang, E.M.; Lee, J.S.; Joo, U.S.; Mook-Jung, I.; Huh, K. Circulating beta amyloid protein is elevated in patients with acute ischemic stroke. J. Neural. Transm. 2005, 112, 1371–1379. [Google Scholar] [CrossRef]
- Qi, J.; Wu, H.; Yang, Y.; Wand, D.; Chen, Y.; Gu, Y.; Liu, T. Cerebral ischemia and Alzheimer’s disease: The expression of amyloid-β and apolipoprotein E in human hippocampus. J. Alzheimers Dis. 2007, 12, 335–341. [Google Scholar] [CrossRef]
- Zetterberg, H.; Mörtberg, E.; Song, L.; Chang, L.; Provuncher, G.K.; Patel, P.P.; Ferrell, E.; Fournier, D.R.; Kan, C.W.; Campbell, T.G.; et al. Hypoxia due to cardiac arrest induces a time-dependent increase in serum amyloid β levels in humans. PLoS ONE 2011, 6, e28263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.H.; Cao, H.Y.; Wang, Y.R.; Jiao, S.S.; Bu, X.L.; Zeng, F.; Wang, G.; Li, J.; Deng, J.; Zhou, H.D.; et al. Serum Aβ is predictive for short-term neurological deficits after acute ischemic stroke. Neurotox. Res. 2015, 27, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Ułamek, M.; Jabłoński, M. Alzheimer’s mechanisms in ischemic brain degeneration. Anat. Rec. 2009, 292, 1863–1881. [Google Scholar] [CrossRef]
- Jabłoński, M.; Maciejewski, R.; Januszewski, S.; Ułamek, M.; Pluta, R. One year follow up in ischemic brain injury and the role of Alzheimer factors. Physiol. Res. 2011, 60 (Suppl. 1), S113–S119. [Google Scholar] [CrossRef] [PubMed]
- Farkas, I.G.; Czigner, A.; Farkas, E.; Dobo, E.; Soos, K.; Penke, B.; Endresz, V.; Mihaly, A. Beta-amyloid peptide-induced blood-brain barrier disruption facilitates T-cell entry into the rat brain. Acta Histochem. 2003, 105, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Paris, D.; Patel, N.; DelleDonne, A.; Quadros, A.; Smeed, R.; Mullan, M. Impaired angiogenesis in a transgenic mouse model of cerebral amyloidosis. Neurosci. Lett. 2004, 366, 80–85. [Google Scholar] [CrossRef]
- Paris, D.; Townsend, K.; Quadros, A.; Humphrey, J.; Sun, J.; Brem, S.; Wotoczek-Obadia, M.; DellaDonne, A.; Patel, N.; Obergon, D.F.; et al. Inhibition of angiogenesis by Aβ peptides. Angiogenesis 2004, 7, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R. Proteins associated with Alzheimer’s disease in conditions predisposing to Alzheimer’s-type neurodegeneration. J. Cereb. Blood Flow Metab. 2001, 21 (Suppl. 1), S424. [Google Scholar]
- Pluta, R.; Januszewski, S.; Ułamek, M. Ischemic blood–brain barrier and amyloid in white matter as etiological factors in leukoaraiosis. Acta Neurochir. Suppl. 2008, 102, 353–356. [Google Scholar]
- Ishikawa, M.; Cooper, D.; Arumugam, T.V.; Zhang, J.H.; Nanda, A.; Granger, D.N. Platelet-leukocyte-endothelial cell interactions after middle cerebral artery occlusion and reperfusion. J. Cereb. Blood Flow Metab. 2004, 24, 907–915. [Google Scholar] [CrossRef] [Green Version]
- Ritter, L.S.; Stempel, K.M.; Coull, B.M.; McDonagh, P.F. Leukocyte-platelet aggregates in rat peripheral blood after ischemic stroke and reperfusion. Biol. Res. Nurs. 2005, 6, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Del Zoppo, G.J.; Schmid-Schonbein, G.W.; Mori, E.; Copeland, B.R.; Chang, C.M. Polymorphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfusion in baboons. Stroke 1991, 22, 1276–1283. [Google Scholar] [CrossRef] [Green Version]
- Kochanek, P.M.; Hallenbeck, J.M. Polymorphonuclear leukocytes and mnocytes/macrophages in the pathogenesis of cerebral ischemia and stroke. Stroke 1992, 23, 1367–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, E.; Del Zoppo, G.J.; Chambers, J.D.; Copeland, B.R.; Arfors, R.E. Inhibition of polymorphonuclear leukocyte adherence suppresses no-reflow after focal cerebral ischemia in baboons. Stroke 1992, 23, 712–718. [Google Scholar] [CrossRef] [Green Version]
- Caceres, M.J.; Schleien, C.L.; Kuluz, J.W.; Gelman, B.; Dietrich, W.D. Early endothelial damage and leukocyte accumulation in piglet brains following cardiac arrest. Acta Neuropathol. 1995, 90, 582–591. [Google Scholar] [CrossRef]
- Gidday, J.M.; Gasche, Y.G.; Copin, J.C.; Shah, A.R.; Perez, R.S.; Shapiro, S.D.; Chan, P.H.; Park, T.S. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H558–H568. [Google Scholar] [CrossRef] [PubMed]
- Ames, A.; Wright, R.L.; Kowada, M.; Thurston, J.M.; Majno, G. Cerebral ischemia. II. The no-reflow phenomenon. Am. J. Pathol. 1968, 52, 437–453. [Google Scholar] [PubMed]
- Saito, K.; Suyama, K.; Nishida, K.; Sei, Y.; Basile, A.S. Early increases in TNF-alpha; IL-6 and IL-1 beta levels following transient cerebral ischemia in gerbil brain. Neurosci. Lett. 1996, 206, 149–152. [Google Scholar] [CrossRef]
- Boutin, H.; LeFeuvre, R.A.; Horai, R.; Asano, M.; Iwakura, Y.; Rothwell, N.J. Role of IL-1α and IL-1β in ischemic brain damage. J. Neurosci. 2001, 21, 5528–5534. [Google Scholar] [CrossRef] [Green Version]
- Hart, M.N.; Sokoll, M.D.; Davies, L.R.; Henriquez, E. Vascular spasm in cat cerebral cortex following ischemia. Stroke 1978, 9, 52–57. [Google Scholar] [CrossRef] [Green Version]
- Nimmo, A.J.; Cernak, I.; Heath, D.L.; Hu, X.; Bennett, C.J.; Vink, R. Neurogenic inflammation is associated with development of edema and functional deficits following traumatic brain injury in rats. Neuropeptides 2004, 38, 40–47. [Google Scholar] [CrossRef]
- Zhang, R.L.; Chopp, M.; Liu, Y.; Zaloga, C.; Jiang, N.; Jones, M.L.; Miyasaka, M.; Ward, P.A. Anti-ICAM-1 antibody reduces ischemic cell damage after transient middle cerebral artery occlusion in the rat. Neurology 1994, 44, 1747–1751. [Google Scholar] [CrossRef]
- Jiang, Y.; Yin, D.; Xu, D.; Men, W.; Cao, R.; Li, B.; Fan, M. Investigating microbleeding in cerebral ischemia rats using susceptibility-weighted imaging. Magn. Reson. Imaging 2015, 33, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Leeuwis, A.E.; Prins, N.D.; Hooghiemstra, A.M.; Benedictus, M.R.; Scheltens, P.; Barkhof, F.; van der Flier, W.M. Microbleeds are associated with depressive symptoms in Alzheimer’s disease. Alzheimers Dement. 2017, 10, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ye, M. Risk factors and their correlation with severity of cerebral microbleed in acute large artery atherosclerotic cerebral infarction patients. Clin. Neurol. Neurosurg. 2022, 221, 107380. [Google Scholar] [CrossRef] [PubMed]
- Hossmann, V.; Hossmann, K.A.; Takagi, S. Effect of intravascular platelet aggregation on blood recirculation following prolonged ischemia of the cat brain. J. Neurol. 1980, 222, 159–170. [Google Scholar] [CrossRef]
- Nortley, R.; Korte, N.; Izquierdo, P.; Hirunpattarasilp, C.; Mishra, A.; Jaunmuktane, Z.; Kyrargyri, V.; Pfeiffer, T.; Khennouf, L.; Madry, C.; et al. Amyloid β oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 2019, 365, eaav9518. [Google Scholar] [CrossRef]
- Niwa, K.; Porter, V.A.; Kazama, K.; Cornfield, D.; Carlsson, G.A.; Iadecola, C. A beta-peptides enhance vasoconstriction in cerebral circulation. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H2417–H2424. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, S.; Kuroiwa, T.; LiYuan, S.; Katsumata, N.; Li, S.; Endo, S.; Mizusawa, H. Long-term cognitive and neuropsychological symptoms after global cerebral ischemia in Mongolian gerbils. Acta Neurochir. Suppl. 2006, 96, 299–302. [Google Scholar]
- de la Tremblaye, P.B.; Plamondon, H. Impaired conditioned emotional response and object recognition are concomitant to neuronal damage in the amygdale and perirhinal cortex in middle-aged ischemic rats. Behav. Brain Res. 2011, 219, 227–233. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.J.; Zhang, M.; Fang, C.Q.; Zhou, H.D. Cerebral ischemia aggravates cognitive impairment in a rat model of Alzheimer’s disease. Life Sci. 2011, 89, 86–92. [Google Scholar] [CrossRef]
- Cohan, C.H.; Neumann, J.T.; Dave, K.R.; Alekseyenko, A.; Binkert, M.; Stransky, K.; Lin, H.W.; Barnes, C.A.; Wright, C.B.; Perez-Pinzon, M.A. Effect of cardiac arrest on cognitive impairment and hippocampal plasticity in middle-aged rats. PLoS ONE 2015, 10, e0124918. [Google Scholar] [CrossRef] [Green Version]
- Rost, N.S.; Brodtmann, A.; Pase, M.P.; van Veluw, S.J.; Biffi, A.; Duering, M.; Hinman, J.D.; Dichgans, M. Post-stroke cognitive impairment and dementia. Circ. Res. 2022, 130, 1252–1271. [Google Scholar] [CrossRef]
- Iwata, A.; Chen, X.H.; McIntosh, T.K.; Browne, K.D.; Smith, D.H. Long-term accumulation of amyloid-beta in axons following brain trauma without persistent upregulation of amyloid precursor protein genes. J. Neuropathol. Exp. Neurol. 2002, 61, 1056–1068. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.R.; Dean, C.; Beiler, S.; Bryan, D.W.; Packard, B.A.; Smulian, A.G.; Linke, M.J.; de Courten-Myers, G.M. Plasma infusions into porcine cerebral white matter induce early edema; oxidative stress; pro-inflammatory cytokine gene expression and DNA fragmentation: Implications for white matter injury with increased blood-brain barrier permeability. Curr. Neurovasc. Res. 2005, 2, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Irving, E.A.; Yatsushiro, K.; McCulloch, J.; Dewar, D. Rapid alteration of tau in oligodendrocytes after focal ischemic injury in the rat: Involvement of free radicals. J. Cereb. Blood Flow Metab. 1997, 17, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Erusalimsky, J.D.; Kurz, D.J. Cellular senescence in vivo: Its relevance in aging and cardiovascular disease. Exp. Gerontol. 2005, 40, 634–642. [Google Scholar] [CrossRef]
- Hayflick, L. The illusion of cell immortality. Br. J. Cancer 2000, 83, 841–846. [Google Scholar] [CrossRef]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Tateno, K.; Kunieda, T.; Komuro, I. Vascular cell senescence and vascular aging. J. Mol. Cell Cardiol. 2004, 36, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef]
- Folin, M.; Baiguera, S.; Tommasini, M.; Guidolin, D.; Conconi, M.T.; De Carlo, E.; Nussdorfer, G.G.; Parnigotto, P.P. Effects of beta-amyloid on rat neuromicrovascular endothelial cells cultured in vitro. Int. J. Mol. Med. 2005, 15, 929–935. [Google Scholar]
- Kokaia, Z.; Lindvall, O. Neurogenesis after ischaemic brain insults. Curr. Opin. Neurobiol. 2003, 13, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Louissaint, A., Jr.; Rao, S.; Leventhal, C.; Goldman, S.A. Coordinated interaction of neurogenesis and angiogenesis in the adult songbird brain. Neuron 2002, 34, 945–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishimaru, H.; Ishikawa, K.; Haga, S.; Shoji, M.; Ohe, Y.; Haga, C.; Sasaki, A.; Takashashi, A.; Maruyama, Y. Accumulation of apolipoprotein E and β-amyloid-like protein in a trace of the hippocampal CA1 pyramidal cell layer after ischaemic delayed neuronal death. NeuroReport 1996, 7, 3063–3067. [Google Scholar] [CrossRef] [PubMed]
- Pogue, A.I.; Lukiw, W.J. Angiogenic signaling in Alzheimer’s disease. NeuroReport 2004, 15, 1507–1510. [Google Scholar] [CrossRef]
- Ohtake, M.; Morino, S.; Kaidoh, T.; Inoue, T. Three-dimensional structural changes in cerebral microvessels after transient focal cerebral ischemia in rats: Scanning electron microscopic study of corrosion casts. Neuropathology 2004, 24, 219–227. [Google Scholar] [CrossRef]
- Hirunpattarasilp, C.; Barkaway, A.; Davis, H.; Pfeiffer, T.; Sethi, H.; Attwell, D. Hyperoxia evokes pericyte-mediated capillary constriction. J. Cereb. Blood Flow Metab. 2022, 42, 2032–2047. [Google Scholar] [CrossRef]
- Wang, W.Z.; Guo, S.Z.; Tsai, T.M.; Anderson, G.L.; Miller, F.N. Platelet-activating factor contributes to postischemic vasospasm. J. Surg. Res. 2000, 89, 139–146. [Google Scholar] [CrossRef]
- Talib, L.L.; Joaquim, H.P.; Forlenza, O.V. Platelet biomarkers in Alzheimer’s disease. World J. Psychiatry 2012, 2, 95–101. [Google Scholar] [CrossRef]
- Lossinsky, A.S.; Badmajew, V.; Robson, J.A.; Moretz, R.C.; Wisniewski, H.M. Sites of egress of inflammatory cells and horseradish peroxidase transport across the blood-brain barrier in a murine model of chronic relapsing experimental allergic encephalomyelitis. Acta Neuropathol. 1989, 78, 359–371. [Google Scholar] [CrossRef]
- Nishijima, K.; Kiryu, J.; Tsujikawa, A.; Miyamoto, K.; Honjo, M.; Tanihara, H.; Nonaka, A.; Yamashiro, K.; Katsuta, H.; Miyahara, S.; et al. Platelets adhering to the vascular wall mediate postischemic leukocyte-endothelial cell interactions in retinal microcirculation. Investig. Ophthalmol. Vis. Sci. 2004, 45, 977–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kniewallner, K.M.; Ehrlich, D.; Kiefer, A.; Marksteiner, J.; Humpel, C. Platelets in the Alzheimer’s disease brain: Do they play a role in cerebral amyloid angiopathy? Curr. Neurovasc. Res. 2015, 12, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Niwa, K.; Younkin, L.; Ebeling, C.; Turner, S.K.; Westaway, D.; Younkin, S.; Ashe, K.H.; Carlson, G.A.; Iadecola, C. Abeta 1-40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc. Natl. Acad. Sci. USA 2000, 97, 9735–9740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, J.M.; Chi, X.; Hellermann, G.; Sutton, E.T. Physiological levels of β-amyloid induce cerebral vessel dysfunction and reduce endothelial nitric oxide production. Neurol. Res. 2001, 23, 506–512. [Google Scholar] [CrossRef]
- Blanc, E.M.; Toborek, M.; Mark, R.J.; Hennig, B.; Mattson, M.P. Amyloid β-peptide induces cell monolayer albumin permeability; impairs glucose transport; and induces apoptosis in vascular endothelial cells. J. Neurochem. 1997, 68, 1870–1881. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, K.; Ho, L.; Younkin, S.G.; Kunkel, D.D.; Ogburn, C.E.; LeBoeuf, R.C.; Furlong, C.E.; Deeb, S.S.; Nochlin, D.; Wegiel, J.; et al. High levels of circulating beta-amyloid peptide do not cause cerebral beta-amyloidosis in transgenic mice. Am. J. Pathol. 1996, 149, 219–227. [Google Scholar] [PubMed]
- De la Torre, J.C. Vascular basis of Alzheimer’s pathogenesis. Ann. N. Y. Acad. Sci. 2002, 977, 196–215. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Montagne, A.; Sagare, A.P.; Nation, D.A.; Schneider, L.S.; Chui, H.C.; Harrington, M.G.; Pa, J.; Law, M.; Wang, D.J.J.; et al. Vascular dysfunction the disregarded partner of Alzheimer’s disease. Alzheimers Dement. 2019, 15, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Van Groen, T.; Puurunen, K.; Mäki, H.M.; Sivenius, J.; Jolkkonen, J. Transformation of diffuse beta-amyloid precursor protein and beta-amyloid deposits to plaques in the thalamus after transient occlusion of the middle cerebral artery in rats. Stroke 2005, 36, 1551–1556. [Google Scholar] [CrossRef] [Green Version]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Woods, A.S.; Cotter, R.J.; Gowing, E.; Ball, M.J. Beta-amyloid- (1-42) is a major component of cerebrovascular amyloid deposits: Implications for the pathology of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 10836–10840. [Google Scholar] [CrossRef] [Green Version]
- Stokes, K.Y.; Granger, D.N. Platelets: A critical link between inflammation and microvascular dysfunction. J. Physiol. 2012, 590, 1023–1034. [Google Scholar] [CrossRef]
- Kalaria, R.N. The role of cerebral ischemia in Alzheimer’s disease. Neurobiol. Aging 2000, 21, 321–330. [Google Scholar] [CrossRef]
- Humpel, C.; Marksteiner, J. Cerebrovascular damage as a cause for Alzheimer’s disease. Curr. Neurovasc. Res. 2005, 2, 341–347. [Google Scholar] [CrossRef]
- Adamczak, J.; Hoehn, M. Poststroke angiogenesis; con: Dark side of angiogenesis. Stroke 2015, 46, e103–e104. [Google Scholar] [CrossRef] [Green Version]
- Bersini, S.; Arrojo, E.; Drigo, R.; Huang, L.; Shokhirev, M.N.; Hetzer, M.W. Transcriptional and functional changes of the human microvasculature during physiological aging and Alzheimer disease. Adv. Biosyst. 2020, 4, e2000044. [Google Scholar] [CrossRef]
- Hatakeyama, M.; Ninomiya, I.; Kanazawa, M. Angiogenesis and neuronal remodeling after ischemic stroke. Neural. Regen. Res. 2020, 15, 16–19. [Google Scholar]
- Grammas, P.; Moore, P.; Weigel, P.H. Microvessels from Alzheimer’s disease brains kill neurons in vitro. Am. J. Pathol. 1999, 154, 337–342. [Google Scholar] [CrossRef]
- Gemmell, E.; Tam, E.; Allan, L.; Hall, R.; Khundakar, A.; Oakley, A.E.; Thomas, A.; Deramecourt, V.; Kalaria, R.J. Neuron volumes in hippocampal subfields in delayed poststroke and aging-related dementias. J. Neuropathol. Exp. Neurol. 2014, 73, 305–311. [Google Scholar] [CrossRef] [Green Version]
- Kuroiwa, T.; Bonnekoh, P.; Hossmann, K.A. Locomotor hyperactivity and hippocampal CA1 injury after transient forebrain ischemia in gerbils. Neurosci. Lett. 1991, 122, 141–144. [Google Scholar] [CrossRef]
- De la Torre, J.C. Is Alzheimer’s disease preceded by neurodegeneration or cerebral hypoperfusion? Ann. Neurol. 2005, 57, 783–784. [Google Scholar] [CrossRef]
- Saver, J.L. Time is brain—Quantified. Stroke 2006, 37, 263–266. [Google Scholar] [CrossRef] [Green Version]
- Nalivaeva, N.N.; Fisk, L.; Kochkina, E.G.; Plesneva, S.A.; Zhuravin, I.A.; Babusikova, E.; Dobrota, D.; Turner, A.J. Effect of hypoxia/ ischemia and hypoxic preconditioning/reperfusion on expression of some amyloid–degrading enzymes. Ann. N. Y. Acad Sci. 2004, 1035, 21–33. [Google Scholar] [CrossRef]
- Armstrong, R.A. Plaques and tangles and the pathogenesis of Alzheimer’s disease. Folia Neuropathol. 2006, 44, 1–11. [Google Scholar]
- Gallego, I.; Villate-Beitia, I.; Saenz-Del-Burgo, L.; Puras, G.; Pedraz, J.L. Therapeutic opportunities and delivery strategies for brain revascularization in stroke; Neurodegeneration; and aging. Pharmacol. Rev. 2022, 74, 439–461. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pluta, R.; Miziak, B.; Czuczwar, S.J. Post-Ischemic Permeability of the Blood–Brain Barrier to Amyloid and Platelets as a Factor in the Maturation of Alzheimer’s Disease-Type Brain Neurodegeneration. Int. J. Mol. Sci. 2023, 24, 10739. https://doi.org/10.3390/ijms241310739
Pluta R, Miziak B, Czuczwar SJ. Post-Ischemic Permeability of the Blood–Brain Barrier to Amyloid and Platelets as a Factor in the Maturation of Alzheimer’s Disease-Type Brain Neurodegeneration. International Journal of Molecular Sciences. 2023; 24(13):10739. https://doi.org/10.3390/ijms241310739
Chicago/Turabian StylePluta, Ryszard, Barbara Miziak, and Stanisław J. Czuczwar. 2023. "Post-Ischemic Permeability of the Blood–Brain Barrier to Amyloid and Platelets as a Factor in the Maturation of Alzheimer’s Disease-Type Brain Neurodegeneration" International Journal of Molecular Sciences 24, no. 13: 10739. https://doi.org/10.3390/ijms241310739