
Generation and Functional Characterization of Anti-CD19 Chimeric Antigen Receptor-Natural Killer Cells from Human Induced Pluripotent Stem Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
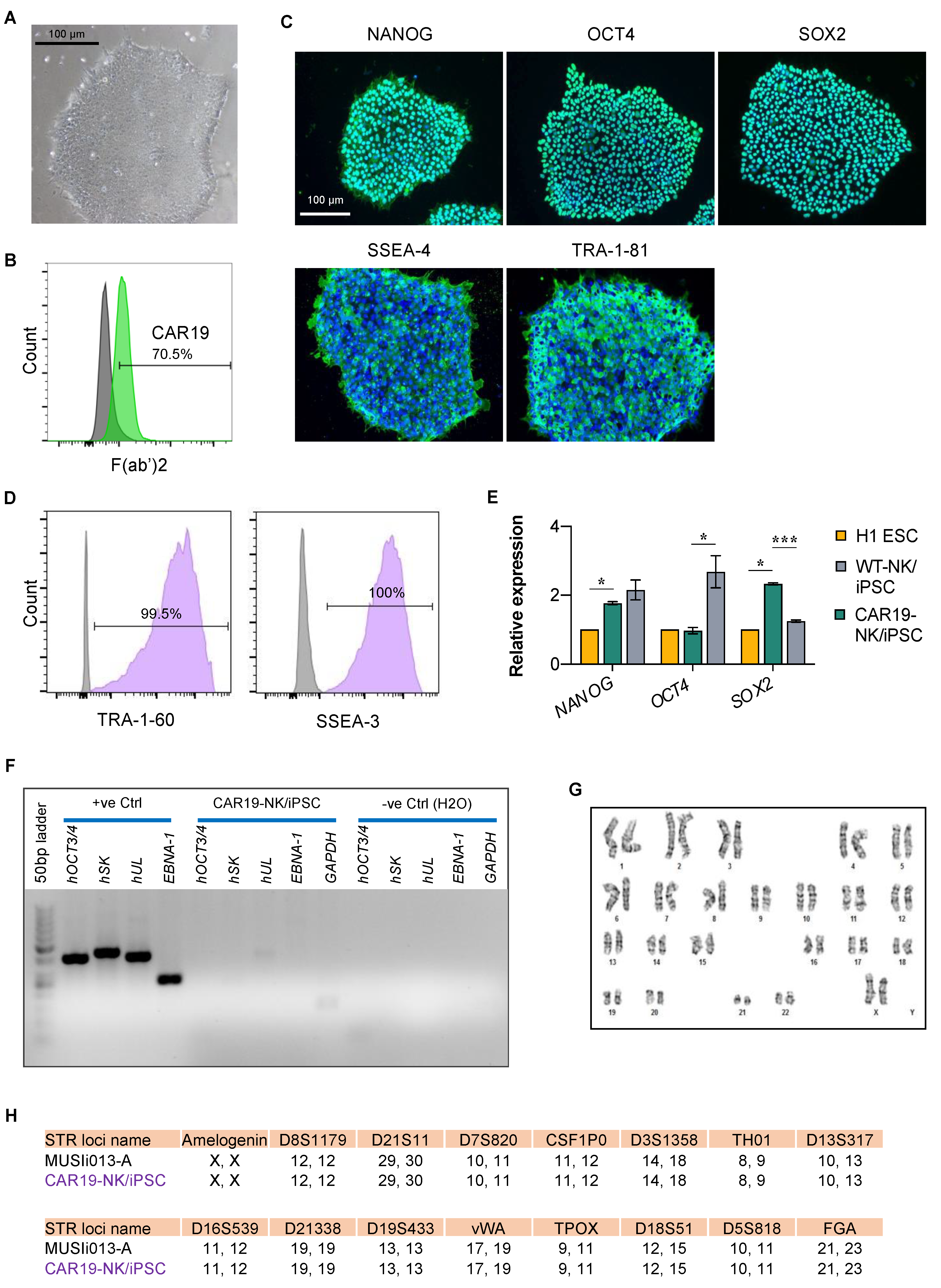
2.1. Engineering the Anti-CD19 CAR-Expressing NK/iPSC Line
2.2. Characterization of CAR19-NK/iPSCs and Its Multilineage Differentiation
2.3. Induction of Hematopoietic Progenitor Cells from CAR19-NK/iPSCs
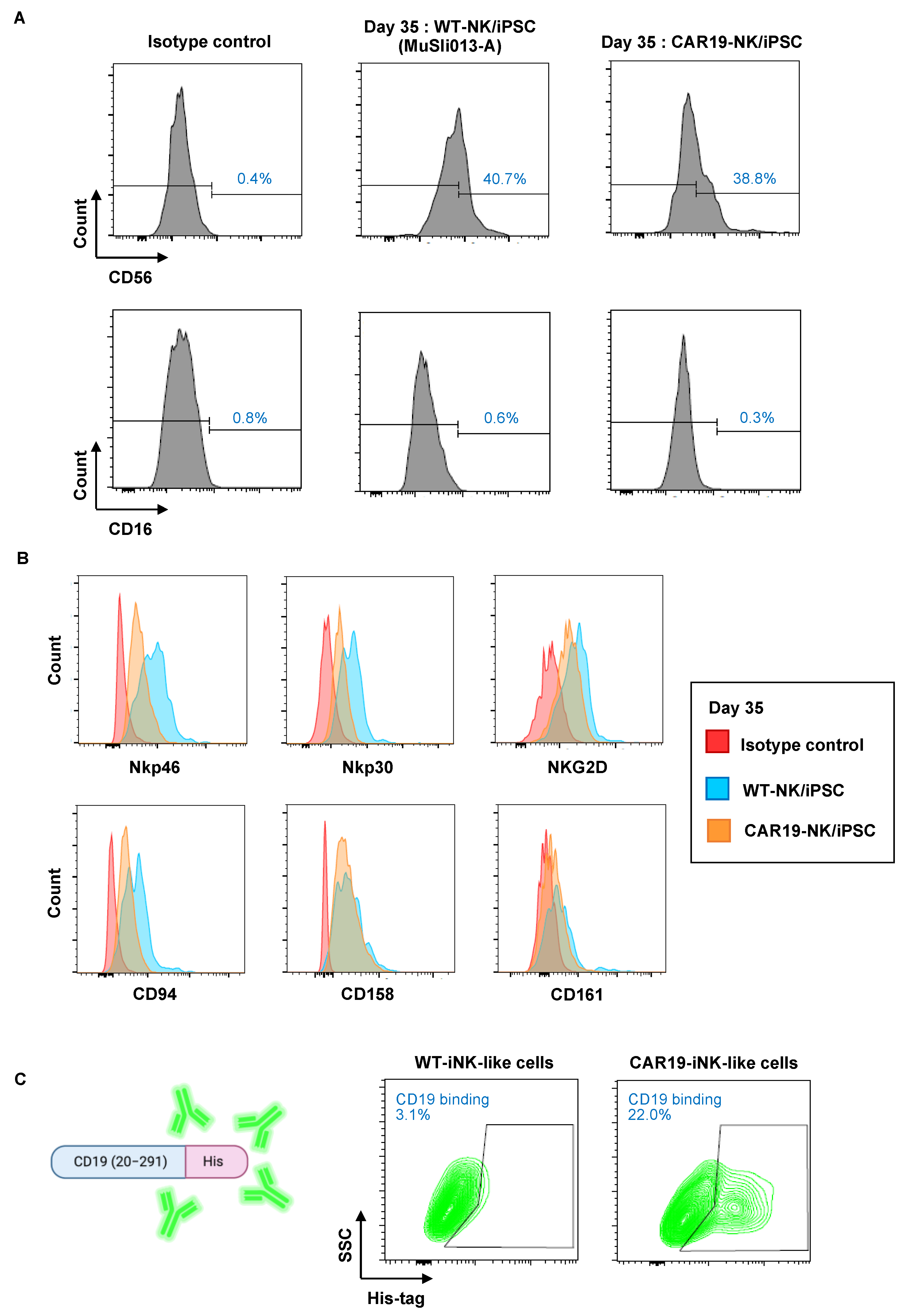
2.4. CAR19-NK/iPSC-Derived HPCs Can Be Differentiated toward the NK-Cell Lineage
2.5. Selective Cytotoxicity of CAR19-NK/iPSC-Derived NK-like Cells against CD19-Positive Hematologic Cancer Cells
2.6. Activation of NK Cell Receptors in CAR19-NK/iPSC-Derived NK-like Cells upon Tumor Exposure
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Lentiviral Particle Production
4.3. CAR Transduction and Single-Cell Clone Isolation
4.4. Genomic DNA Sequencing
4.5. Evaluation of CAR Expression
4.6. Immunofluorescence Staining
4.7. Flow Cytometry
4.8. qPCR Analysis
4.9. Karyotyping
4.10. STR Analysis
4.11. Spontaneous In Vitro Differentiation via EB Formation
4.12. NK Cell Differentiation
4.13. NK Cell Cytotoxicity Assay
4.14. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Song, W.; Rubinstein, M.; Liu, D. Recent updates in cancer immunotherapy: A comprehensive review and perspective of the 2018 China Cancer Immunotherapy Workshop in Beijing. J. Hematol. Oncol. 2018, 11, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target Ther. 2021, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Chavda, V.P.; Bezbaruah, R.; Dhamne, H.; Yang, D.H.; Zhao, H.B. CAR T-cell therapy for the management of mantle cell lymphoma. Mol. Cancer 2023, 22, 67. [Google Scholar] [CrossRef]
- Pfefferle, A.; Huntington, N.D. You have got a fast CAR: Chimeric antigen receptor NK Cells in cancer therapy. Cancers 2020, 12, 706. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Lal, G. The molecular mechanism of natural killer cells function and its importance in cancer immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zheng, H.; Diao, Y. Natural killer cells and current applications of chimeric antigen receptor-modified NK-92 cells in tumor immunotherapy. Int. J. Mol. Sci. 2019, 20, 20317. [Google Scholar] [CrossRef] [Green Version]
- Tonn, T.; Schwabe, D.; Klingemann, H.G.; Becker, S.; Esser, R.; Koehl, U.; Suttorp, M.; Seifried, E.; Ottmann, O.G.; Bug, G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013, 15, 1563–1570. [Google Scholar] [CrossRef]
- Myers, J.A.; Miller, J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 85–100. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Czerwińska, P.; Mazurek, S.; Kołodziejczak, I.; Wiznerowicz, M. Gene delivery methods and genome editing of human pluripotent stem cells. Rep. Pract. Oncol. Radiother. 2019, 24, 180–187. [Google Scholar] [CrossRef]
- Alhaji, S.Y.; Nordin, N.; Ngai, S.C.; Al Abbar, A.; Mei, L., I; Abdullah, S. Lack of methylation on transgene leads to high level and persistent transgene expression in induced pluripotent stem cells. Gene 2020, 758, 144958. [Google Scholar] [CrossRef]
- Arias, J.; Yu, J.; Varshney, M.; Inzunza, J.; Nalvarte, I. Hematopoietic stem cell- and induced pluripotent stem cell-derived CAR-NK cells as reliable cell-based therapy solutions. Stem Cells Transl. Med. 2021, 10, 987–995. [Google Scholar] [CrossRef]
- Pratumkaew, P.; Issaragrisil, S.; Luanpitpong, S. Induced pluripotent stem cells as a tool for modeling hematologic disorders and as a potential source for cell-based therapies. Cells 2021, 10, 3250. [Google Scholar] [CrossRef]
- Luanpitpong, S.; Poohadsuan, J.; Klaihmon, P.; Issaragrisil, S. Selective cytotoxicity of single and dual anti-CD19 and anti-CD138 chimeric antigen receptor-natural killer cells against hematologic malignancies. J. Immunol. Res. 2021, 2021, 5562630. [Google Scholar] [CrossRef]
- Klaihmon, P.; Luanpitpong, S.; Lorthongpanich, C.; Laowtammathron, C.; Waeteekul, S.; Terbto, P.; Issaragrisil, S. Episomal vector reprogramming of human umbilical cord blood natural killer cells to an induced pluripotent stem cell line MUSIi013-A. Stem Cell Res. 2021, 55, 102472. [Google Scholar] [CrossRef]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Ohi, Y.; Qin, H.; Hong, C.; Blouin, L.; Polo, J.M.; Guo, T.; Qi, Z.; Downey, S.L.; Manos, P.D.; Rossi, D.J.; et al. Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human iPS cells. Nat. Cell Biol. 2011, 13, 541–549. [Google Scholar] [CrossRef]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Nianias, A.; Themeli, M. Induced pluripotent stem cell (iPSC)-derived lymphocytes for adoptive cell immunotherapy: Recent advances and challenges. Curr. Hematol. Malig. Rep. 2019, 14, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Valamehr, B.; Abujarour, R.; Robinson, M.; Le, T.; Robbins, D.; Shoemaker, D.; Flynn, P. A novel platform to enable the high-throughput derivation and characterization of feeder-free human iPSCs. Sci. Rep. 2012, 2, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupo, K.B.; Moon, J.I.; Chambers, A.M.; Matosevic, S. Differentiation of natural killer cells from induced pluripotent stem cells under defined, serum- and feeder-free conditions. Cytotherapy 2021, 23, 939–952. [Google Scholar] [CrossRef] [PubMed]
- Huntington, N.D.; Vosshenrich, C.A.; Di Santo, J.P. Developmental pathways that generate natural-killer-cell diversity in mice and humans. Nat. Rev. Immunol. 2007, 7, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, X.; Yuan, X.; Wang, W.; Wang, Y. Chimeric antigen receptor-engineered NK cells: New weapons of cancer immunotherapy with great potential. Exp. Hematol. Oncol. 2022, 11, 85. [Google Scholar] [CrossRef]
- Gong, J.H.; Maki, G.; Klingemann, H.G. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 1994, 8, 652–658. [Google Scholar]
- Uphoff, C.C.; Denkmann, S.A.; Steube, K.G.; Drexler, H.G. Detection of EBV, HBV, HCV, HIV-1, HTLV-I and -II, and SMRV in human and other primate cell lines. J. Biomed. Biotechnol. 2010, 2010, 904767. [Google Scholar] [CrossRef]
- Reina-Ortiz, C.; Constantinides, M.; Fayd-Herbe-de-Maudave, A.; Présumey, J.; Hernandez, J.; Cartron, G.; Giraldos, D.; Díez, R.; Izquierdo, I.; Azaceta, G.; et al. Expanded NK cells from umbilical cord blood and adult peripheral blood combined with daratumumab are effective against tumor cells from multiple myeloma patients. Oncoimmunology 2020, 10, 1853314. [Google Scholar] [CrossRef]
- Karagiannis, P.; Kim, S.I. iPSC-derived natural killer cells for cancer immunotherapy. Mol. Cells. 2021, 44, 541–548. [Google Scholar] [CrossRef]
- Du, W.; Cui, L.; Zhang, J.; Zhang, H.; Liu, R.; Yang, W.; Zhang, Y. Generation of universal natural killer cells from a cryopreserved cord blood mononuclear cell-derived induced pluripotent stem cell library. FEBS Open Bio. 2022, 12, 1771–1781. [Google Scholar] [CrossRef]
- Schmidt, P.; Raftery, M.J.; Pecher, G. Engineering NK cells for CAR therapy-recent advances in gene transfer methodology. Front. Immunol. 2021, 11, 611163. [Google Scholar] [CrossRef]
- Guillerey, C.; Huntington, N.D.; Smyth, M.J. Targeting natural killer cells in cancer immunotherapy. Nat. Immunol. 2016, 17, 1025–1036. [Google Scholar] [CrossRef]
- Mehta, R.S.; Rezvani, K. Chimeric antigen receptor expressing natural killer cells for the immunotherapy of cancer. Front. Immunol. 2018, 9, 283. [Google Scholar] [CrossRef] [Green Version]
- Hermanson, D.L.; Bendzick, L.; Pribyl, L.; McCullar, V.; Vogel, R.I.; Miller, J.S.; Geller, M.A.; Kaufman, D.S. Induced pluripotent stem cell-derived natural killer cells for treatment of ovarian cancer. Stem Cells 2016, 34, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Chadwick, K.; Wang, L.; Li, L.; Menendez, P.; Murdoch, B.; Rouleau, A.; Bhatia, M. Cytokines and BMP-4 promote hematopoietic differentiation of human embryonic stem cells. Blood 2003, 102, 906–915. [Google Scholar] [CrossRef]
- Kennedy, M.; D’Souza, S.L.; Lynch-Kattman, M.; Schwantz, S.; Keller, G. Development of the hemangioblast defines the onset of hematopoiesis in human ES cell differentiation cultures. Blood 2007, 109, 2679–2687. [Google Scholar] [CrossRef] [Green Version]
- Pick, M.; Azzola, L.; Mossman, A.; Stanley, E.G.; Elefanty, A.G. Differentiation of human embryonic stem cells in serum-free medium reveals distinct roles for bone morphogenetic protein 4, vascular endothelial growth factor, stem cell factor, and fibroblast growth factor 2 in hematopoiesis. Stem Cells 2007, 25, 2206–2214. [Google Scholar] [CrossRef]
- Pearson, S.; Sroczynska, P.; Lacaud, G.; Kouskoff, V. The stepwise specification of embryonic stem cells to hematopoietic fate is driven by sequential exposure to Bmp4, activin A, bFGF and VEGF. Development 2008, 135, 1525–1535. [Google Scholar] [CrossRef] [Green Version]
- Luanpitpong, S.; Poohadsuan, J.; Klaihmon, P.; Kang, X.; Tangkiettrakul, K.; Issaragrisil, S. Metabolic sensor O-GlcNAcylation regulates megakaryopoiesis and thrombopoiesis through c-Myc stabilization and integrin perturbation. Stem Cells 2021, 39, 787–802. [Google Scholar] [CrossRef]
- Luanpitpong, S.; Kang, X.; Janan, M.; Thumanu, K.; Li, J.; Kheolamai, P.; Issaragrisil, S. Metabolic sensor O-GlcNAcylation regulates erythroid differentiation and globin production via BCL11A. Stem Cell Res. Ther. 2022, 13, 274. [Google Scholar] [CrossRef]
- Luanpitpong, S.; Rodboon, N.; Samart, P.; Janan, M.; Klaihmon, P.; Lorthongpanich, C.; U-Pratya, Y.; Issaragrisil, S. Inhibition of O-GlcNAcase inhibits hematopoietic and leukemic stem cell self-renewal and drives dendritic cell differentiation via STAT3/5 signaling. Stem Cells 2022, 40, 1078–1093. [Google Scholar] [CrossRef]
- Stewart, S.A.; Dykxhoorn, D.M.; Palliser, D.; Mizuno, H.; Yu, E.Y.; An, D.S.; Sabatini, D.M.; Chen, I.S.; Hahn, W.C.; Sharp, P.A.; et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 2003, 9, 493–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klaihmon, P.; Kang, X.; Issaragrisil, S.; Luanpitpong, S. Generation and Functional Characterization of Anti-CD19 Chimeric Antigen Receptor-Natural Killer Cells from Human Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2023, 24, 10508. https://doi.org/10.3390/ijms241310508
Klaihmon P, Kang X, Issaragrisil S, Luanpitpong S. Generation and Functional Characterization of Anti-CD19 Chimeric Antigen Receptor-Natural Killer Cells from Human Induced Pluripotent Stem Cells. International Journal of Molecular Sciences. 2023; 24(13):10508. https://doi.org/10.3390/ijms241310508
Chicago/Turabian StyleKlaihmon, Phatchanat, Xing Kang, Surapol Issaragrisil, and Sudjit Luanpitpong. 2023. "Generation and Functional Characterization of Anti-CD19 Chimeric Antigen Receptor-Natural Killer Cells from Human Induced Pluripotent Stem Cells" International Journal of Molecular Sciences 24, no. 13: 10508. https://doi.org/10.3390/ijms241310508