

Synthesis and Anticancer Activity of A-Ring-Modified Derivatives of Dihydrobetulin

Abstract
:1. Introduction
2. Results and Discussion
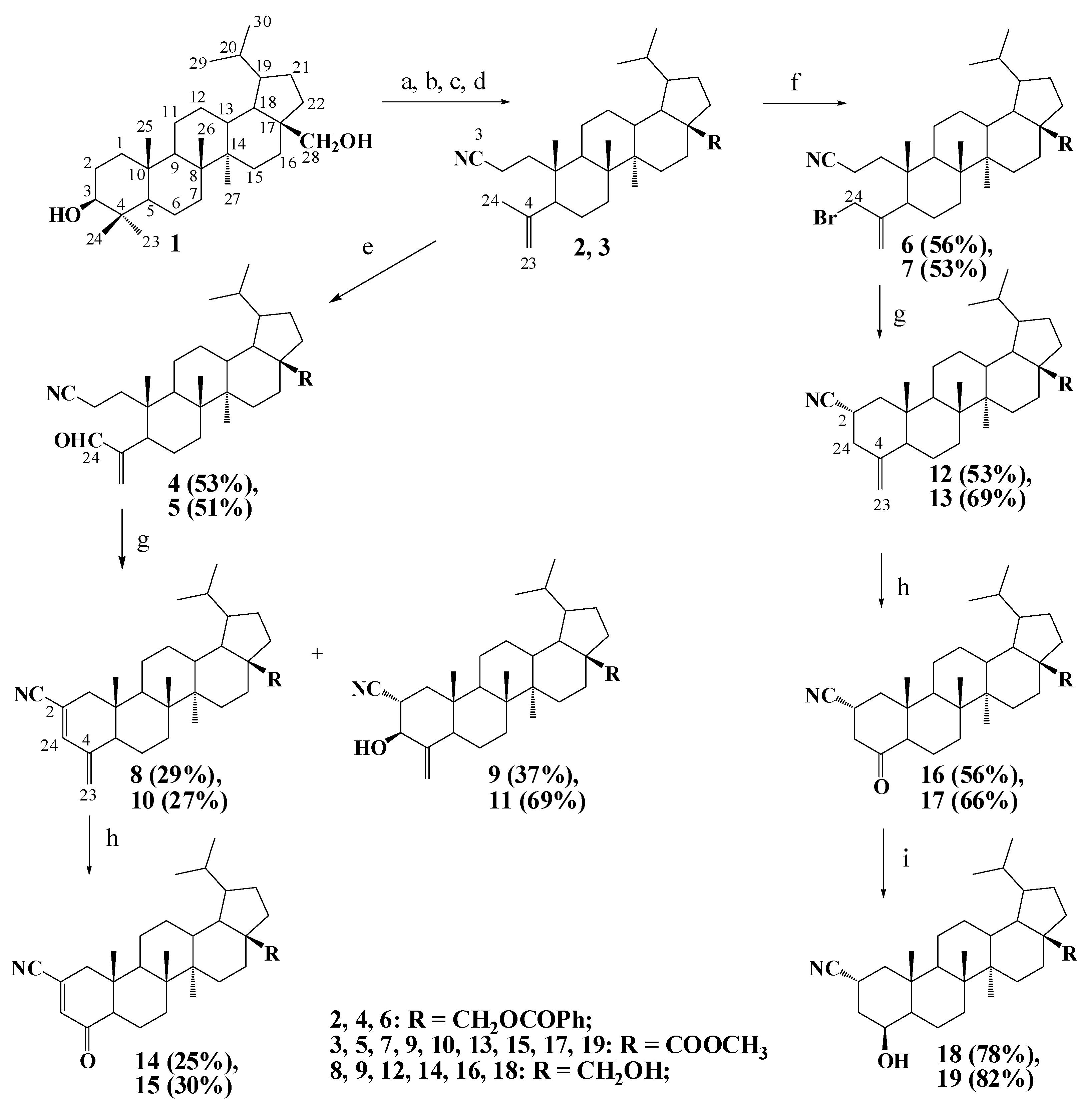
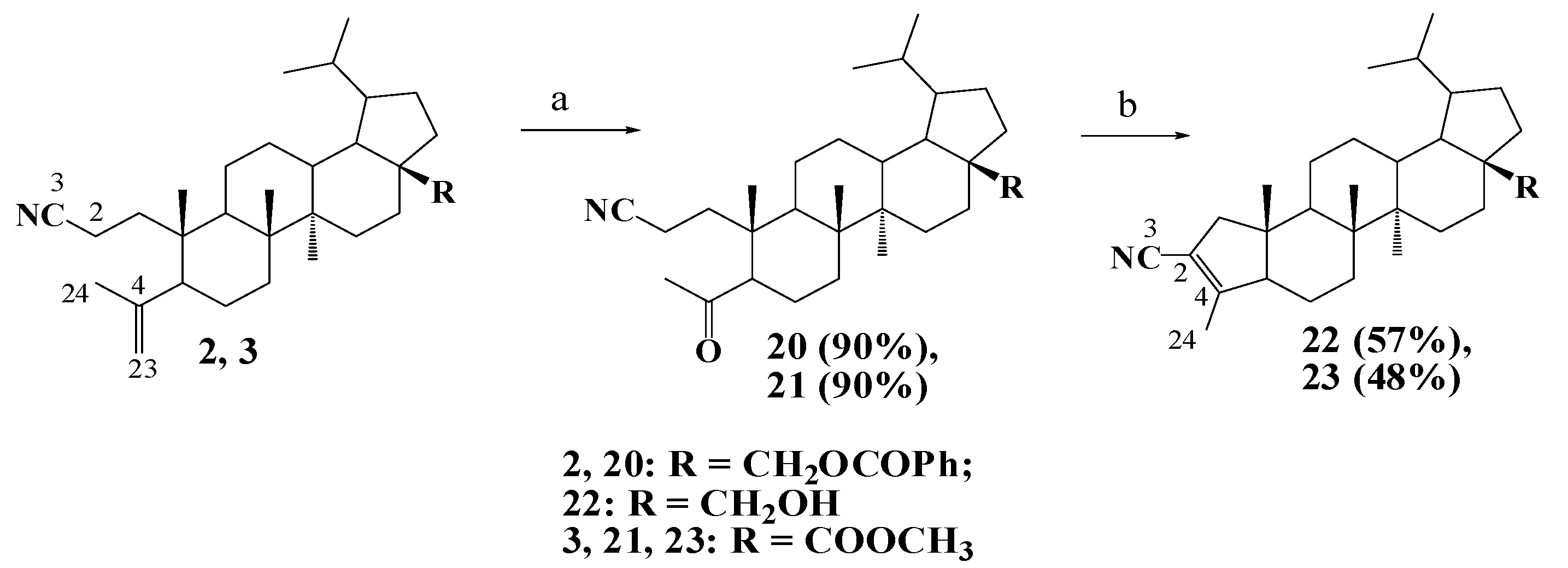
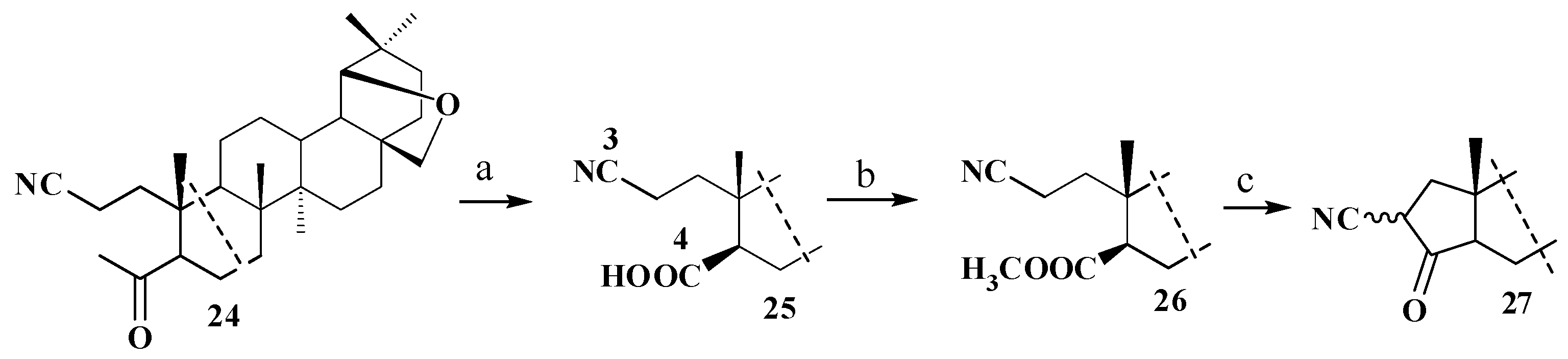
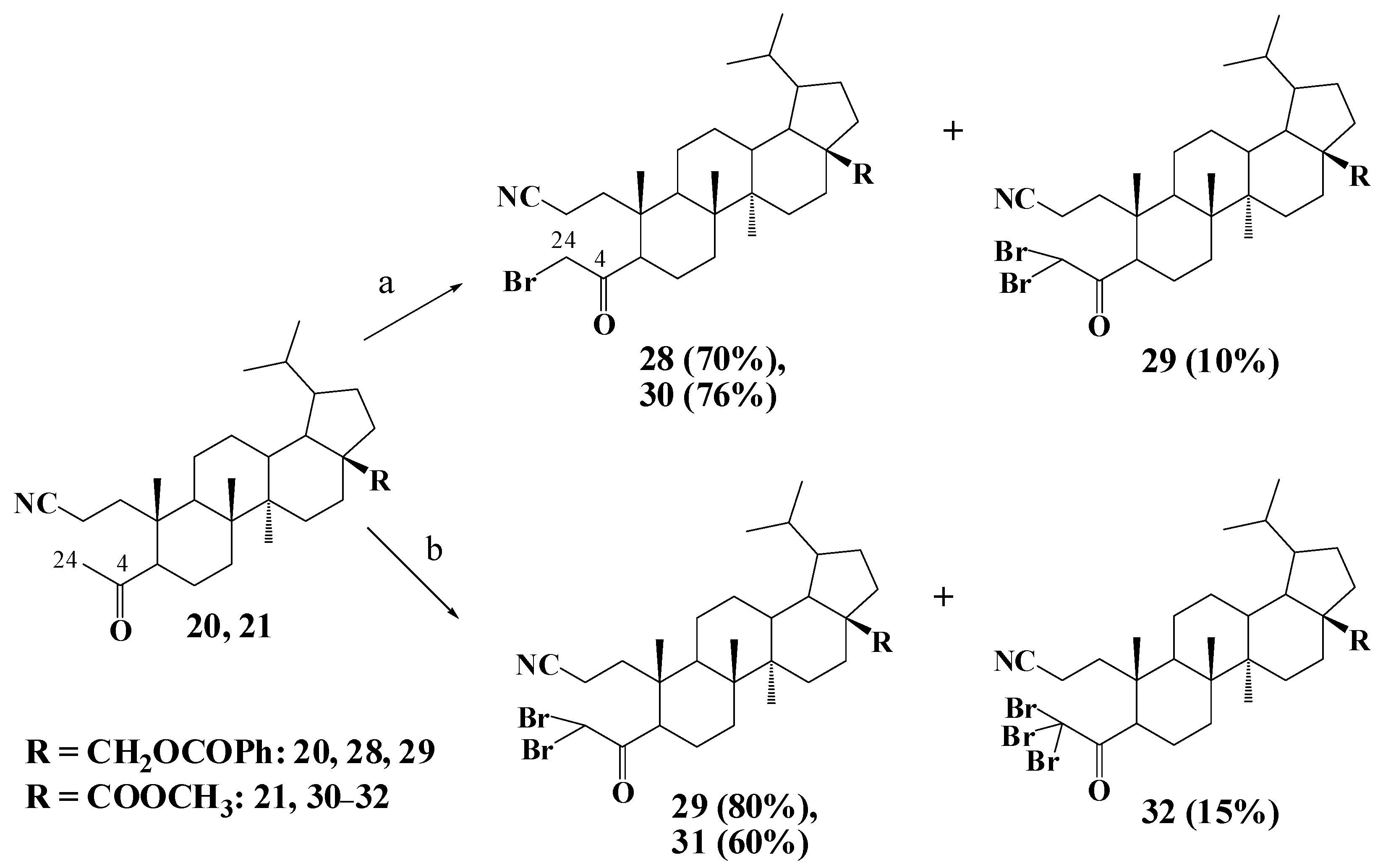
2.1. Chemistry
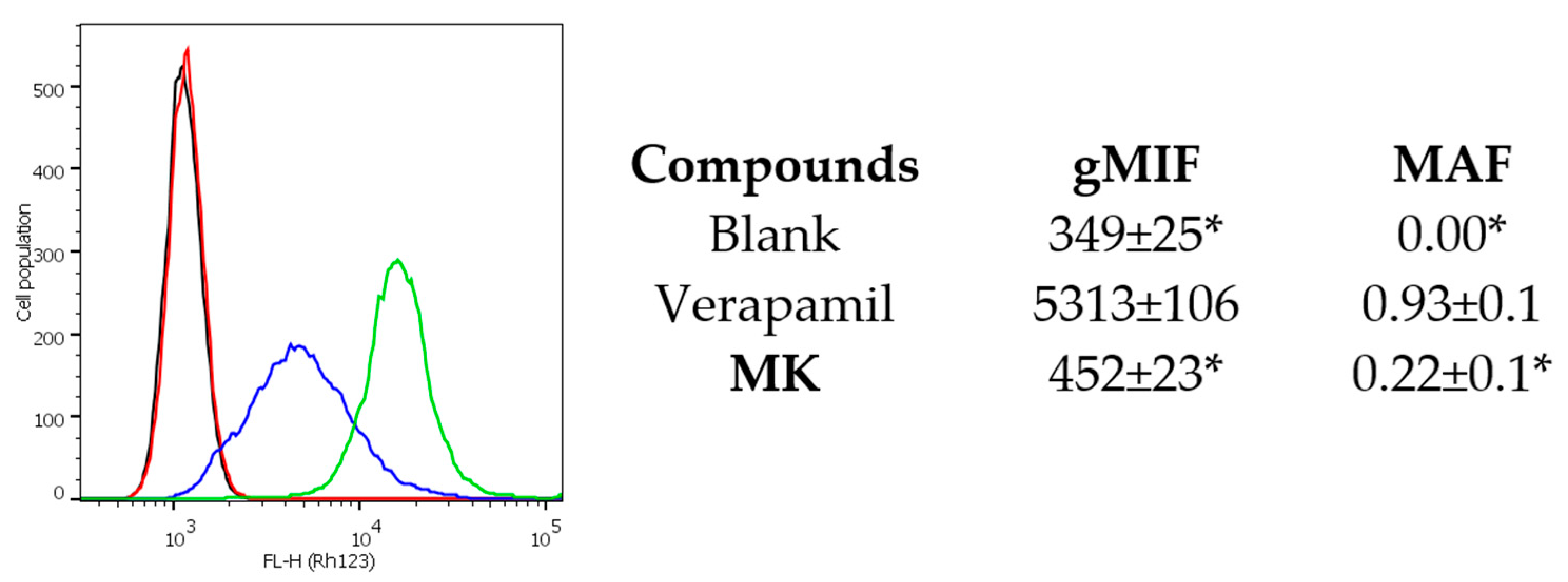
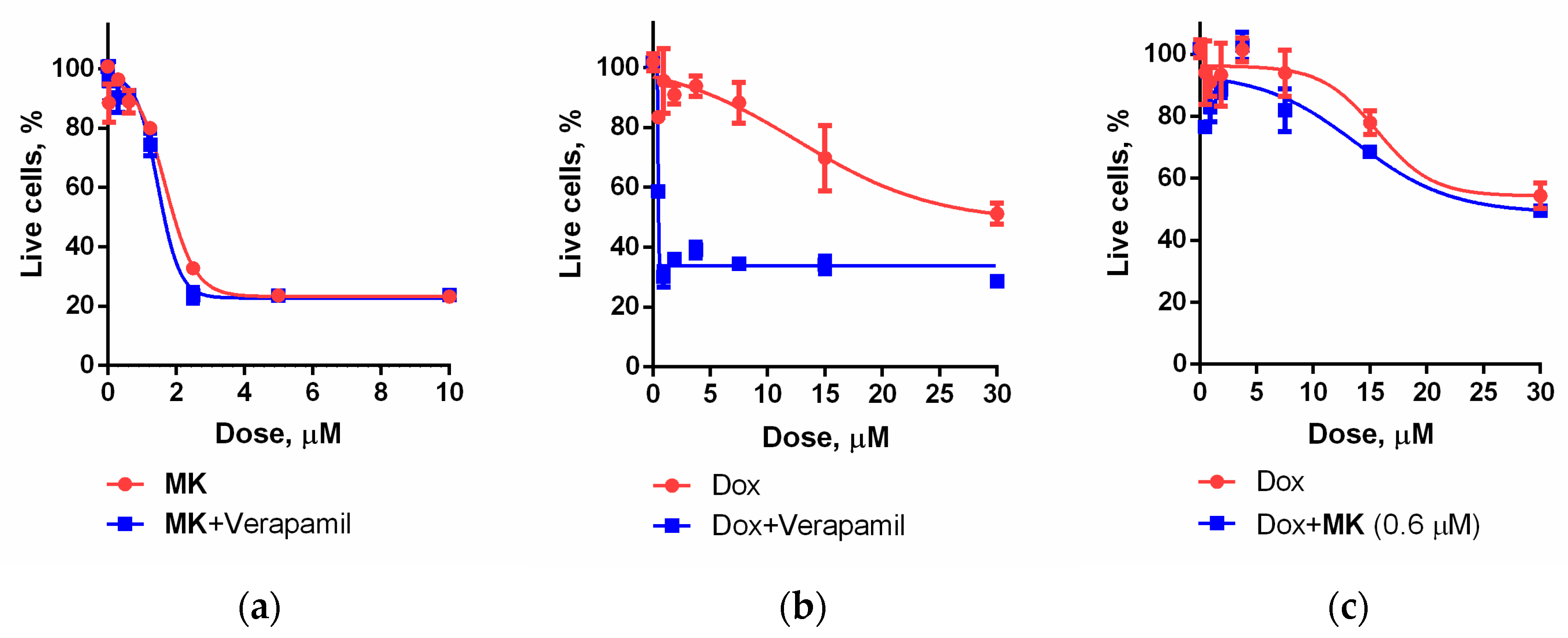
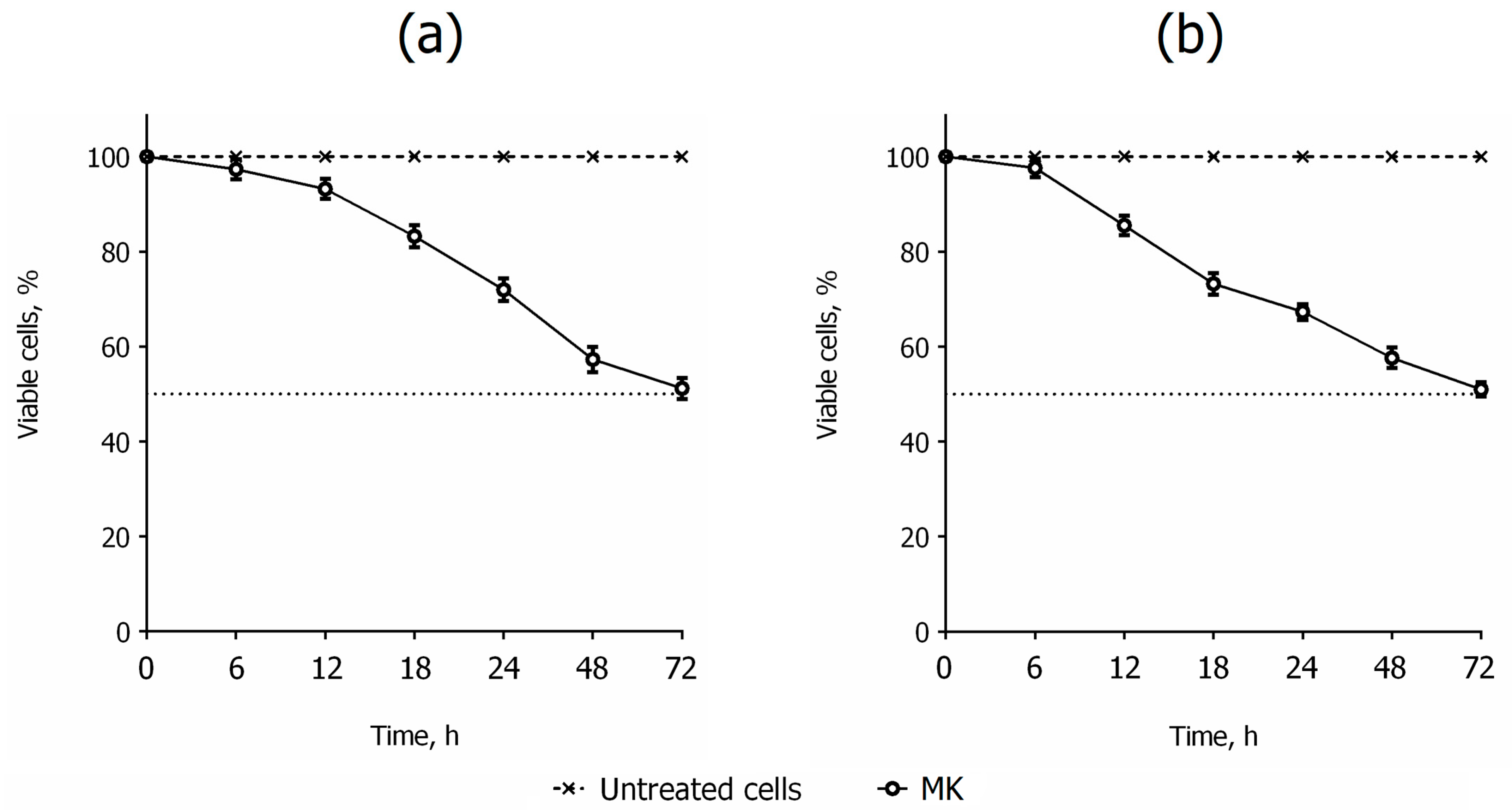
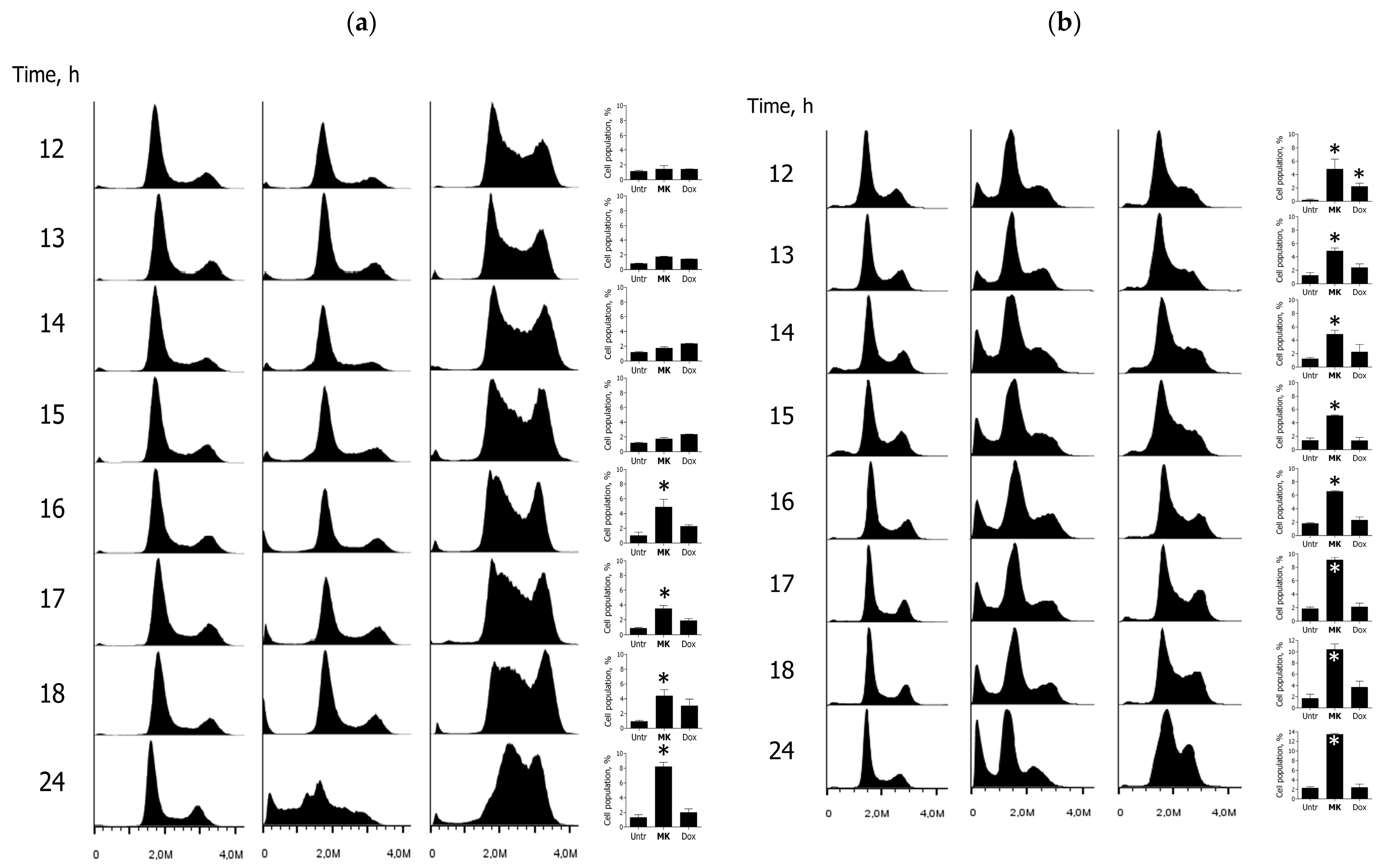
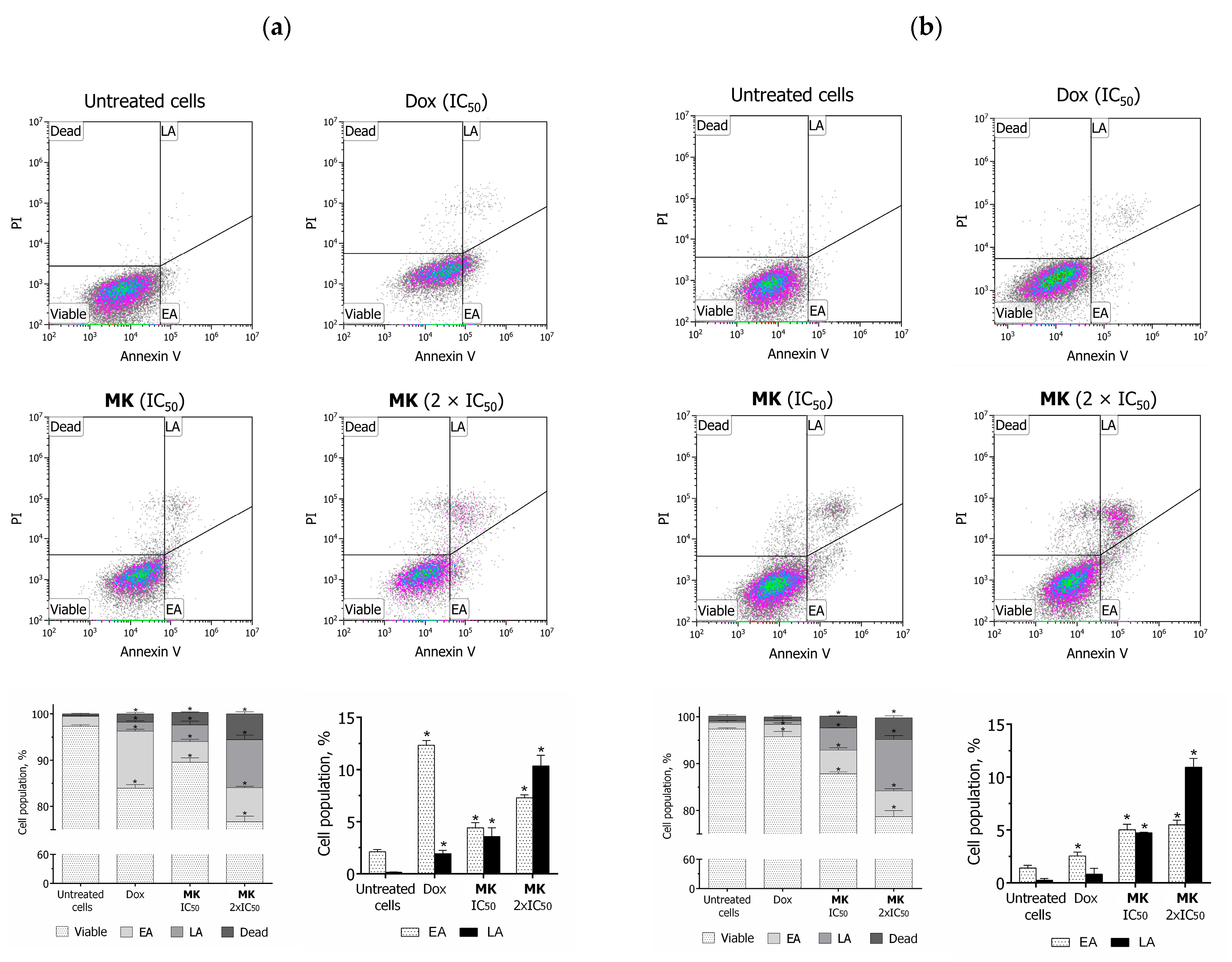
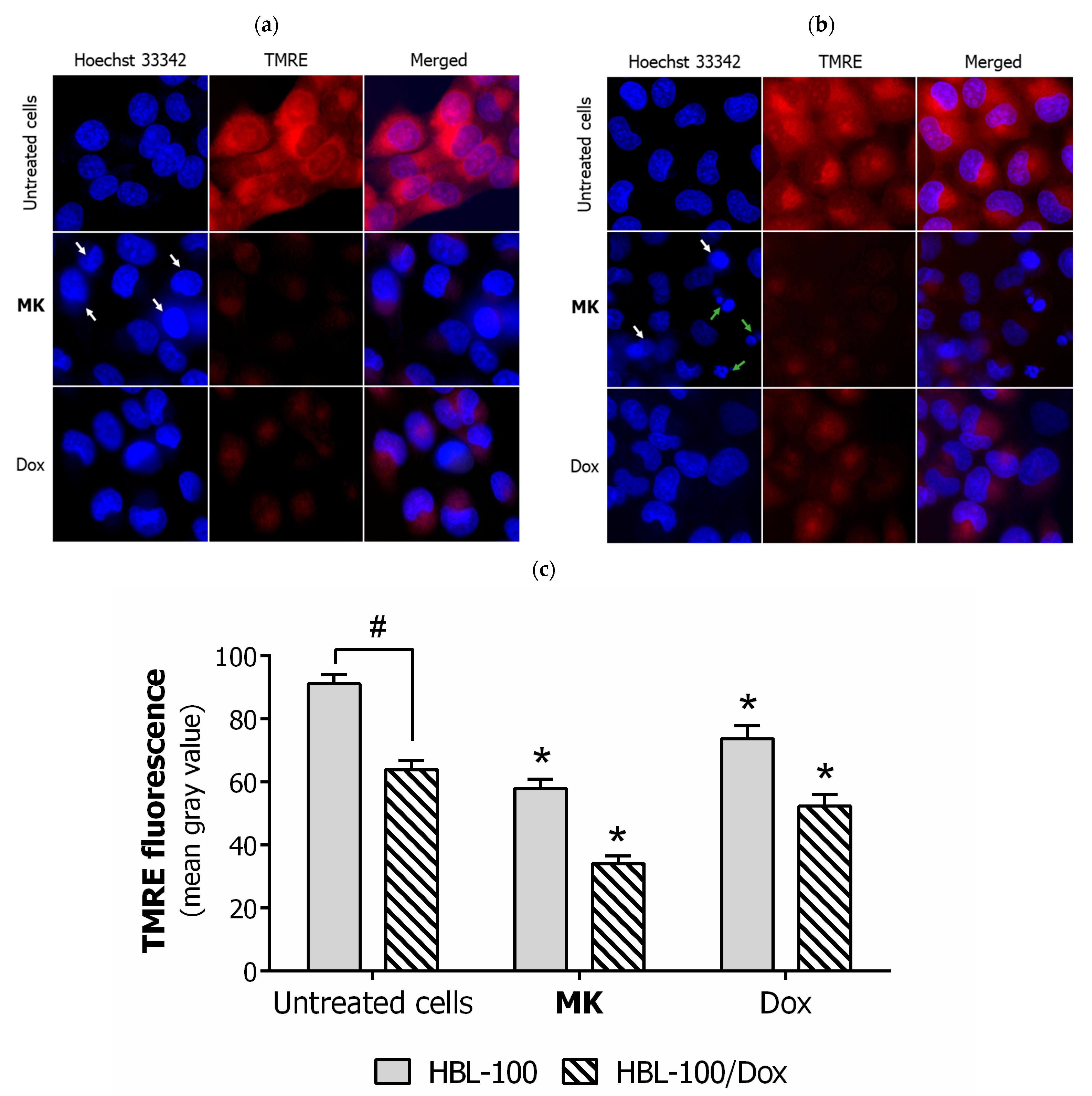
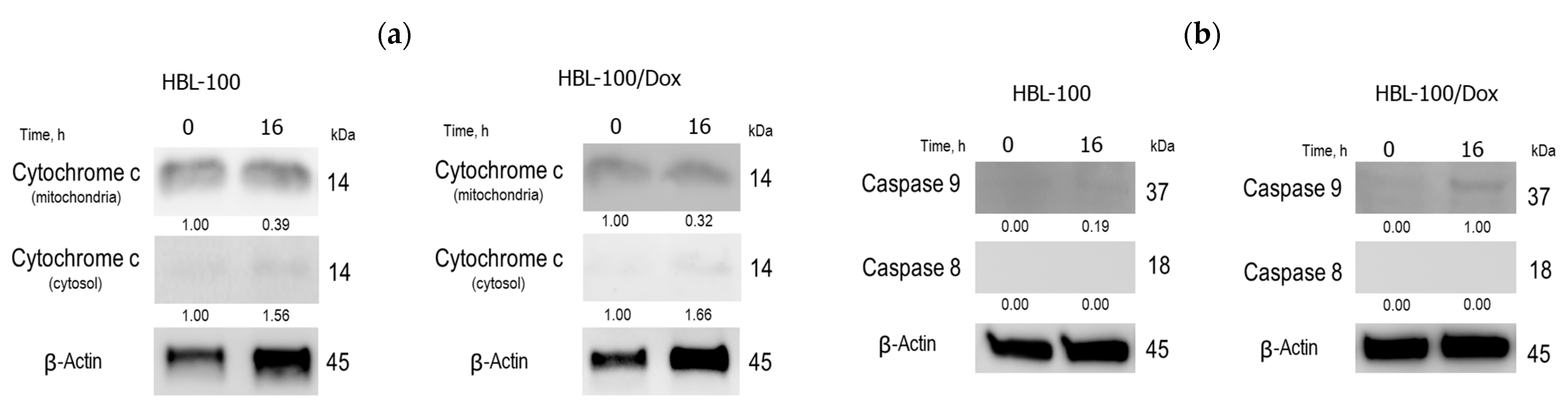
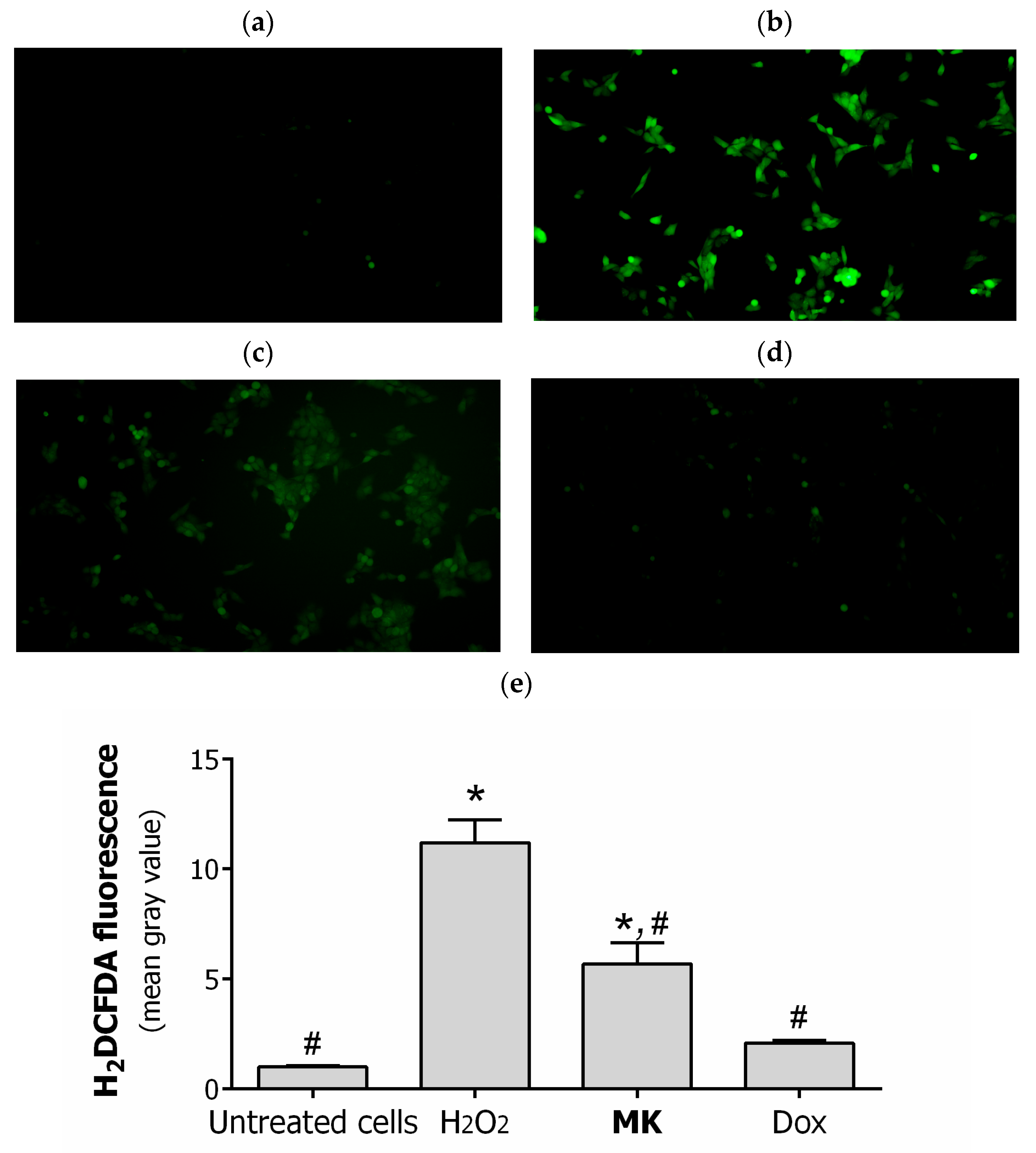
2.2. Biology
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for Synthesis of Compounds 4, 5
3.1.2. General Procedure for Synthesis of Compounds 6, 7
3.1.3. General Procedure for Synthesis of Compounds 8–13, 22, 23
3.1.4. General Procedure for Synthesis of Compounds 14–17, 20, 21
3.1.5. General Procedure for Synthesis of Compounds 18, 19
3.1.6. General Procedure for Synthesis of Compounds 28–30
3.1.7. General Procedure for Synthesis of Compounds 29, 31, 32
3.2. Biology
3.2.1. Cell Lines and Culture Conditions
3.2.2. MTT Cell Viability Assay
3.2.3. In Silico P-gp Substrate and P-gp Inhibitor Prediction
3.2.4. Evaluation of P-gp Functional Activity
3.2.5. Combined Action of Compounds
3.2.6. Cell Cycle Analysis
3.2.7. Annexin V-FITC/PI Assay
3.2.8. Fluorescent Microscopy
3.2.9. Western Blotting
3.2.10. Flow Cytometric Analysis of Active Caspase-3
3.2.11. Statistics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottesman, M.M.; Lavi, O.; Hall, M.D.; Gillet, J.-P. Toward a Better Understanding of the Complexity of Cancer Drug Resistance. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Catalano, A.; Iacopetta, D.; Ceramella, J.; Scumaci, D.; Giuzio, F.; Saturnino, C.; Aquaro, S.; Rosano, C.; Sinicropi, M.S. Multidrug Resistance (MDR): A Widespread Phenomenon in Pharmacological Therapies. Molecules 2022, 27, 616. [Google Scholar] [CrossRef]
- Gottesman, M.M. Mechanisms of Cancer Drug Resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Qin, Z.; Zhang, W.D.; Cheng, G.; Yehuda, A.G.; Ashby, C.R.; Chen, Z.S.; Cheng, X.D.; Qin, J.J. Medicinal Chemistry Strategies to Discover P-Glycoprotein Inhibitors: An Update. Drug Resist. Updat. 2020, 49, 100681. [Google Scholar] [CrossRef]
- Thomas, H.; Coley, H.M. Overcoming Multidrug Resistance in Cancer: An Update on the Clinical Strategy of Inhibiting P-Glycoprotein. Cancer Control 2003, 10, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Dantzic, D.; Noel, P.; Merien, F.; Liu, D.-X.; Lu, J.; Han, H.; McKeage, M.; Li, Y. The Effects of Synthetically Modified Natural Compounds on ABC Transporters. Pharmaceutics 2018, 10, 127. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, B.M.F.; Cardoso, D.S.P.; Ferreira, M.J.U. Overcoming Multidrug Resistance: Flavonoid and Terpenoid Nitrogen-Containing Derivatives as ABC Transporter Modulators. Molecules 2020, 25, 3364. [Google Scholar] [CrossRef]
- Csuk, R. Betulinic Acid and Its Derivatives: A Patent Review (2008–2013). Expert Opin. Ther. Pat. 2014, 24, 913–923. [Google Scholar] [CrossRef]
- Zhang, D.-M.; Xu, H.-G.; Wang, L.; Li, Y.-J.; Sun, P.-H.; Wu, X.-M.; Wang, G.-J.; Chen, W.-M.; Ye, W.-C. Betulinic Acid and Its Derivatives as Potential Antitumor Agents. Med. Res. Rev. 2015, 35, 1127–1155. [Google Scholar] [CrossRef]
- Lombrea, A.; Scurtu, A.D.; Avram, S.; Pavel, I.Z.; Turks, M.; Lugiņina, J.; Peipiņš, U.; Dehelean, C.A.; Soica, C.; Danciu, C. Anticancer Potential of Betulonic Acid Derivatives. Int. J. Mol. Sci. 2021, 22, 3676. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Li, X.; Dong, S.; Zhou, W. Betulinic Acid in the Treatment of Tumour Diseases: Application and Research Progress. Biomed. Pharmacother. 2021, 142, 111990. [Google Scholar] [CrossRef] [PubMed]
- Nistor, G.; Trandafirescu, C.; Prodea, A.; Milan, A.; Cristea, A.; Ghiulai, R.; Racoviceanu, R.; Mioc, A.; Mioc, M.; Ivan, V.; et al. Semisynthetic Derivatives of Pentacyclic Triterpenes Bearing Heterocyclic Moieties with Therapeutic Potential. Molecules 2022, 27, 6552. [Google Scholar] [CrossRef] [PubMed]
- Laiolo, J.; Barbieri, C.L.; Joray, M.B.; Lanza, P.A.; Palacios, S.M.; Vera, D.M.A.; Carpinella, M.C. Plant Extracts and Betulin from Ligaria Cuneifolia Inhibit P-Glycoprotein Function in Leukemia Cells. Food Chem. Toxicol. 2021, 147, 111922. [Google Scholar] [CrossRef]
- Saeed, M.E.M.; Mahmoud, N.; Sugimoto, Y.; Efferth, T.; Abdel-Aziz, H. Betulinic Acid Exerts Cytotoxic Activity against Multidrug-Resistant Tumor Cells via Targeting Autocrine Motility Factor Receptor (AMFR). Front. Pharmacol. 2018, 9, 481. [Google Scholar] [CrossRef]
- Zhang, D.-M.; Shu, C.; Chen, J.-J.; Sodani, K.; Wang, J.; Bhatnagar, J.; Lan, P.; Ruan, Z.-X.; Xiao, Z.-J.; Ambudkar, S.V.; et al. BBA, a Derivative of 23-Hydroxybetulinic Acid, Potently Reverses ABCB1-Mediated Drug Resistance In Vitro and In Vivo. Mol. Pharm. 2012, 9, 3147–3159. [Google Scholar] [CrossRef]
- Zhang, D.M.; Li, Y.J.; Shu, C.; Ruan, Z.X.; Chen, W.M.; Yiu, A.; Peng, Y.H.; Wang, J.; Lan, P.; Yao, Z.; et al. Bipiperidinyl Derivatives of 23-Hydroxybetulinic Acid Reverse Resistance of HepG2/ADM and MCF-7/ADR Cells. Anticancer Drugs 2013, 24, 441–454. [Google Scholar] [CrossRef]
- Rybalkina, E.Y.; Moiseeva, N.I.; Karamysheva, A.F.; Eroshenko, D.V.; Konysheva, A.V.; Nazarov, A.V.; Grishko, V.V. Triterpenoids with Modified A-Ring as Modulators of P-Gp-Dependent Drug-Resistance in Cancer Cells. Chem. Biol. Interact. 2021, 348, 109645. [Google Scholar] [CrossRef]
- Moiseeva, N.; Eroshenko, D.; Laletina, L.; Rybalkina, E.; Susova, O.; Karamysheva, A.; Tolmacheva, I.; Nazarov, M.; Grishko, V. The Molecular Mechanisms of Oleanane Aldehyde-β-Enone Cytotoxicity against Doxorubicin-Resistant Cancer Cells. Biology 2023, 12, 415. [Google Scholar] [CrossRef]
- Grishko, V.V.; Galaiko, N.V.; Tolmacheva, I.A.; Kucherov, I.I.; Eremin, V.F.; Boreko, E.I.; Savinova, O.V.; Slepukhin, P.A. Functionalization, Cyclization and Antiviral Activity of A-Secotriterpenoids. Eur. J. Med. Chem. 2014, 83, 601–608. [Google Scholar] [CrossRef]
- Tolmacheva, I.A.; Nazarov, A.V.; Eroshenko, D.V.; Grishko, V.V. Synthesis, cytotoxic evaluation, and molecular docking studies of the semisynthetic “triterpenoid-steroid” hybrids. Steroids 2018, 140, 131–143. [Google Scholar] [CrossRef]
- Nazarov, A.V.; Tolmacheva, I.A.; Zhukova, A.E.; Grishko, V.V. Synthetic Modification and Cytotoxic Evaluation of 2-Cyano-3,4-Secotriterpenic Methylketones. Chem. Pap. 2019, 73, 1767–1775. [Google Scholar] [CrossRef]
- Dračínský, M.; Hybelbauerová, S.; Sejbal, J.; Buděšínský, M. Preparation and Conformational Study of B-Ring Substituted Lupane Derivatives. Collect. Czechoslov. Chem. Commun. 2006, 71, 1131–1160. [Google Scholar] [CrossRef]
- Klinot, J.; Šumanová, V.; Vystrčil, A. Triterpenes. XXI. 3,4-Seco Derivatives of Betulinic Acid. Collect. Czechoslov. Chem. Commun. 1972, 37, 603–609. [Google Scholar] [CrossRef]
- Valterová, I.; Klinot, J.; Vystrčil, A. Preparation and Antibacterial Activity of Di-, Tri- and Tetraoic Acids Derived from 3,4-Secolupane. Collect. Czechoslov. Chem. Commun. 1983, 48, 649–661. [Google Scholar] [CrossRef]
- Saint-Ruf, C.; Nardeux, P.; Estrade, S.; Brouty-Boye, D.; Lavialle, C.; Rhim, J.S.; Cassingena, R. Accelerated Malignant Conversion of Human HBL-100 Cells by the v-Ki-Ras Oncogene. Exp. Cell Res. 1988, 176, 60–67. [Google Scholar] [CrossRef]
- Martins, A.; Vasas, A.; Schelz, Z.S.; Viveiros, M.; Molnár, J.; Hohmann, J.; Amaral, L. Constituents of Carpobrotus Edulis Inhibit P-Glycoprotein of MDR1-Transfected Mouse Lymphoma Cells. Anticancer Res. 2010, 30, 829–836. [Google Scholar]
- El-Readi, M.Z.; Al-Abd, A.M.; Althubiti, M.A.; Almaimani, R.A.; Al-Amoodi, H.S.; Ashour, M.L.; Wink, M.; Eid, S.Y. Multiple Molecular Mechanisms to Overcome Multidrug Resistance in Cancer by Natural Secondary Metabolites. Front. Pharmacol. 2021, 12, 658513. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, X.; Duan, Z.; Wang, X. An Update on Circumventing Multidrug Resistance in Cancer by Targeting P-Glycoprotein. Curr. Cancer Drug Targets 2017, 18, 677–696. [Google Scholar] [CrossRef]
- Wang, R.B.; Kuo, C.L.; Lien, L.L.; Lien, E.J. Structure-Activity Relationship: Analyses of P-Glycoprotein Substrates and Inhibitors. J. Clin. Pharm. Ther. 2003, 28, 203–228. [Google Scholar] [CrossRef]
- Guéniche, N.; Huguet, A.; Bruyere, A.; Habauzit, D.; Le Hégarat, L.; Fardel, O. Comparative in Silico Prediction of P-Glycoprotein-Mediated Transport for 2010–2020 US FDA-Approved Drugs Using Six Web-Tools. Biopharm. Drug Dispos. 2021, 42, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Jouan, E.; Le Vée, M.; Mayati, A.; Denizot, C.; Parmentier, Y.; Fardel, O. Evaluation of P-Glycoprotein Inhibitory Potential Using a Rhodamine 123 Accumulation Assay. Pharmaceutics 2016, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Saneja, A.; Panda, A.K. An Annexin V-FITC—Propidium Iodide-Based Method for Detecting Apoptosis in a Non-Small Cell Lung Cancer Cell Line. In Methods in Molecular Biology; Humana: New York, NY, USA, 2021; Volume 2279, pp. 213–223. [Google Scholar] [CrossRef]
- Crowley, L.C.; Marfell, B.J.; Waterhouse, N.J. Analyzing Cell Death by Nuclear Staining with Hoechst 33342. Cold Spring Harb. Protoc. 2016, 2016, 778–781. [Google Scholar] [CrossRef]
- De, P.; Carlson, J.H.; Leyland-Jones, B.; Williams, C.; Dey, N. Triple Fluorescence Staining to Evaluate Mechanism-Based Apoptosis Following Chemotherapeutic and Targeted Anti-Cancer Drugs in Live Tumor Cells. Sci. Rep. 2018, 8, 13192. [Google Scholar] [CrossRef] [Green Version]
- Jin, P.; Jiang, J.; Zhou, L.; Huang, Z.; Nice, E.C.; Huang, C.; Fu, L. Mitochondrial Adaptation in Cancer Drug Resistance: Prevalence, Mechanisms, and Management. J. Hematol. Oncol. 2022, 15, 97. [Google Scholar] [CrossRef]
- Giacomini, I.; Cortini, M.; Tinazzi, M.; Baldini, N.; Cocetta, V.; Ragazzi, E.; Avnet, S.; Montopoli, M. Contribution of Mitochondrial Activity to Doxorubicin-Resistance in Osteosarcoma Cells. Cancers 2023, 15, 1370. [Google Scholar] [CrossRef] [PubMed]
- Brentnall, M.; Rodriguez-Menocal, L.; De Guevara, R.L.; Cepero, E.; Boise, L.H. Caspase-9, Caspase-3 and Caspase-7 Have Distinct Roles during Intrinsic Apoptosis. BMC Cell Biol. 2013, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Huang, Y. Mitochondrial Targeted Strategies and Their Application for Cancer and Other Diseases Treatment. J. Pharm. Investig. 2020, 50, 271–293. [Google Scholar] [CrossRef]
- Serafim, T.L.; Carvalho, F.S.; Bernardo, T.C.; Pereira, G.C.; Perkins, E.; Holy, J.; Krasutsky, D.A.; Kolomitsyna, O.N.; Krasutsky, P.A.; Oliveira, P.J. New Derivatives of Lupane Triterpenoids Disturb Breast Cancer Mitochondria and Induce Cell Death. Bioorg. Med. Chem. 2014, 22, 6270–6287. [Google Scholar] [CrossRef]
- Dzhemileva, L.U.; Tuktarova, R.A.; Dzhemilev, U.M.; D’yakonov, V.A. Pentacyclic Triterpenoids-Based Ionic Compounds: Synthesis, Study of Structure–Antitumor Activity Relationship, Effects on Mitochondria and Activation of Signaling Pathways of Proliferation, Genome Reparation and Early Apoptosis. Cancers 2023, 15, 756. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Semenova, A.A.; Ilzorkina, A.I.; Mikheeva, I.B.; Yashin, V.A.; Penkov, N.V.; Vydrina, V.A.; Ishmuratov, G.Y.; Sharapov, V.A.; Khoroshavina, E.I.; et al. Effect of Betulin and Betulonic Acid on Isolated Rat Liver Mitochondria and Liposomes. Biochim. Biophys. Acta (BBA)-Biomembr. 2020, 1862, 183383. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhang, X.; Zhao, M.; Wang, Y.; Cheng, X.; Wang, D.; Xu, Y.; Du, Z.; Yu, X. Celastrol Targets Mitochondrial Respiratory Chain Complex I to Induce Reactive Oxygen Species-Dependent Cytotoxicity in Tumor Cells. BMC Cancer 2011, 11, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valencia-Chan, L.S.; Estrada-Alfaro, N.; Ceballos-Cruz, J.J.; Torres-Tapia, L.W.; Peraza-Sánchez, S.R.; Moo-Puc, R.E. Lupane Triterpene Derivatives Improve Antiproliferative Effect on Leukemia Cells through Apoptosis Induction. Molecules 2022, 27, 8263. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An Integrated Online Platform for Accurate and Comprehensive Predictions of ADMET Properties. Nucleic Acids Res. 2021, 49, 5–14. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Wang, P.-H.; Tu, Y.-S.; Tseng, Y.J. PgpRules: A Decision Tree Based Prediction Server for P-Glycoprotein Substrates and Inhibitors. Bioinformatics 2019, 35, 4535. [Google Scholar] [CrossRef] [Green Version]
- Stromskaya, T.P.; Rybalkina, Y.Y.; Turkina, A.G.; Zabotina, T.N.; Logacheva, N.P.; Zakharova, Y.S.; Mechetner, Y.B.; Baryshnikov, A.Y.; Khoroshko, N.D.; Stavrovskaya, A.A. Functional Activity and Expression of P-Glycoprotein in Chronic Myeloid Leukemia. Ter. Arkhiv 2001, 73, 20–25. [Google Scholar]
- Homolya, L.; Holló, M.; Müller, M.; Mechetner, E.B.; Sarkadi, B. A New Method for Quantitative Assessment of P-Glycoprotein-Related Multidrug Resistance in Tumour Cells. Br. J. Cancer 1996, 73, 849–855. [Google Scholar] [CrossRef] [Green Version]
- Shihan, M.H.; Novo, S.G.; Le Marchand, S.J.; Wang, Y.; Duncan, M.K. A Simple Method for Quantitating Confocal Fluorescent Images. Biochem. Biophys. Rep. 2021, 25, 100916. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | IC50 Values (Mean ± SD), µM | |||||
|---|---|---|---|---|---|---|
| 12 | 14 | 19 | 30 | 31 (MK) | Dox | |
| HEK293 | 76.5 ± 2.5 | 77.8 ± 2.9 | 34.6 ± 1.1 | 24.2 ± 0.5 | 11.5 ± 0.6 | 0.4 ± 0.1 |
| MCF-7 | 64.2 ± 3.0 | 56.7 ± 3.7 | 47.9 ± 0.3 | 12.0 ± 0.1 | 7.1 ± 0.7 | 0.4 ± 0.1 |
| HCT116 | 51.0 ± 3.7 | 49.9 ± 0.1 | 28.2 ± 2.1 | 13.5 ± 0.3 | 3.9 ± 0.1 | 1.3 ± 0.3 |
| RD TE32 | 98.6 ± 2.3 | 49.1 ± 1.4 | 25.2 ± 0.1 | 7.1 ± 0.9 | 0.7 ± 0.1 | 1.3 ± 0.1 |
| MS | >200 | 69.1 ± 1.0 | 22.2 ± 2.0 | 45.9 ± 2.2 | 16.5 ± 1.5 | 1.3 ± 0.2 |
| A549 | 44.4 ± 2.4 | 63.2 ± 1.4 | 25.9 ± 1.8 | 27.7 ± 0.3 | 7.0 ± 0.2 | 2.0 ± 0.2 |
| PC-3 | 55.7 ± 4.9 | 28.3 ± 2.3 | 17.2 ± 0.7 | 34.0 ± 1.3 | 10.4 ± 1.3 | 12.6 ± 0.2 |
| HEpG2 | >100 | >100 | 31.8 ± 2.7 | 49.6 ± 0.5 | 16.6 ± 0.4 | 1.8 ± 0.3 |
| HBL-100 | 36.6 ± 1.4 | 46.9 ± 2.2 | 16.0 ± 0.9 | 3.6 ± 0.3 | 1.0 ± 0.1 | 0.4 ± 0.1 |
| HBL-100/Dox | 63.0 ± 1.9 | 54.0 ± 1.8 | 21.1 ± 2.2 | 4.5 ± 0.2 | 1.8 ± 0.1 | 29.6 ± 0.9 |
| Compounds | Web-Tools | Substrate | Inhibitor | MlogP |
|---|---|---|---|---|
| MK | ADMETlab 2.0 1 | −−− | +++ | 5.98 |
| pkCSM | − | + | 8.07 | |
| PgpRules | − | + | 8.07 | |
| Verapamil | ADMETlab 2.0 1 | +++ | +++ | 3.33 |
| pkCSM | + | + | 5.09 | |
| PgpRules | + | + | 3.13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolmacheva, I.; Beloglazova, Y.; Nazarov, M.; Gagarskikh, O.; Grishko, V. Synthesis and Anticancer Activity of A-Ring-Modified Derivatives of Dihydrobetulin. Int. J. Mol. Sci. 2023, 24, 9863. https://doi.org/10.3390/ijms24129863
Tolmacheva I, Beloglazova Y, Nazarov M, Gagarskikh O, Grishko V. Synthesis and Anticancer Activity of A-Ring-Modified Derivatives of Dihydrobetulin. International Journal of Molecular Sciences. 2023; 24(12):9863. https://doi.org/10.3390/ijms24129863
Chicago/Turabian StyleTolmacheva, Irina, Yulia Beloglazova, Mikhail Nazarov, Olga Gagarskikh, and Victoria Grishko. 2023. "Synthesis and Anticancer Activity of A-Ring-Modified Derivatives of Dihydrobetulin" International Journal of Molecular Sciences 24, no. 12: 9863. https://doi.org/10.3390/ijms24129863