
Extended Depth of Focus Two-Photon Light-Sheet Microscopy for In Vivo Fluorescence Imaging of Large Multicellular Organisms at Cellular Resolution

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
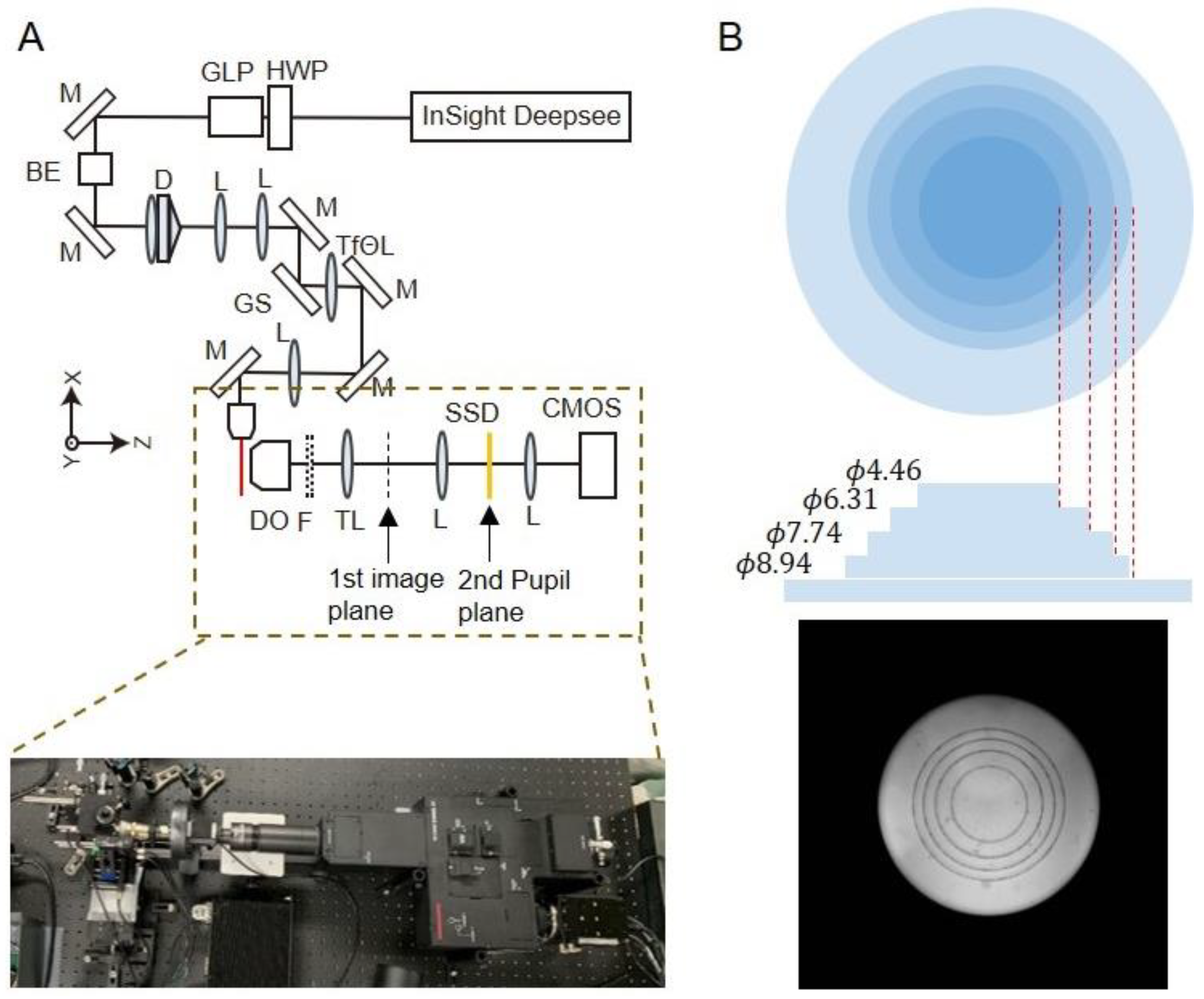
2.1. Construction of Two-Photon Light-Sheet Microscopy with a DOF Device
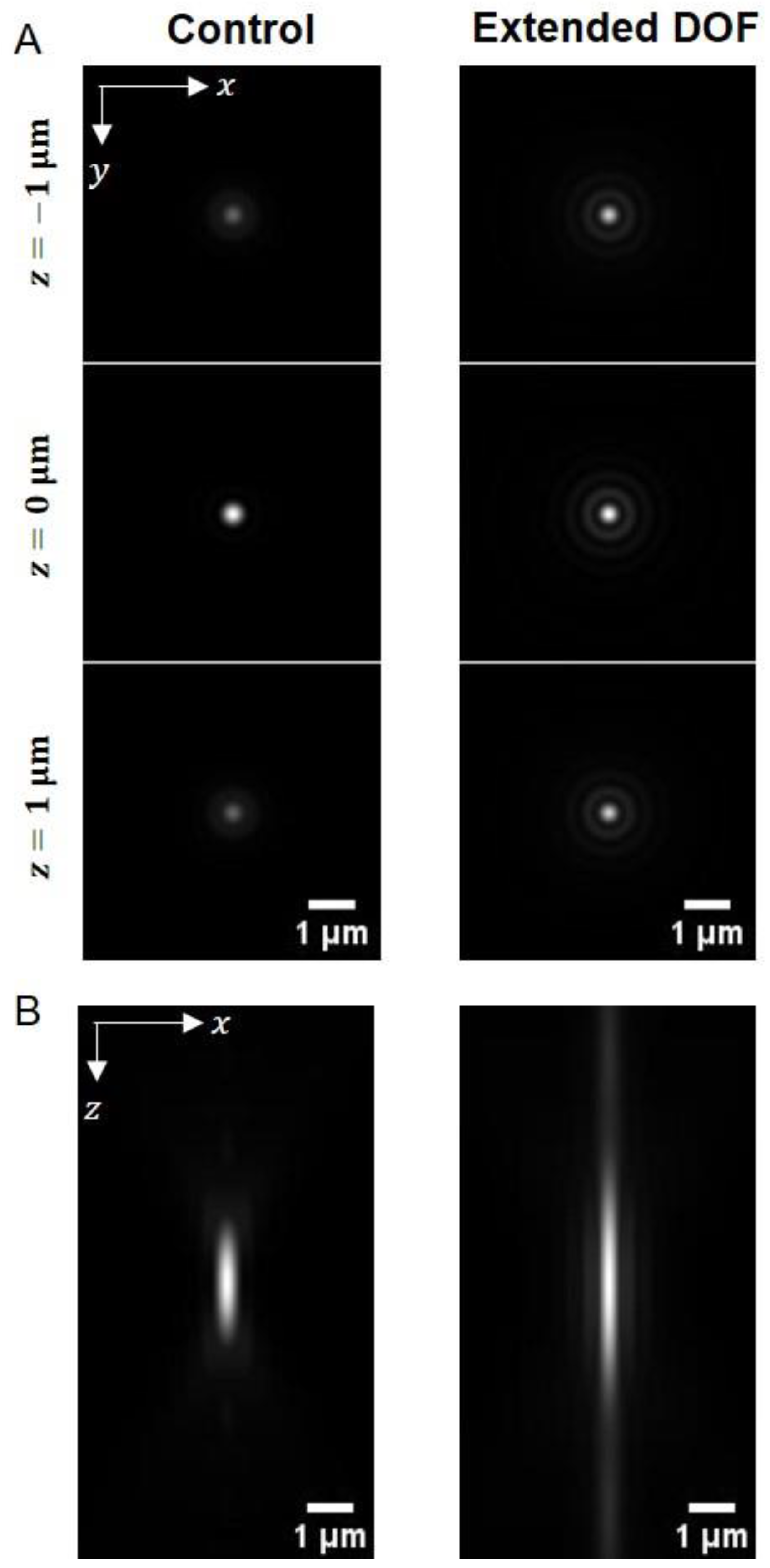
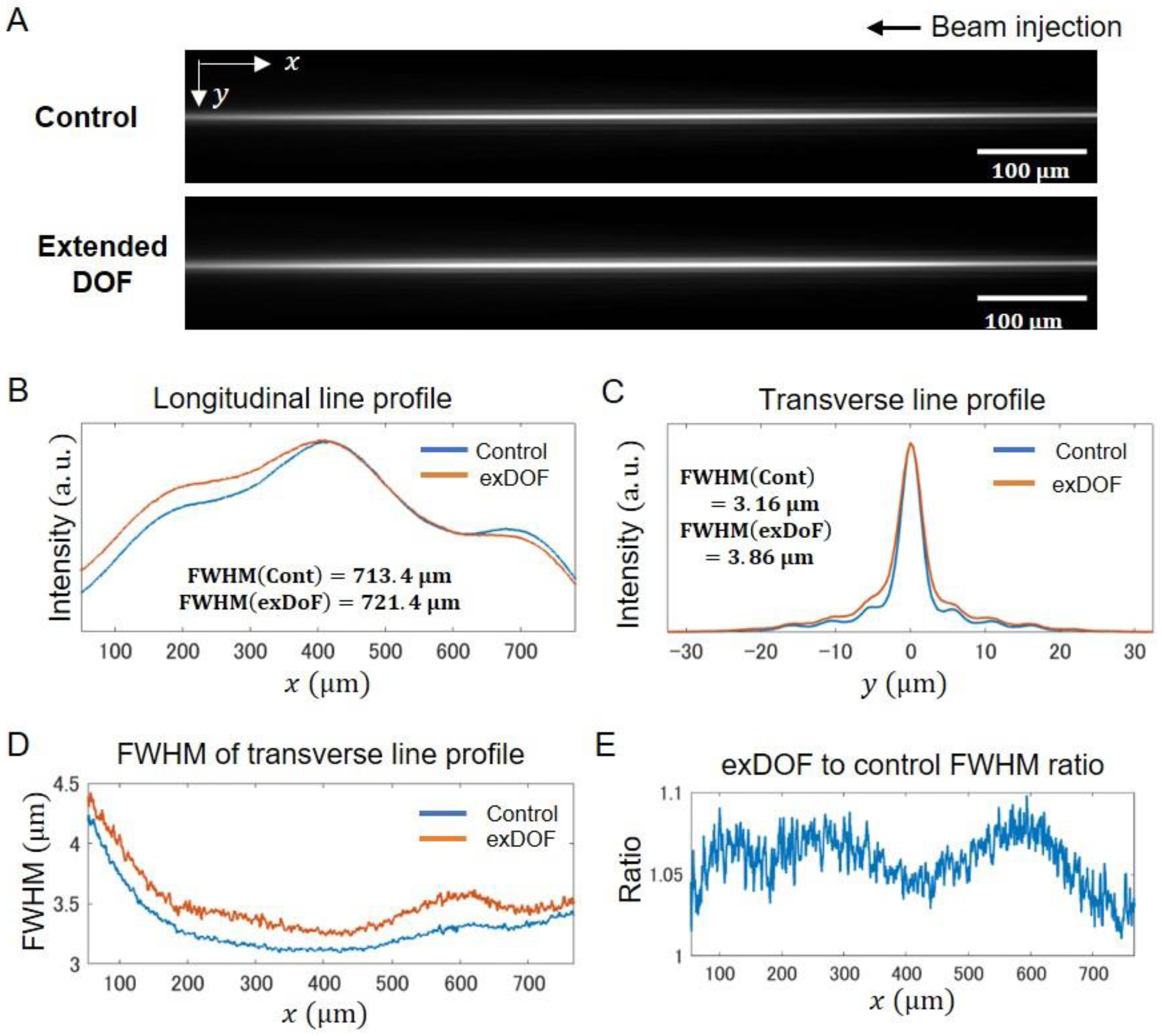
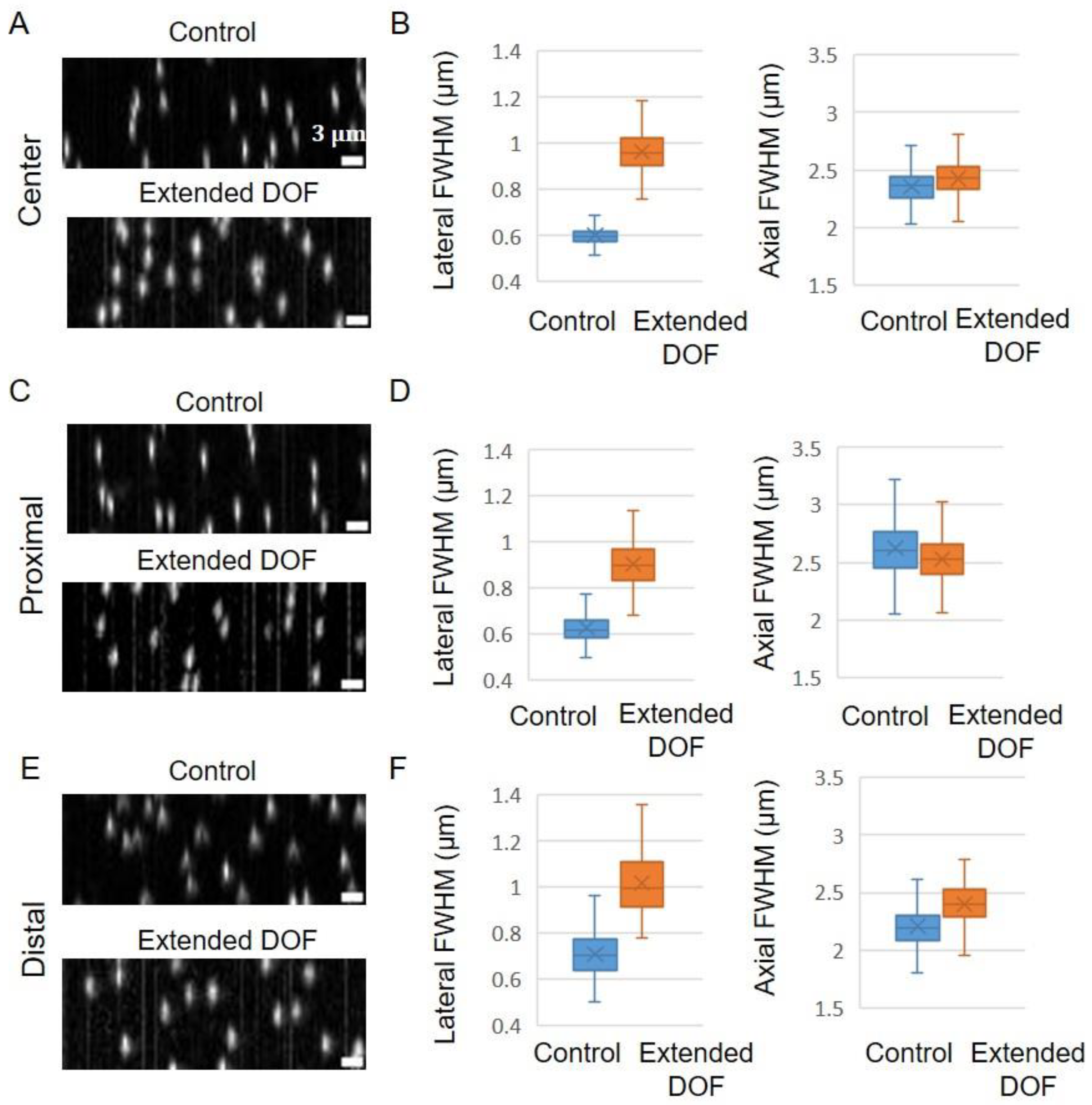
2.2. Performance of Extended DOF Two-Photon Light-Sheet Microscopy
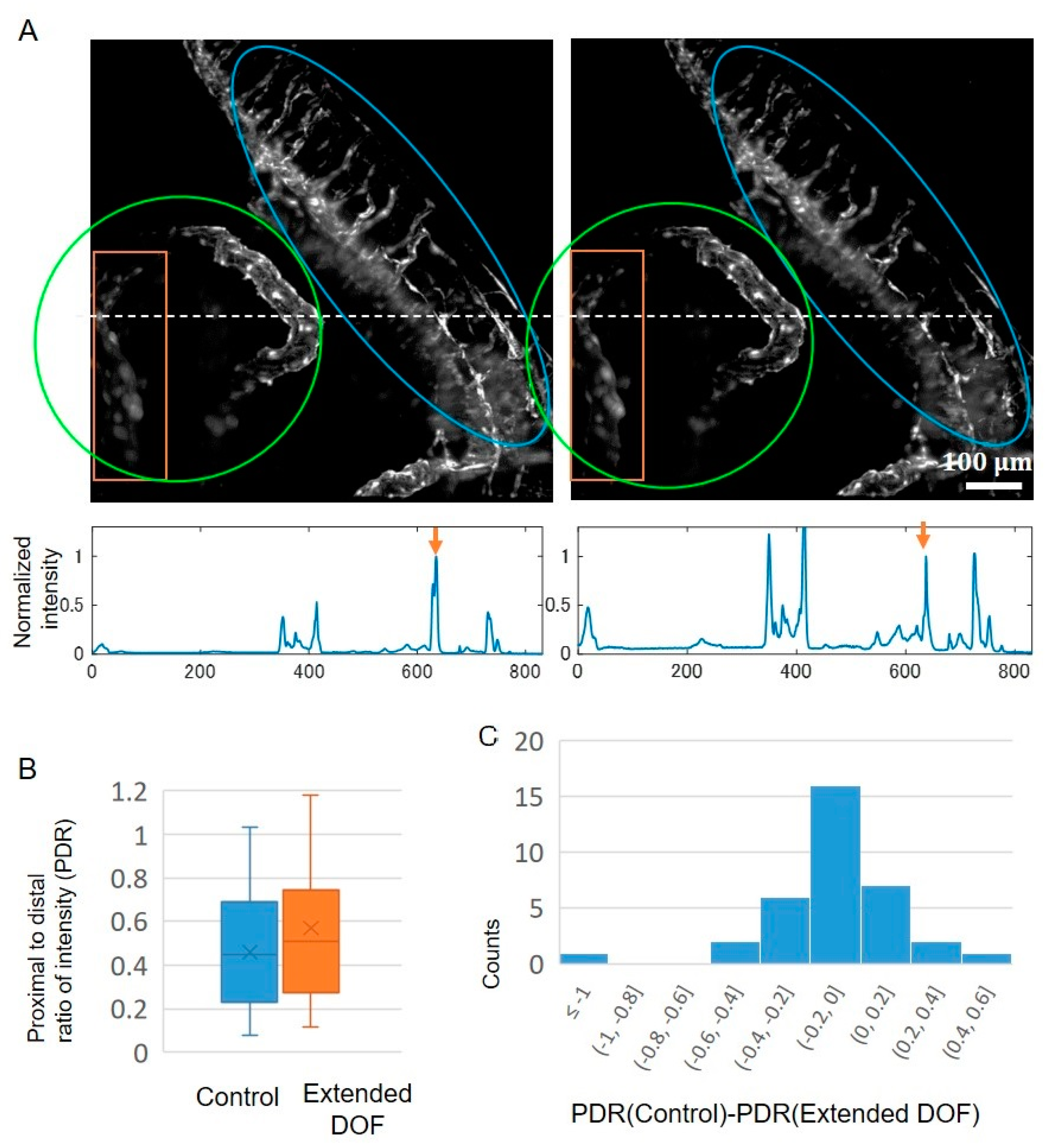
2.3. In Vivo Imaging Application of the Extended DOF Two-Photon Light-Sheet Microscopy
3. Discussion
3.1. Design of Light-Sheet Microscopy
3.2. Application to In Vivo Imaging
3.3. Future Application to High Throughput Analysis
4. Materials and Methods
4.1. Optical System
4.2. Measurement of Optical Properties
4.3. Fish Husbandry
4.4. Preparation of Medaka Embryos
4.5. Image Processing
4.6. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Computer Simulation of the Point Spread Function
References
- Huisken, J.; Swoger, J.; Del Bene, F.; Wittbrodt, J.; Stelzer, E.H. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science 2004, 305, 1007–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Power, R.; Huisken, J. A guide to light-sheet fluorescence microscopy for multiscale imaging. Nat. Methods 2017, 14, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Keller, P.J. Imaging Morphogenesis: Technological Advances and Biological Insights. Science 2013, 340, 1234168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahrbach, F.O.; Gurchenkov, V.; Alessandri, K.; Nassoy, P.; Rohrbach, A. Light-sheet microscopy in thick media using scanned Bessel beams and two-photon fluorescence excitation. Opt. Express 2013, 21, 13824–13839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahrbach, F.O.; Rohrbach, A. Propagation stability of self-reconstructing Bessel beams enables contrast-enhanced imaging in thick media. Nat. Commun. 2012, 3, 632. [Google Scholar] [CrossRef] [PubMed]
- Olarte, O.E.; Licea-Rodriguez, J.; Palero, J.A.; Gualda, E.J.; Artigas, D.; Mayer, J.; Swoger, J.; Sharpe, J.; Rocha-Mendoza, I.; Rangel-Rojo, R.; et al. Image formation by linear and nonlinear digital scanned light-sheet fluorescence microscopy with Gaussian and Bessel beam profiles. Biomed. Opt. Express 2012, 3, 1492–1505. [Google Scholar] [CrossRef] [Green Version]
- Welf, E.S.; Driscoll, M.K.; Dean, K.M.; Schäfer, C.; Chu, J.; Davidson, M.W.; Lin, M.Z.; Danuser, G.; Fiolka, R. Quantitative Multiscale Cell Imaging in Controlled 3D Microenvironments. Dev. Cell 2016, 36, 462–475. [Google Scholar] [CrossRef] [Green Version]
- Vettenburg, T.; Dalgarno, H.I.C.; Nylk, J.; Coll-Lladó, C.; Ferrier, D.; Cizmar, T.; Gunn-Moore, F.J.; Dholakia, K. Light-sheet microscopy using an Airy beam. Nat. Methods 2014, 11, 541–544. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.-C.; Legant, W.R.; Wang, K.; Shao, L.; Milkie, D.E.; Davidson, M.W.; Janetopoulos, C.; Wu, X.S.; Hammer, J.A., III; Liu, Z.; et al. Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science 2014, 346, 1257998. [Google Scholar] [CrossRef] [Green Version]
- Planchon, T.A.; Gao, L.; Milkie, D.; Davidson, M.W.; Galbraith, J.A.; Galbraith, C.G.; Betzig, E. Rapid three-dimensional isotropic imaging of living cells using Bessel beam plane illumination. Nat. Methods 2011, 8, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Denk, W.; Strickler, J.H.; Webb, W.W. Two-photon laser scanning fluorescence microscopy. Science 1990, 248, 73–76. [Google Scholar] [CrossRef] [Green Version]
- Zipfel, W.R.; Williams, R.M.; Webb, W.W. Nonlinear magic: Multiphoton microscopy in the biosciences. Nat. Biotechnol. 2003, 21, 1369–1377. [Google Scholar] [CrossRef]
- Truong, T.; Supatto, W.; Koos, D.S.; Choi, J.M.; Fraser, S. Deep and fast live imaging with two-photon scanned light-sheet microscopy. Nat. Methods 2011, 8, 757–760. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Zhang, H.; Li, Y.; Ashok, A.; Liang, R.; Zhou, W.; Peng, L. Cellular imaging of deep organ using two-photon Bessel light-sheet nonlinear structured illumination microscopy. Biomed. Opt. Express 2014, 5, 1296–1308. [Google Scholar] [CrossRef] [Green Version]
- Takanezawa, S.; Saitou, T.; Imamura, T. Wide field light-sheet microscopy with lens-axicon controlled two-photon Bessel beam illumination. Nat. Commun. 2021, 12, 2979. [Google Scholar] [CrossRef]
- Abrahamsson, S.; Usawa, S.; Gustafsson, M. A new approach to extended focus for high-speed high-resolution biological microscopy. In Three-Dimensional and Multidimensional Microscopy: Image Acquisition and Processing XIII; SPIE: Washington, DC, USA, 2006. [Google Scholar]
- Chen, B.; Chakraborty, T.; Daetwyler, S.; Manton, J.D.; Dean, K.; Fiolka, R. Extended depth of focus multiphoton microscopy via incoherent pulse splitting. Biomed. Opt. Express 2020, 11, 3830–3842. [Google Scholar] [CrossRef]
- Gu, M. Advanced Optical Imaging Theory; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2000; Volume 75. [Google Scholar]
- Deguchi, T.; Fujimori, K.E.; Kawasaki, T.; Maruyama, K.; Yuba, S. In vivo visualization of the lymphatic vessels in pFLT4-EGFP transgenic medaka. Genes 2012, 50, 625–634. [Google Scholar] [CrossRef]
- Keomanee-Dizon, K.; Jones, M.; Luu, P.; Fraser, S.E.; Truong, T.V. Extended depth-of-field light-sheet microscopy improves imaging of large volumes at high numerical aperture. Appl. Phys. Lett. 2022, 121, 163701. [Google Scholar] [CrossRef] [PubMed]
- Olarte, O.E.; Andilla, J.; Artigas, D.; Loza-Alvarez, P. Decoupled illumination detection in light sheet microscopy for fast volumetric imaging. Optica 2015, 2, 702–705. [Google Scholar] [CrossRef]
- Tomer, R.; Lovett-Barron, M.; Kauvar, I.; Andalman, A.; Burns, V.M.; Sankaran, S.; Grosenick, L.; Broxton, M.; Yang, S.; Deisseroth, K. SPED Light Sheet Microscopy: Fast Mapping of Biological System Structure and Function. Cell 2015, 163, 1796–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.-Y.; Hwang, S.-P.L.; Lee, C.-H.; Chen, B.-C. Two-photon scanned light sheet fluorescence microscopy with axicon imaging for fast volumetric imaging. J. Biomed. Opt. 2021, 26, 116503. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, W.R.; Williams, R.M.; Christie, R.; Nikitin, A.Y.; Hyman, B.T.; Webb, W.W. Live tissue intrinsic emission microscopy using multiphoton-excited native fluorescence and second harmonic generation. Proc. Natl. Acad. Sci. USA 2003, 100, 7075–7080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitou, T.; Kiyomatsu, H.; Imamura, T. Quantitative Morphometry for Osteochondral Tissues Using Second Harmonic Generation Microscopy and Image Texture Information. Sci. Rep. 2018, 8, 2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitou, T.; Takanezawa, S.; Ninomiya, H.; Watanabe, T.; Yamamoto, S.; Hiasa, Y.; Imamura, T. Tissue Intrinsic Fluorescence Spectra-Based Digital Pathology of Liver Fibrosis by Marker-Controlled Segmentation. Front. Med. 2018, 5, 350. [Google Scholar] [CrossRef]
- Kobayashi-Taguchi, K.; Saitou, T.; Kamei, Y.; Murakami, A.; Nishiyama, K.; Aoki, R.; Kusakabe, E.; Noda, H.; Yamashita, M.; Kitazawa, R.; et al. Computer-Aided Detection of Quantitative Signatures for Breast Fibroepithelial Tumors Using Label-Free Multi-Photon Imaging. Molecules 2022, 27, 3340. [Google Scholar] [CrossRef]
- Theer, P.; Mongis, C.; Knop, M. PSFj: Know your fluorescence microscope. Nat. Methods 2014, 11, 981–982. [Google Scholar] [CrossRef]
- Kinoshita, M.; Murata, K.; Naruse, K.; Tanaka, M. Medaka: Biology, Management, and Experimental Protocols; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saitou, T.; Imamura, T. Extended Depth of Focus Two-Photon Light-Sheet Microscopy for In Vivo Fluorescence Imaging of Large Multicellular Organisms at Cellular Resolution. Int. J. Mol. Sci. 2023, 24, 10186. https://doi.org/10.3390/ijms241210186
Saitou T, Imamura T. Extended Depth of Focus Two-Photon Light-Sheet Microscopy for In Vivo Fluorescence Imaging of Large Multicellular Organisms at Cellular Resolution. International Journal of Molecular Sciences. 2023; 24(12):10186. https://doi.org/10.3390/ijms241210186
Chicago/Turabian StyleSaitou, Takashi, and Takeshi Imamura. 2023. "Extended Depth of Focus Two-Photon Light-Sheet Microscopy for In Vivo Fluorescence Imaging of Large Multicellular Organisms at Cellular Resolution" International Journal of Molecular Sciences 24, no. 12: 10186. https://doi.org/10.3390/ijms241210186