
Design, Synthesis, Antiproliferative Actions, and DFT Studies of New Bis–Pyrazoline Derivatives as Dual EGFR/BRAFV600E Inhibitors
 , and
, and
Abstract
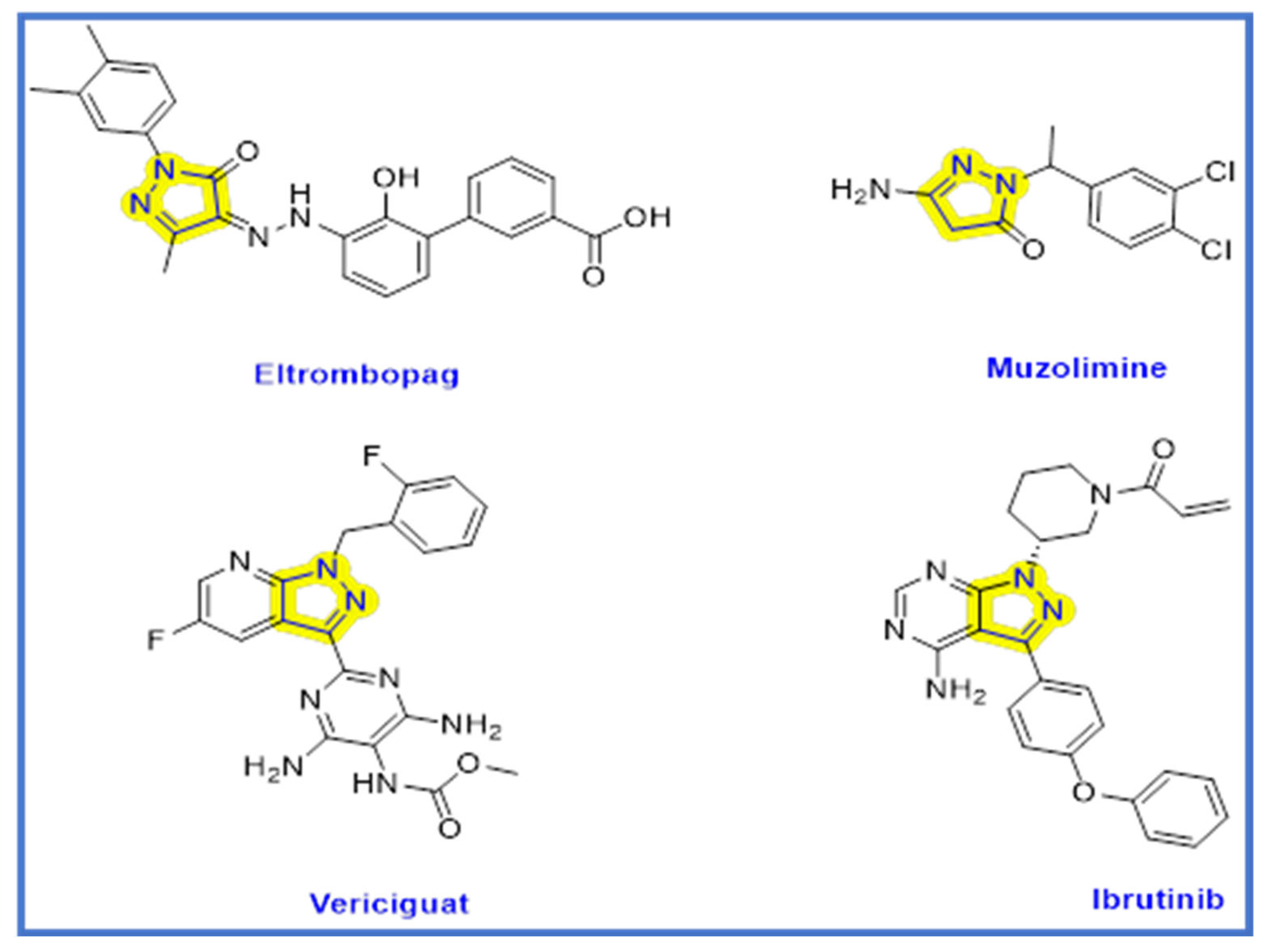
:1. Introduction
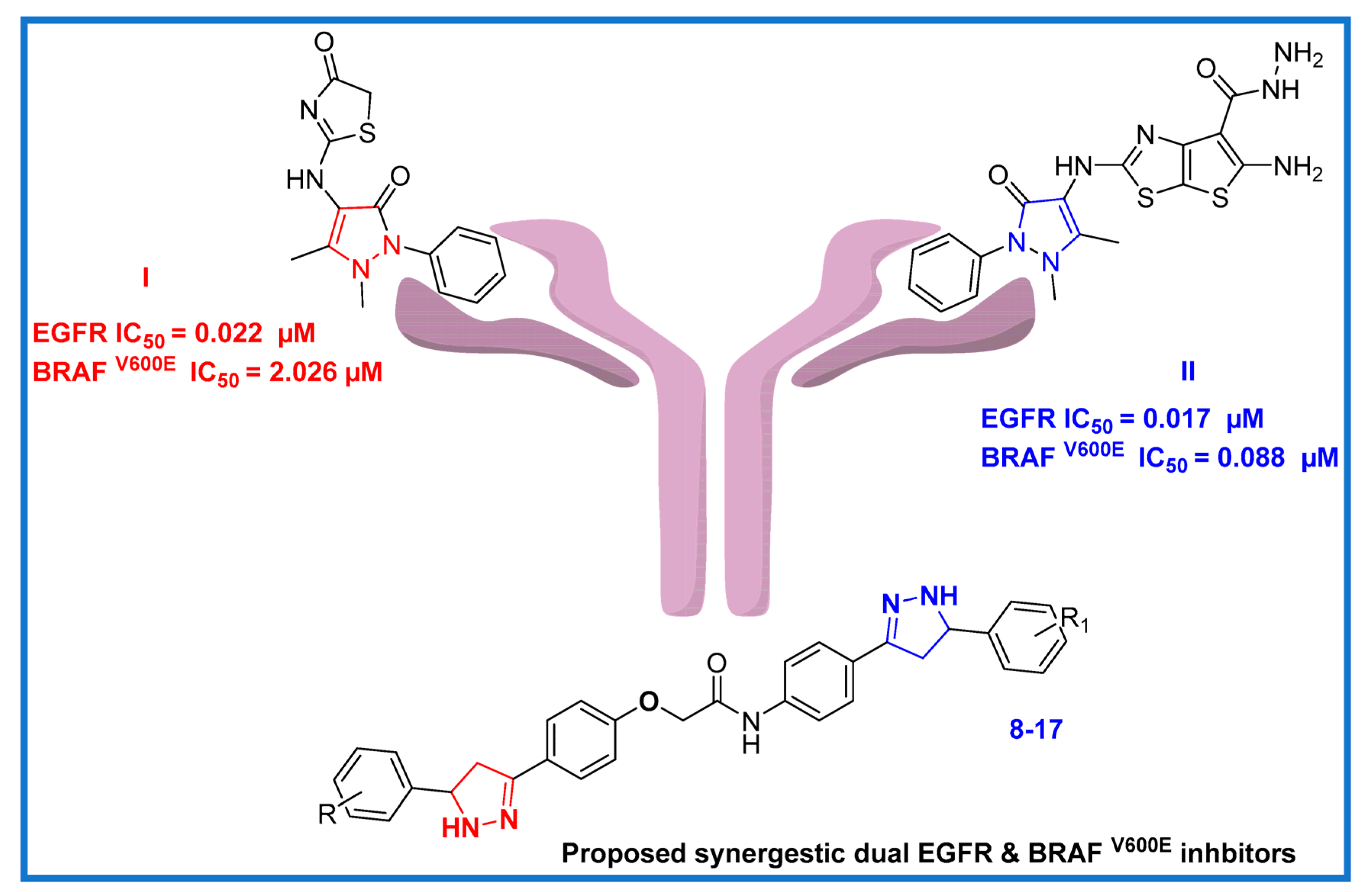
2. Results and Discussion
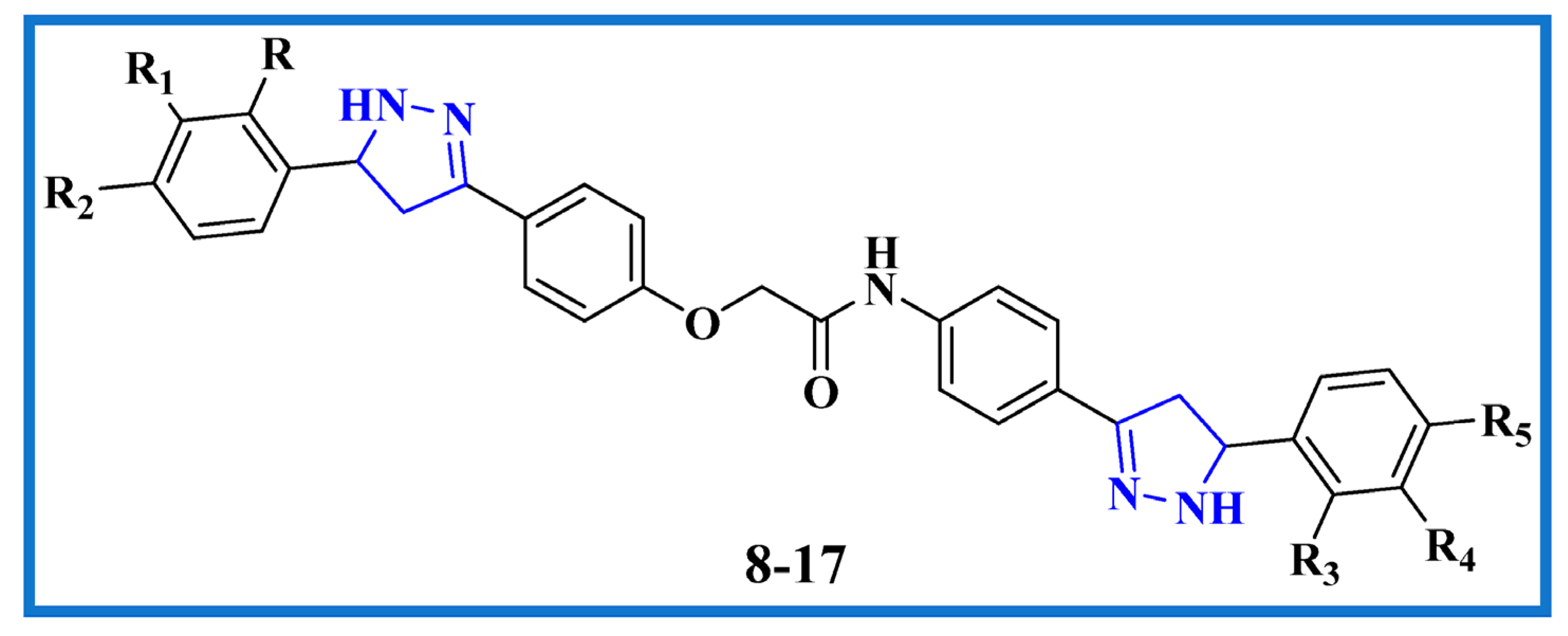
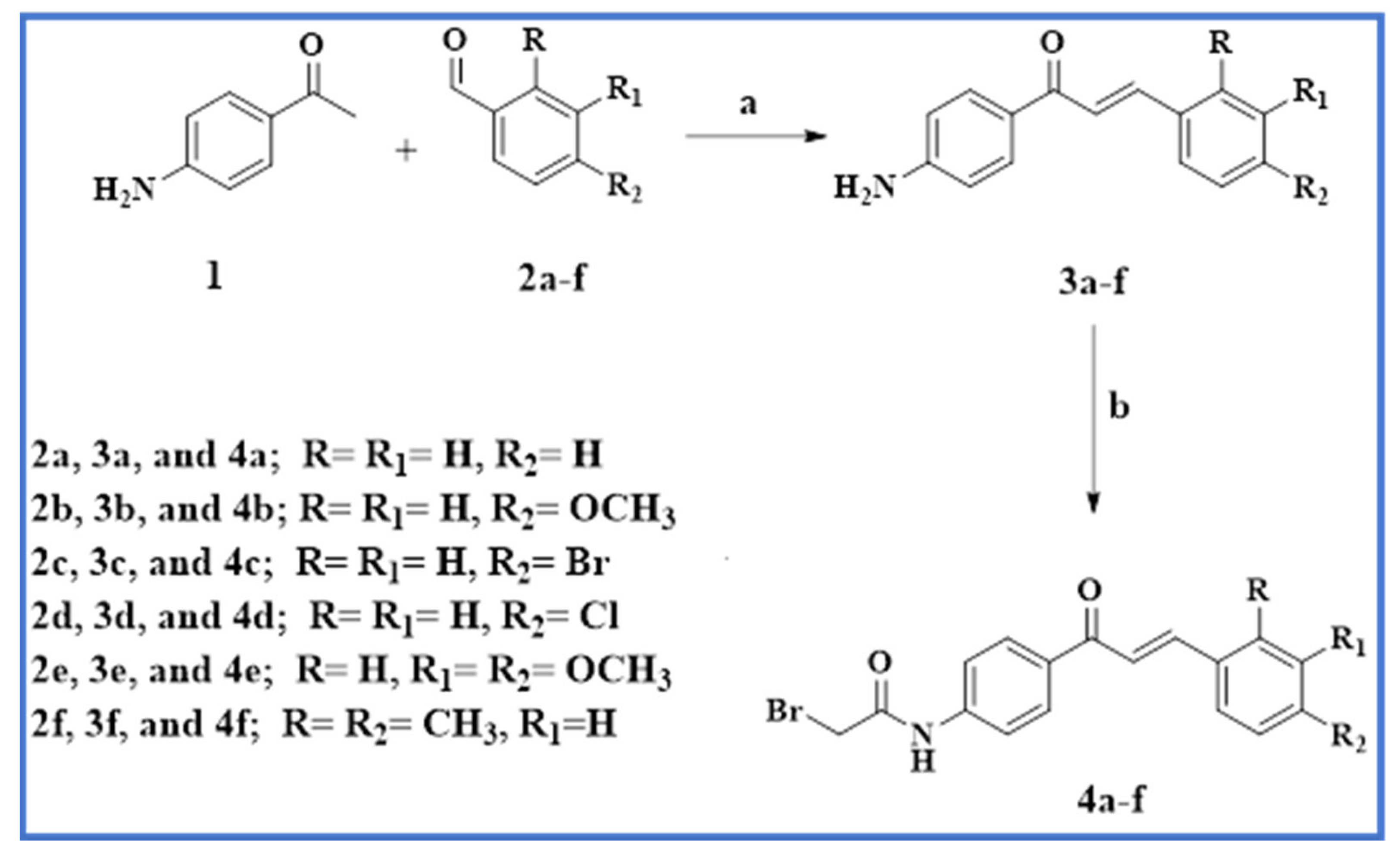
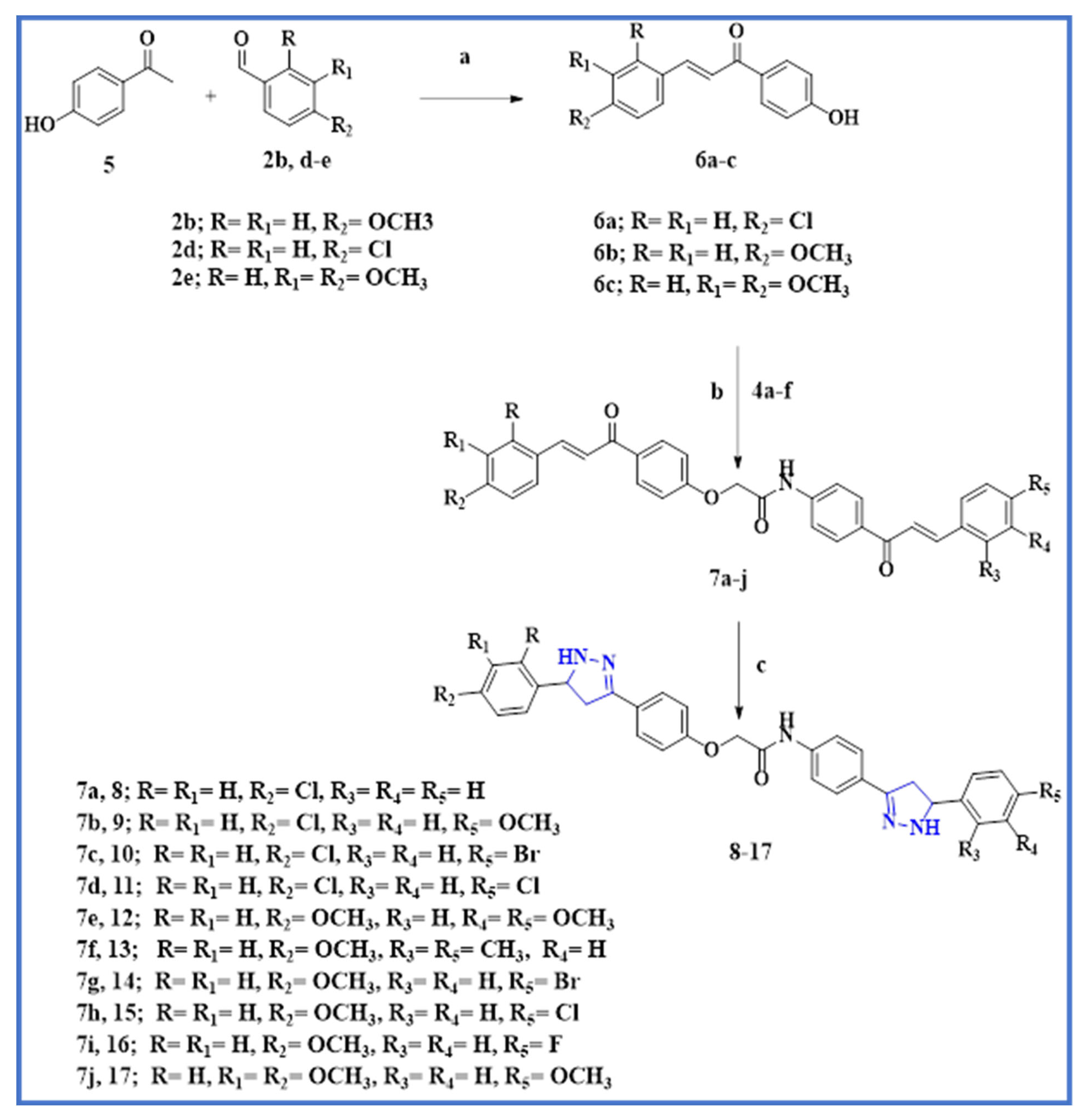
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Cell Viability Assay
2.2.2. Antiproliferative Assay
2.2.3. Structure–Activity Relationship (SAR) Investigation
- The presence and number of methoxy groups attached to the molecule can significantly impact the antiproliferative activity of the compounds. It is possible that the methoxy group(s) may be contributing to the activity by affecting the interaction of the molecule with its target.
- The substituent at the phenyl ring attached to the ring B pyrazoline also plays an important role in determining the antiproliferative activity of the compounds. This suggests that the location and nature of the substituent may be affecting the ability to interact with the target and elicit an antiproliferative effect.
- The activity of the compounds increases in the following order: OCH3 > Cl > Br > F > H > CH3, indicating that the nature of the substituent plays a critical role in modulating the activity of the compounds.
- The OCH3 group is an electron-donating group, which can increase the electron density of the molecule and enhance its interaction with the target, potentially leading to higher activity. Additionally, the size and shape of the OCH3 group may allow it to form favorable interactions with the target, further enhancing its activity.
- The Cl, Br, and F groups are electron-withdrawing groups, which can decrease the electron density of the molecule and affect its electronic properties and interaction with the target. However, the size and electronegativity of the halogen substituents can also play a role. The larger size of Br and Cl compared to F may allow them to form stronger interactions with the target, potentially leading to higher activity. Additionally, the mild electronegativity of Cl and Br may be better suited for interacting with the target compared to F.
- The CH3 group and H atom are non-polar and relatively small, which may not significantly affect the electronic properties of the molecule or its interaction with the target. However, the size and shape of the substituents can also play a role in steric effects, which can affect the interaction of the molecule with the target. In this case, the CH3 group is bulkier than the H group, which may lead to steric hindrance and reduce the ability of the molecule to interact with the target.
2.2.4. EGFR Inhibitory Assay
2.2.5. BRAFV600E Inhibitory Assay
2.2.6. Apoptotic Marker Activation Assay
Caspase 3 Activation Assay
Caspase-8, Bax, and Bcl-2 Levels Assay
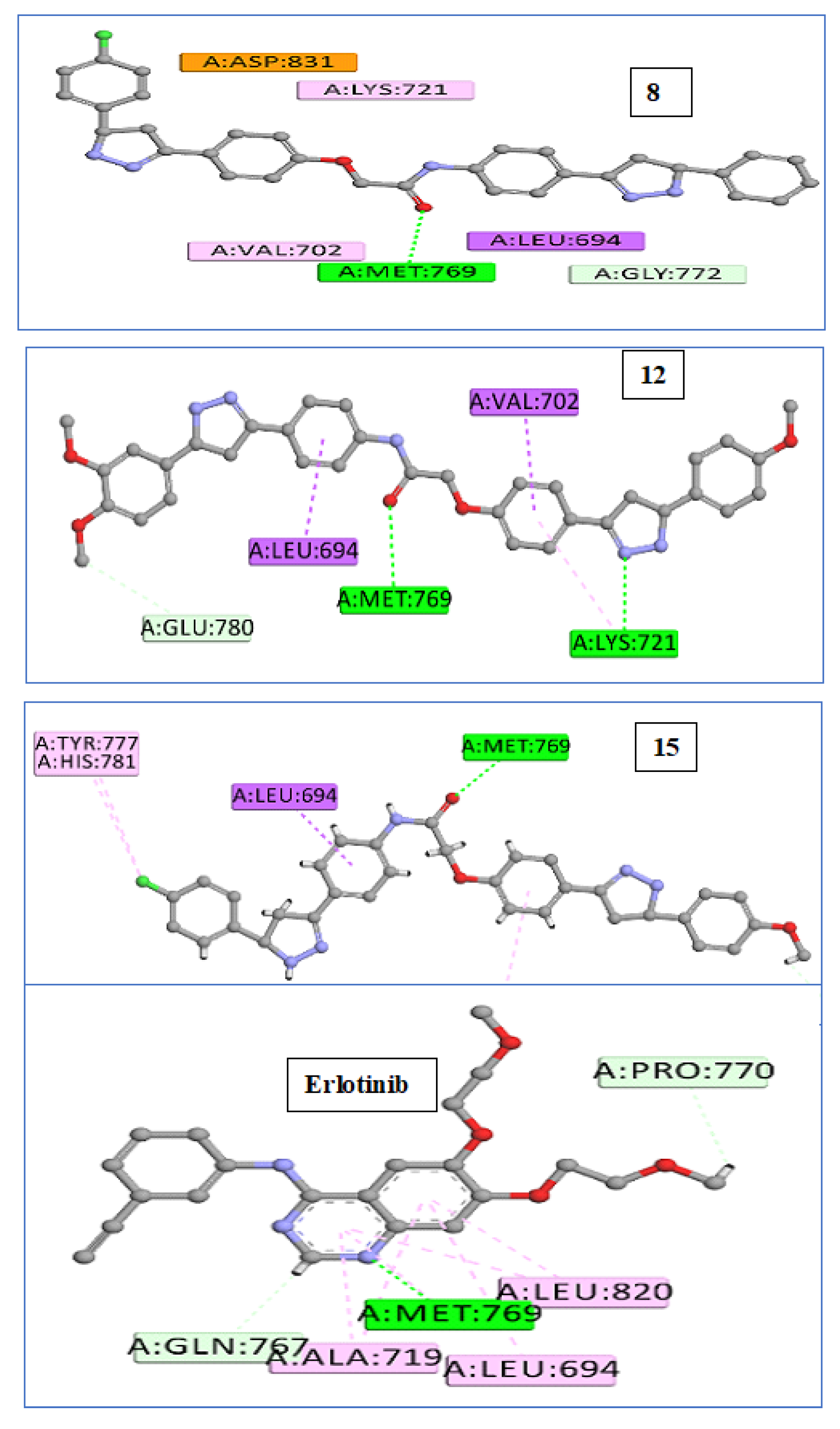
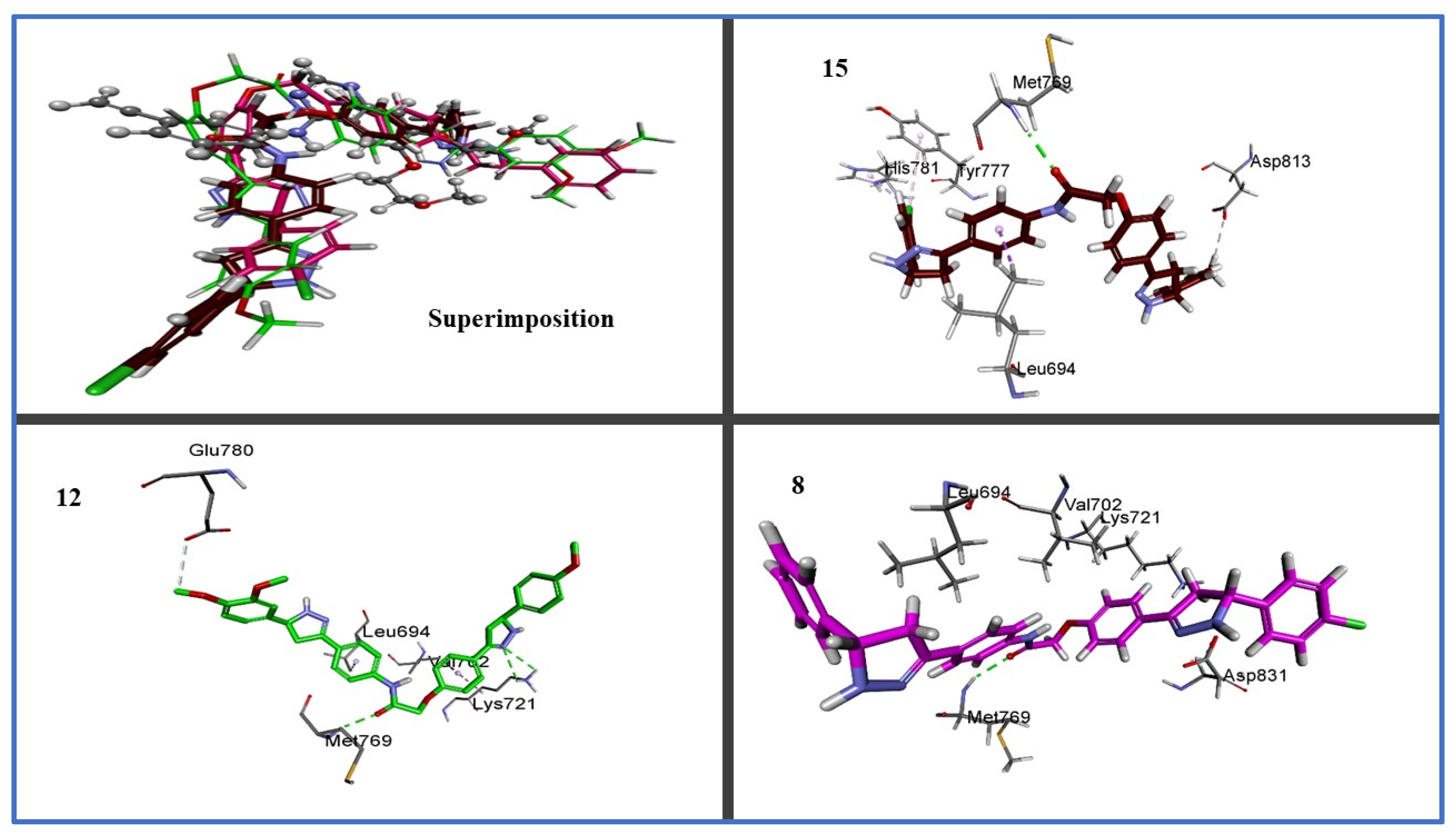
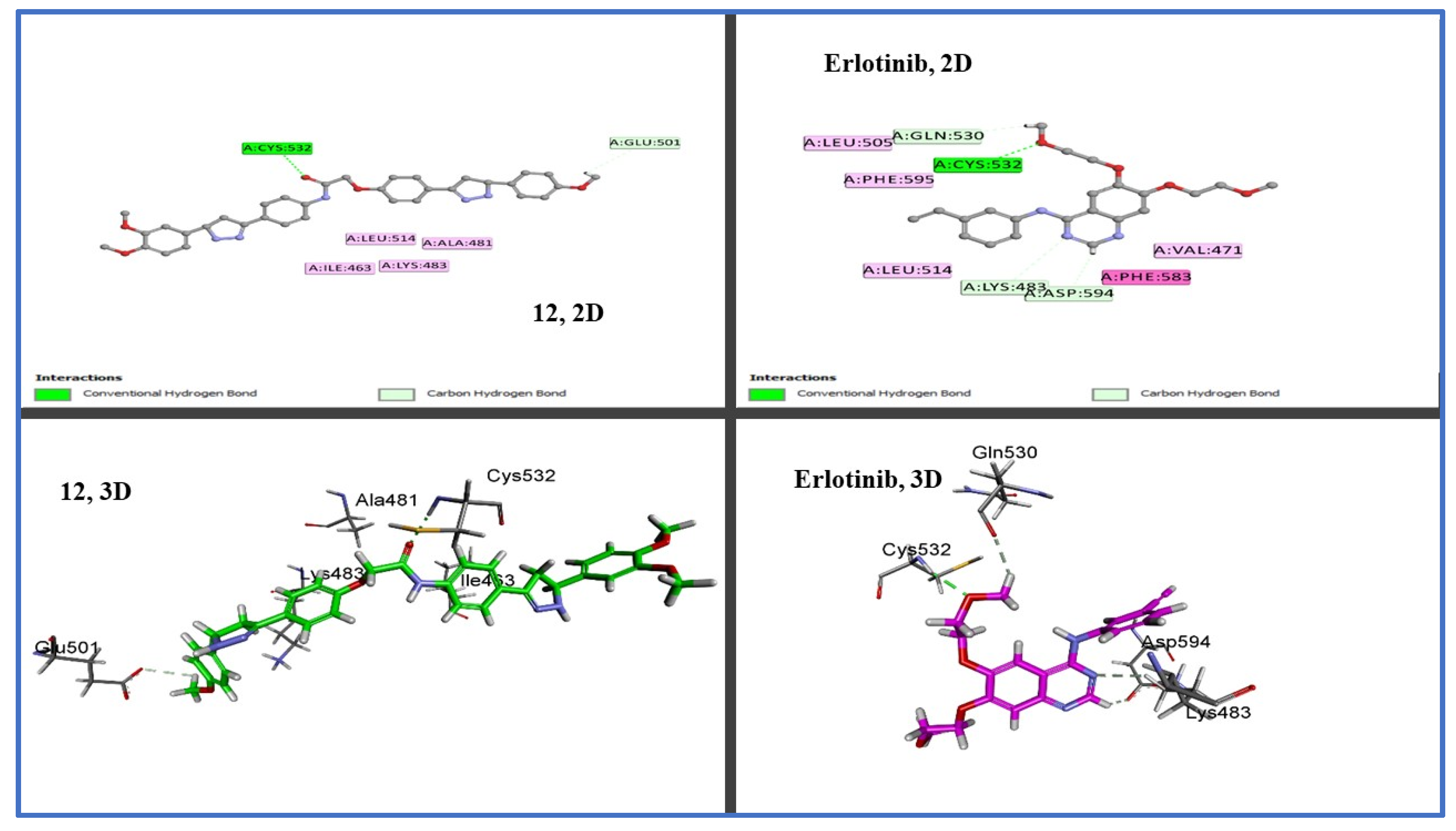
2.3. In Silico Studies
2.3.1. Docking Study
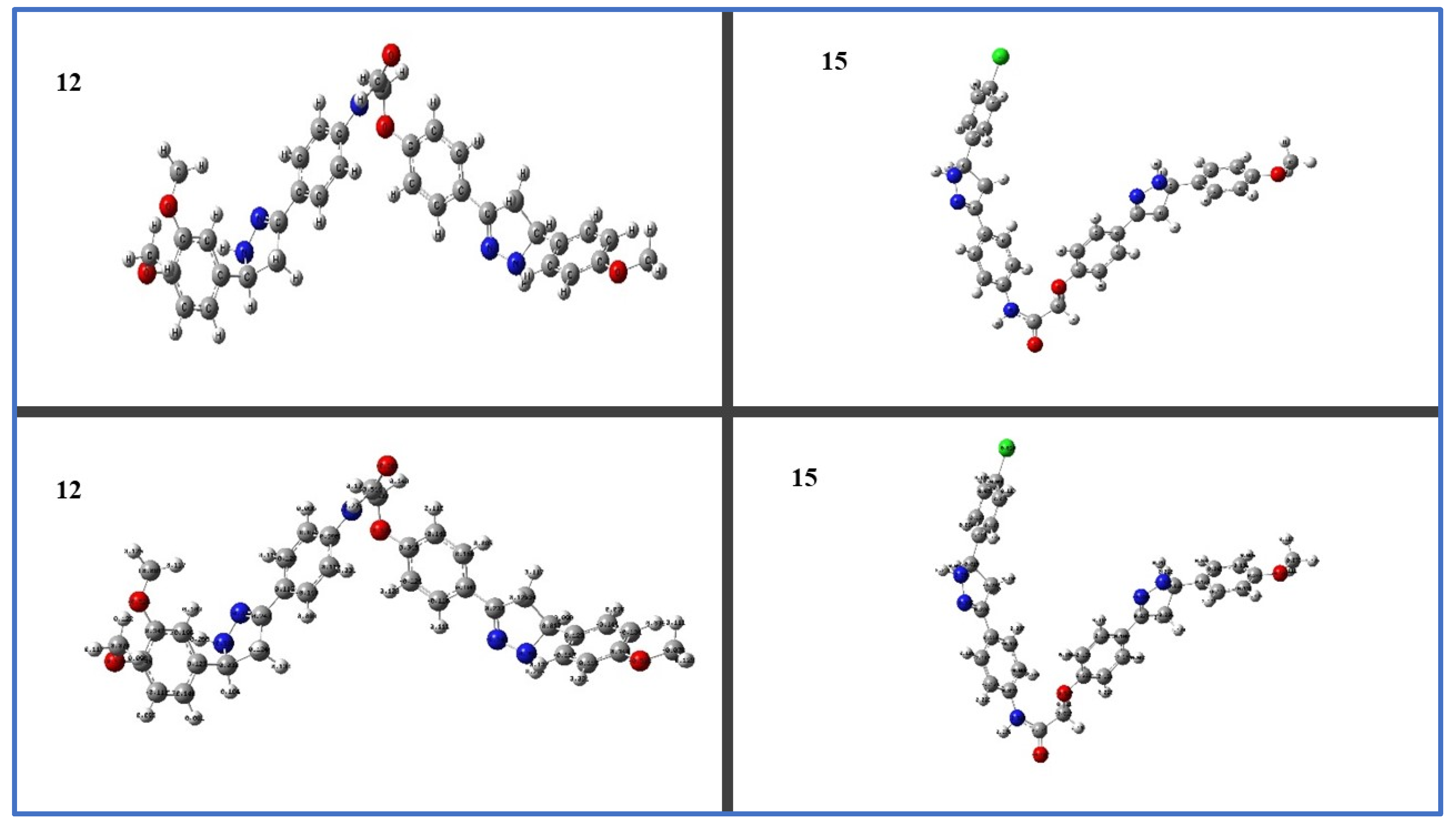
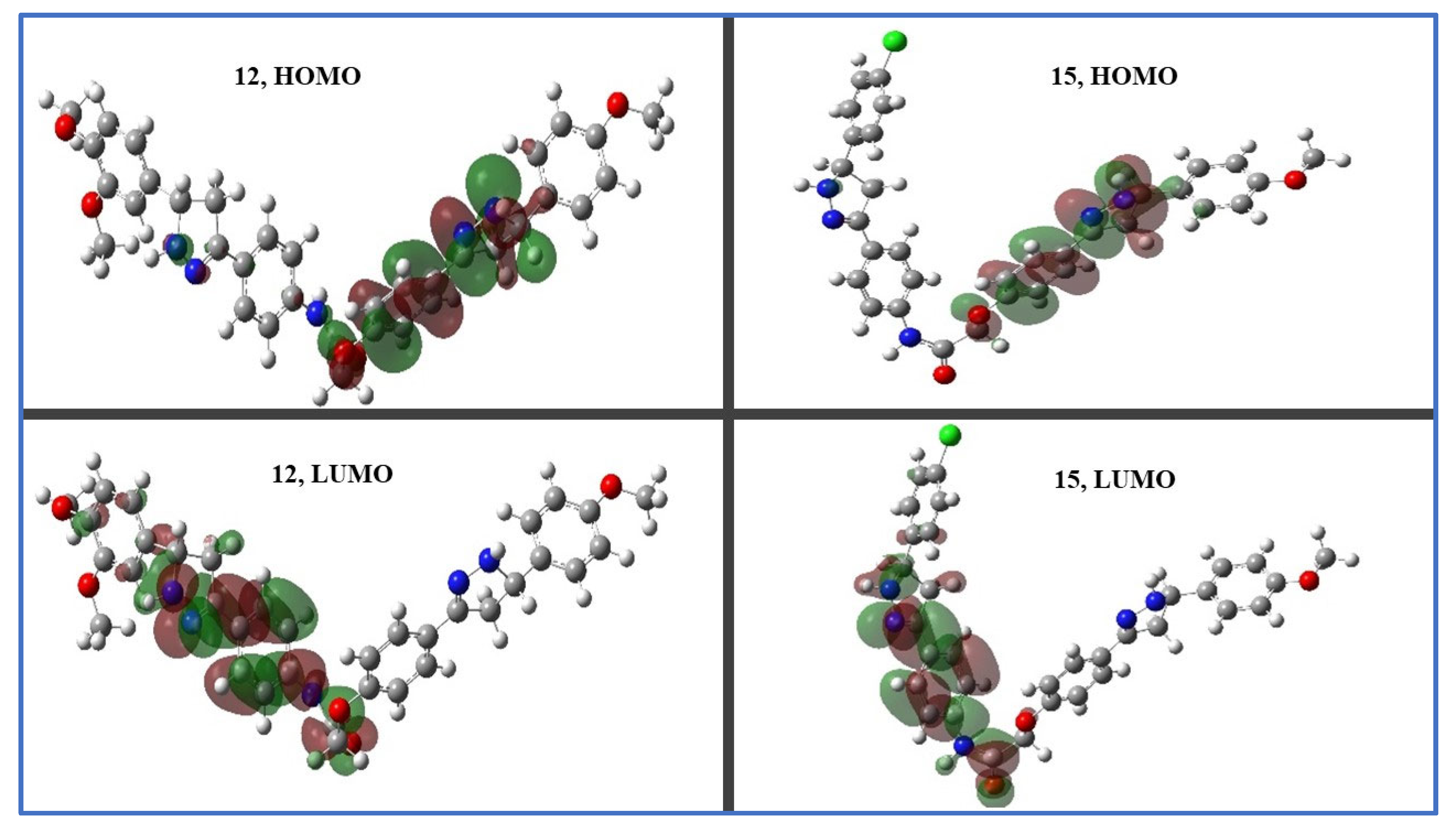
2.3.2. Density Functional Theory (DFT) Studies
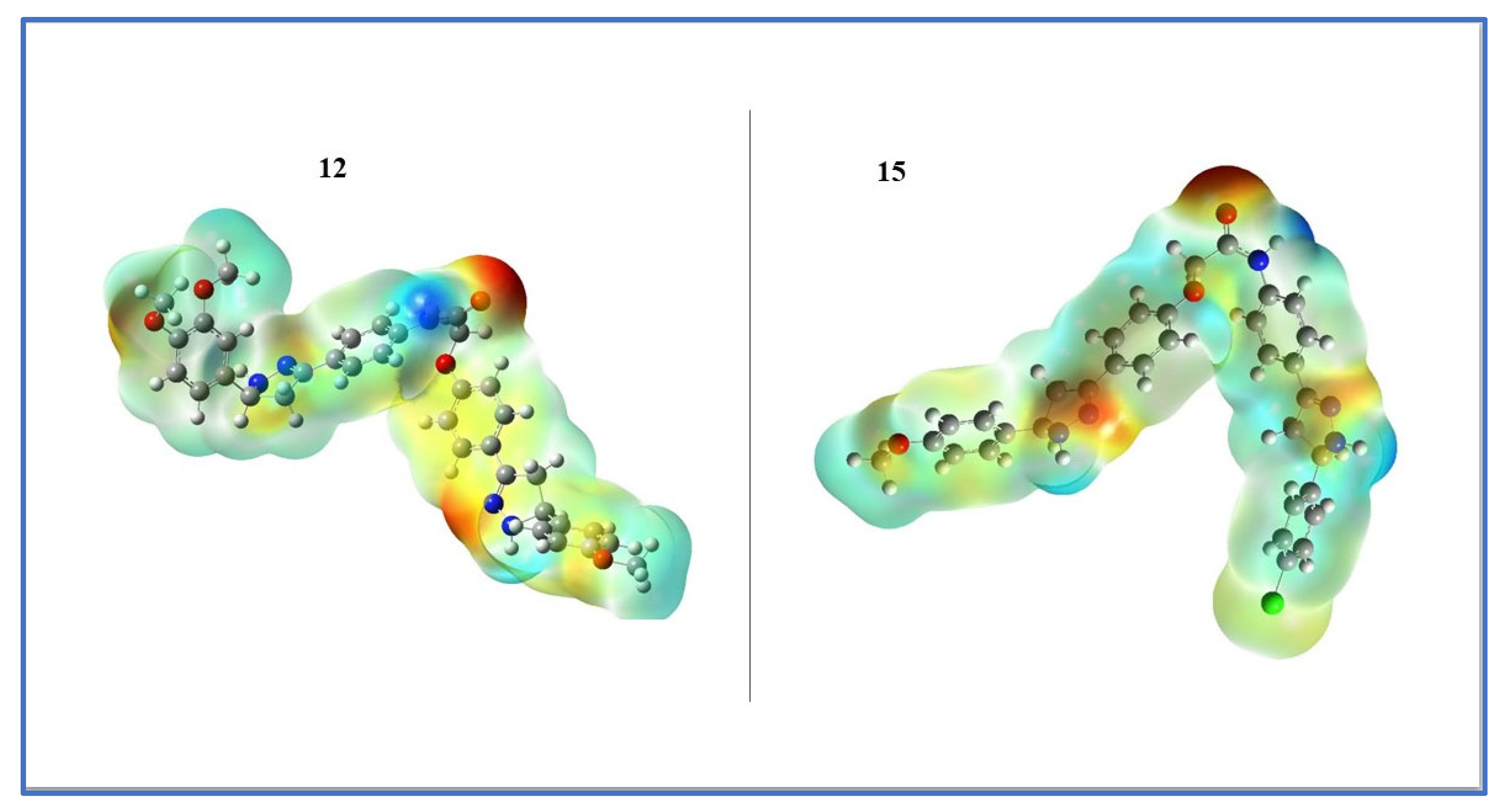
2.3.3. Frontier Molecular Orbitals (FMOs) and Electrostatic Potential of Molecules (MEP)
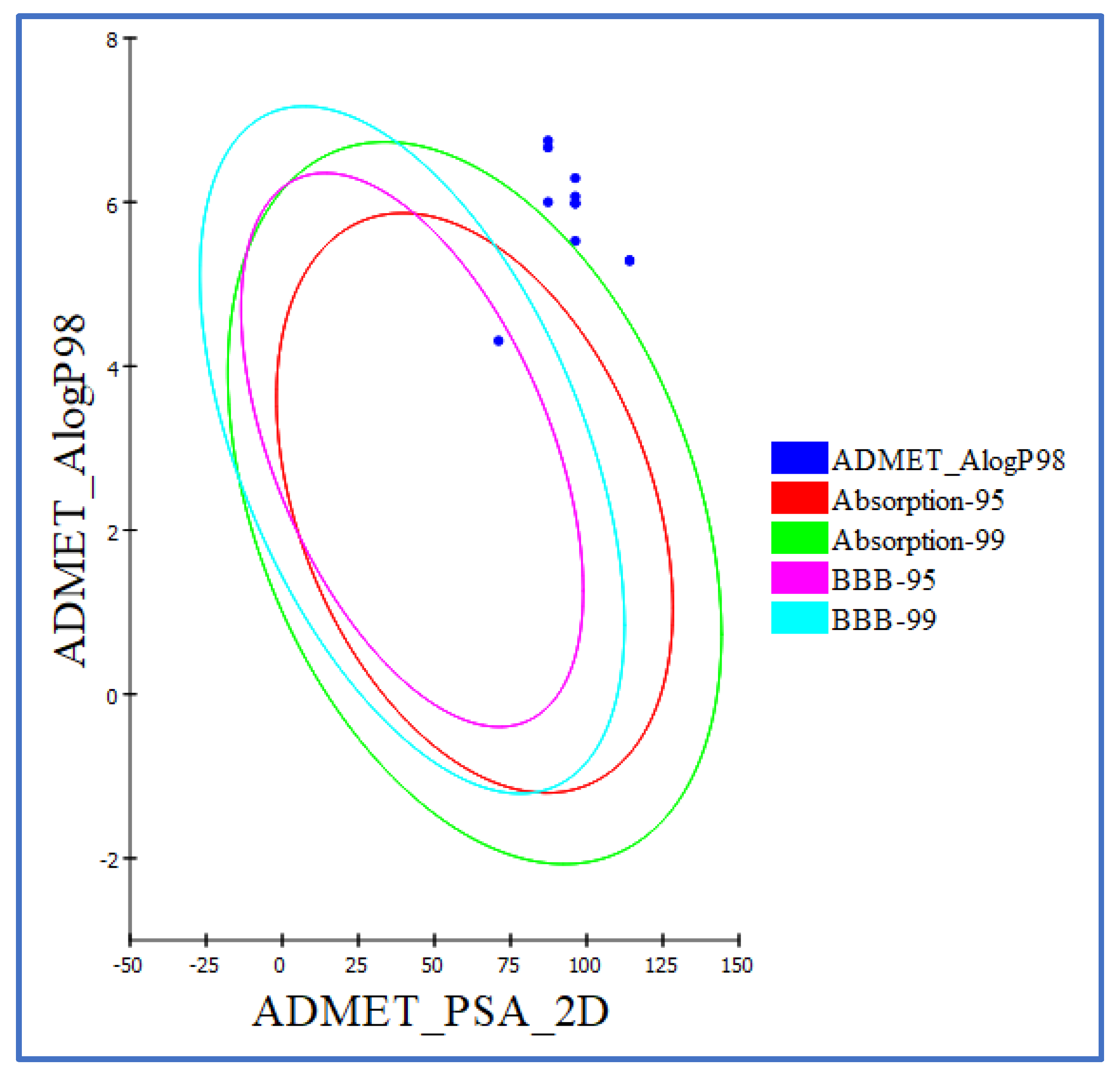
2.4. ADMET Evaluation
2.5. In Silico Toxicity Predictions
3. Materials and Methods
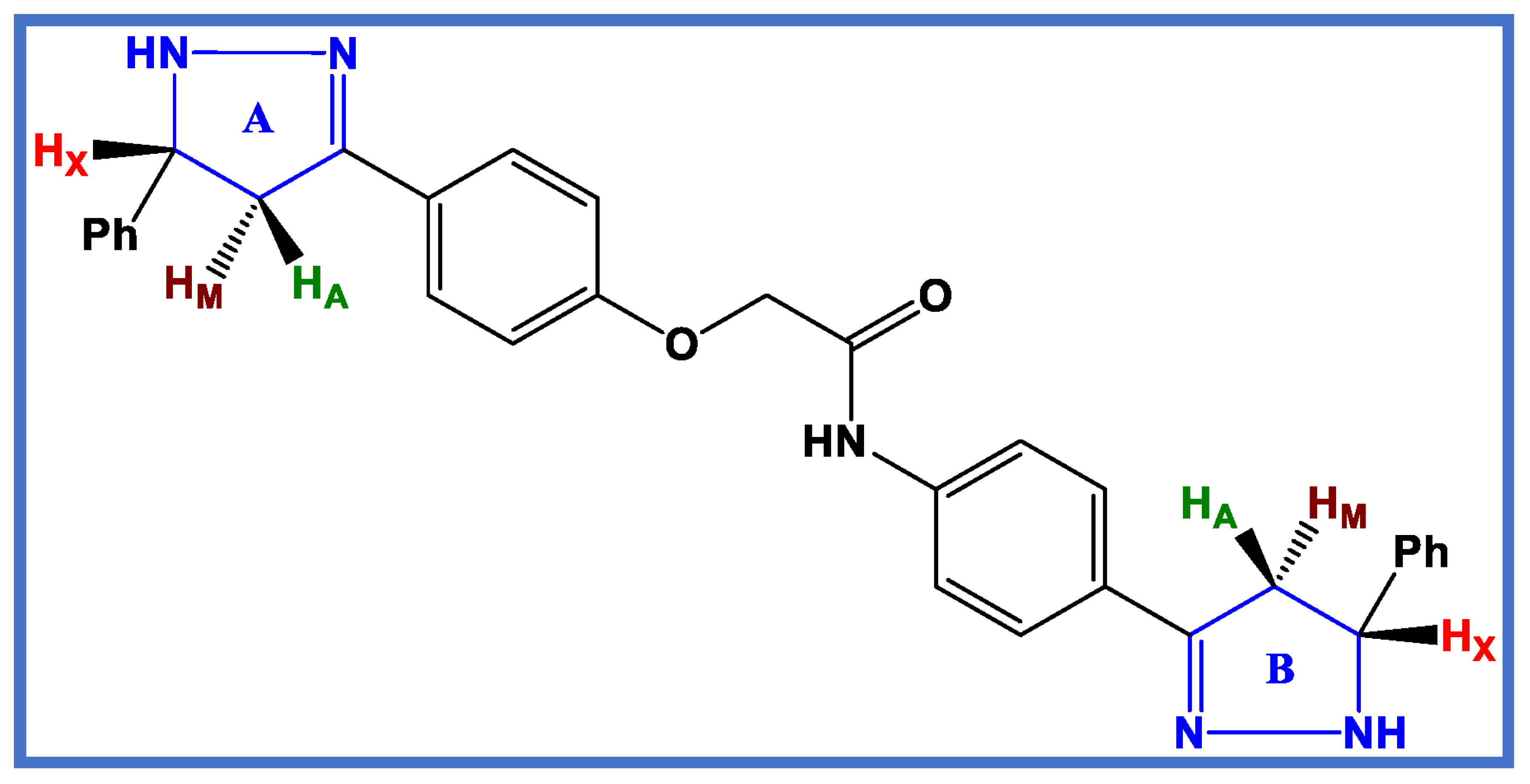
3.1. Chemistry
3.1.1. General Details: See Appendix SA (Supplementary File)
3.1.2. General Procedure for the Synthesis of 2-[4-(E)-3-Arylacryloylphenoxy]-N-[4-(E)-3-arylacryloylphenyl]Acetamides (7a-j)
3.1.3. General Procedure for the Synthesis of Bis-Pyrazoline Hybrid Hits (8–17)
3.2. Biology
3.2.1. Cell Viability Assay
3.2.2. Antiproliferative Assay
3.2.3. EGFR Inhibitory Assay
3.2.4. BRAFV600E Inhibitory Assay
3.2.5. Apoptotic Marker Assay
Caspase 3 Activation Assay
Caspase-8, Bax, and Bcl-2 Level Assays
3.3. Statistical Analysis
3.4. Docking Study
3.5. Quantum Chemical Calculations
3.6. In Silico ADMET Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jassem, A.M.; Dhumad, A.M.; Salim, J.K.; Jabir, H.A. An alternative technique for cyclization synthesis, in vitro anti-esophageal cancer evaluation, and molecular docking of novel thiazolidin-4-one derivatives. J. Mol. Struct. 2023, 1280, 135079. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef]
- Zaki, R.M.; Wani, M.Y.; Mohammed, A.; El-Said, W.A. Design, synthesis and evaluation of novel Se-alkylated pyrazoles and their cyclized analogs as potential anticancer agents. J. Mol. Struct. 2023, 1276, 134670. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Bedard, P.L.; Hyman, D.M.; Davids, M.S.; Siu, L.L. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet 2020, 395, 1078–1088. [Google Scholar] [CrossRef]
- Raghavendra, N.M.; Pingili, D.; Kadasi, S.; Mettu, A.; Prasad, S. Dual or multi-targeting inhibitors: The next generation anticancer agents. Eur. J. Med. Chem. 2018, 143, 1277–1300. [Google Scholar] [CrossRef]
- Elwaie, T.A.; Abbas, S.E.; Aly, E.I.; George, R.F.; Ali, H.; Kraiouchkine, N.; Abdelwahed, K.S.; Fandy, T.E.; El Sayed, K.A.; Abd Elmageed, Z.Y. HER2 kinase-targeted breast cancer therapy: Design, synthesis, and in vitro and in vivo evaluation of novel lapatinib congeners as selective and potent HER2 inhibitors with favorable metabolic stability. J. Med. Chem. 2020, 63, 15906–15945. [Google Scholar] [CrossRef]
- Liang, C.; Ma, Y.; Yong, L.; Yang, C.; Wang, P.; Liu, X.; Zhu, B.; Zhou, H.; Liu, X.; Liu, Z. Y-box binding protein-1 promotes tumorigenesis and progression via the epidermal growth factor receptor/AKT pathway in spinal chordoma. Cancer Sci. 2019, 110, 166–179. [Google Scholar] [CrossRef]
- Crispo, F.; Notarangelo, T.; Pietrafesa, M.; Lettini, G.; Storto, G.; Sgambato, A.; Maddalena, F.; Landriscina, M. BRAF inhibitors in thyroid cancer: Clinical impact, mechanisms of resistance and future perspectives. Cancers 2019, 11, 1388. [Google Scholar] [CrossRef]
- Cheng, W.-L.; Feng, P.-H.; Lee, K.-Y.; Chen, K.-Y.; Sun, W.-L.; Van Hiep, N.; Luo, C.-S.; Wu, S.-M. The role of EREG/EGFR pathway in tumor progression. Int. J. Mol. Sci. 2021, 22, 12828. [Google Scholar] [CrossRef]
- Keller, S.; Schmidt, M.H. EGFR and EGFRvIII promote angiogenesis and cell invasion in glioblastoma: Combination therapies for an effective treatment. Int. J. Mol. Sci. 2017, 18, 1295. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Castanaro, C.; Luan, B.; Yang, K.; Fan, L.; Fairhurst, J.L.; Rafique, A.; Potocky, T.B.; Shan, J.; Delfino, F.J. ERBB3/HER2 Signaling Promotes Resistance to EGFR Blockade in Head and Neck and Colorectal Cancer ModelsERBB3 Signaling Promotes Resistance to EGFR Blockade. Mol. Cancer Ther. 2014, 13, 1345–1355. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Tamosaitis, L.; Kvederaviciute, K.; Tarvydas, R.; Staniute, G.; Kalyan, K.; Meskinyte-Kausiliene, E.; Stankevicius, V.; Valius, M. KRAS, NRAS and BRAF mutations in colorectal cancer and melanoma. Med. Oncol. 2017, 34, 26. [Google Scholar] [CrossRef] [PubMed]
- Koelblinger, P.; Thuerigen, O.; Dummer, R. Development of encorafenib for BRAF-mutated advanced melanoma. Curr. Opin. Oncol. 2018, 30, 125. [Google Scholar] [CrossRef]
- Garcia-Carbonero, N.; Martinez-Useros, J.; Li, W.; Orta, A.; Perez, N.; Carames, C.; Hernandez, T.; Moreno, I.; Serrano, G.; Garcia-Foncillas, J. KRAS and BRAF mutations as prognostic and predictive biomarkers for standard chemotherapy response in metastatic colorectal cancer: A single institutional study. Cells 2020, 9, 219. [Google Scholar] [CrossRef]
- Fiskus, W.; Mitsiades, N. B-Raf inhibition in the clinic: Present and future. Annu. Rev. Med. 2016, 67, 29–43. [Google Scholar] [CrossRef]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF (V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef]
- Notarangelo, T.; Sisinni, L.; Condelli, V.; Landriscina, M. Dual EGFR and BRAF blockade overcomes resistance to vemurafenib in BRAF mutated thyroid carcinoma cells. Cancer Cell Int. 2017, 17, 86. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, H.; Lirussi, F.; Garrido, C.; Ye, X.-Y.; Xie, T. Dual inhibitors of histone deacetylases and other cancer-related targets: A pharmacological perspective. Biochem. Pharmacol. 2020, 182, 114224. [Google Scholar] [CrossRef]
- Tan, L.; Zhang, J.; Wang, Y.; Wang, X.; Wang, Y.; Zhang, Z.; Shuai, W.; Wang, G.; Chen, J.; Wang, C. Development of dual inhibitors targeting epidermal growth factor receptor in cancer therapy. J. Med. Chem. 2022, 65, 5149–5183. [Google Scholar] [CrossRef]
- Tandon, R.; Kapoor, S.; Vali, S.; Senthil, V.; Nithya, D.; Venkataramanan, R.; Sharma, A.; Talwadkar, A.; Ray, A.; Bhatnagar, P.K. Dual epidermal growth factor receptor (EGFR)/insulin-like growth factor-1 receptor (IGF-1R) inhibitor: A novel approach for overcoming resistance in anticancer treatment. Eur. J. Pharmacol. 2011, 667, 56–65. [Google Scholar] [CrossRef]
- Gomaa, H.A.; Shaker, M.E.; Alzarea, S.I.; Hendawy, O.; Mohamed, F.A.; Gouda, A.M.; Ali, A.T.; Morcoss, M.M.; Abdelrahman, M.H.; Trembleau, L. Optimization and SAR investigation of novel 2,3-dihydropyrazino [1,2-a] indole-1, 4-dione derivatives as EGFR and BRAFV600E dual inhibitors with potent antiproliferative and antioxidant activities. Bioorg. Chem. 2022, 120, 105616. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S. Apoptosis mediated by Fas and its related diseases. Nihon Ika Daigaku Zasshi 1997, 64, 459. [Google Scholar] [CrossRef] [PubMed]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Giaccia, A.J.; Kastan, M.B. The complexity of p53 modulation: Emerging patterns from divergent signals. Genes Dev. 1998, 12, 2973–2983. [Google Scholar] [CrossRef]
- Farooq, S.; Ngaini, Z. One-pot and two-pot synthesis of chalcone based mono and bis-pyrazolines. Tetrahedron Lett. 2020, 61, 151416. [Google Scholar] [CrossRef]
- Yamali, C.; Gul, H.I.; Kazaz, C.; Levent, S.; Gulcin, I. Synthesis, structure elucidation, and in vitro pharmacological evaluation of novel polyfluoro substituted pyrazoline type sulfonamides as multi-target agents for inhibition of acetylcholinesterase and carbonic anhydrase I and II enzymes. Bioorg. Chem. 2020, 96, 103627. [Google Scholar] [CrossRef]
- Matiadis, D.; Sagnou, M. Pyrazoline hybrids as promising anticancer agents: An up-to-date overview. Int. J. Mol. Sci. 2020, 21, 5507. [Google Scholar] [CrossRef]
- Ansari, A.; Ali, A.; Asif, M. Biologically active pyrazole derivatives. New J. Chem. 2017, 41, 16–41. [Google Scholar] [CrossRef]
- Eid, N.M.; George, R.F. Facile synthesis of some pyrazoline-based compounds with promising anti-inflammatory activity. Future Med. Chem. 2018, 10, 183–199. [Google Scholar] [CrossRef]
- Saleh, N.M.; El-Gazzar, M.G.; Aly, H.M.; Othman, R.A. Novel anticancer fused pyrazole derivatives as EGFR and VEGFR-2 dual TK inhibitors. Front. Chem. 2020, 7, 917. [Google Scholar] [CrossRef] [PubMed]
- Shanafelt, T.D.; Wang, X.V.; Kay, N.E.; Hanson, C.A.; O’Brien, S.; Barrientos, J.; Jelinek, D.F.; Braggio, E.; Leis, J.F.; Zhang, C.C. Ibrutinib–rituximab or chemoimmunotherapy for chronic lymphocytic leukemia. N. Engl. J. Med. 2019, 381, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Garnock-Jones, K.P. Eltrombopag. Drugs 2011, 71, 1333–1353. [Google Scholar] [CrossRef] [PubMed]
- Hampel, P.; Römermann, K.; MacAulay, N.; Löscher, W. Azosemide is more potent than bumetanide and various other loop diuretics to inhibit the sodium-potassium-chloride-cotransporter human variants hNKCC1A and hNKCC1B. Sci. Rep. 2018, 8, 9877. [Google Scholar] [CrossRef] [PubMed]
- Berliner, D.; Hänselmann, A.; Bauersachs, J. The treatment of heart failure with reduced ejection fraction. Dtsch. Ärzteblatt Int. 2020, 117, 376. [Google Scholar] [CrossRef]
- Ibraheem, F.; Ahmad, M.; Ashfaq, U.A.; Aslam, S.; Ali Khan, Z.; Sultan, S. Synthesis, molecular docking and anti-diabetic studies of novel benzimidazole-pyrazoline hybrid molecules. Pak. J. Pharm. Sci. 2020, 33, 847–855. [Google Scholar]
- Rana, M.; Arif, R.; Khan, F.I.; Maurya, V.; Singh, R.; Faizan, M.I.; Yasmeen, S.; Dar, S.H.; Alam, R.; Sahu, A. Pyrazoline analogs as potential anticancer agents and their apoptosis, molecular docking, MD simulation, DNA binding and antioxidant studies. Bioorg. Chem. 2021, 108, 104665. [Google Scholar] [CrossRef]
- Beyhan, N.; Kocyigit-Kaymakcioglu, B.; Gümrü, S.; Aricioglu, F. Synthesis and anticonvulsant activity of some 2-pyrazolines derived from chalcones. Arab. J. Chem. 2017, 10, S2073–S2081. [Google Scholar] [CrossRef]
- Revanasiddappa, B.; Jisha, M.; Kumar, M.V.; Kumar, H. Synthesis, Antibacterial and Antifungal Evlaution of Novel Pyrazoline Derivatives. Dhaka Univ. J. Pharm. Sci. 2018, 17, 221–226. [Google Scholar] [CrossRef]
- Malvar, D.d.C.; Ferreira, R.T.; de Castro, R.A.; de Castro, L.L.; Freitas, A.C.C.; Costa, E.A.; Florentino, I.F.; Mafra, J.C.M.; de Souza, G.E.P.; Vanderlinde, F.A. Antinociceptive, anti-inflammatory and antipyretic effects of 1.5-diphenyl-1H-Pyrazole-3-carbohydrazide, a new heterocyclic pyrazole derivative. Life Sci. 2014, 95, 81–88. [Google Scholar] [CrossRef]
- Kumar, G.; Tanwar, O.; Kumar, J.; Akhter, M.; Sharma, S.; Pillai, C.; Alam, M.M.; Zama, M. Pyrazole-pyrazoline as promising novel antimalarial agents: A mechanistic study. Eur. J. Med. Chem. 2018, 149, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Dong, M.; Zhao, J.; Zhang, X.; Mei, X. Synthesis and antifungal activity of novel pyrazolines and isoxazolines derived from cuminaldehyde. J. Pestic. Sci. 2019, 44, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Othman, I.M.; Alamshany, Z.M.; Tashkandi, N.Y.; Gad-Elkareem, M.A.; Abd El-Karim, S.S.; Nossier, E.S. Synthesis and biological evaluation of new derivatives of thieno-thiazole and dihydrothiazolo-thiazole scaffolds integrated with a pyrazoline nucleus as anticancer and multi-targeting kinase inhibitors. RSC Adv. 2022, 12, 561–577. [Google Scholar] [CrossRef] [PubMed]
- Kerru, N.; Singh, P.; Koorbanally, N.; Raj, R.; Kumar, V. Recent advances (2015–2016) in anticancer hybrids. Eur. J. Med. Chem. 2017, 142, 179–212. [Google Scholar] [CrossRef] [PubMed]
- Abou-Zied, H.A.; Beshr, E.A.; Gomaa, H.A.; Mostafa, Y.A.; Youssif, B.G.; Hayallah, A.M.; Abdel-Aziz, M. Discovery of new cyanopyridine/chalcone hybrids as dual inhibitors of EGFR/BRAFV600E with promising antiproliferative properties. Arch. Der Pharm. 2022, 356, e2200464. [Google Scholar] [CrossRef] [PubMed]
- Abdelbaset, M.S.; Abdel-Aziz, M.; Abuo-Rahma, G.E.D.A.; Abdelrahman, M.H.; Ramadan, M.; Youssif, B.G. Novel quinoline derivatives carrying nitrones/oximes nitric oxide donors: Design, synthesis, antiproliferative and caspase-3 activation activities. Arch. Der Pharm. 2019, 352, 1800270. [Google Scholar] [CrossRef] [PubMed]
- Al-Sanea, M.M.; Gotina, L.; Mohamed, M.F.; Gomaa, H.A.; Mathew, B.; Youssif, B.G.; Alharbi, K.S.; Elsayed, Z.M.; Abdelgawad, M.A.; Eldehna, W.M. Design, synthesis and biological evaluation of new hdac1 and hdac2 inhibitors endowed with ligustrazine as a novel cap moiety. Drug Des. Dev. Ther. 2020, 14, 497. [Google Scholar] [CrossRef]
- Mohassab, A.M.; Hassan, H.A.; Abdelhamid, D.; Gouda, A.M.; Youssif, B.G.M.; Tateishi, H.; Fujita, M.; Otsuka, M.; Abdel-Aziz, M. Design and synthesis of novel quinoline/chalcone/1,2,4-triazole hybrids as potent antiproliferative agent targeting EGFR and BRAFV600E kinases. Bioorg. Chem. 2021, 106, 104510. [Google Scholar] [CrossRef]
- Mahmoud, M.A.; Mohammed, A.F.; Salem, O.I.; Gomaa, H.A.; Youssif, B.G. New 1,3,4-oxadiazoles linked with the 1,2,3-triazole moiety as antiproliferative agents targeting the EGFR tyrosine kinase. Arch. Der Pharm. 2022, 355, 2200009. [Google Scholar] [CrossRef]
- Gomaa, H.A.; El-Sherief, H.A.; Hussein, S.; Gouda, A.M.; Salem, O.I.; Alharbi, K.S.; Hayallah, A.M.; Youssif, B.G. Novel 1,2,4-triazole derivatives as apoptotic inducers targeting p53: Synthesis and antiproliferative activity. Bioorg. Chem. 2020, 105, 104369. [Google Scholar] [CrossRef]
- Mohamed, F.A.; Gomaa, H.A.; Hendawy, O.; Ali, A.T.; Farghaly, H.S.; Gouda, A.M.; Abdelazeem, A.H.; Abdelrahman, M.H.; Trembleau, L.; Youssif, B.G. Design, synthesis, and biological evaluation of novel EGFR inhibitors containing 5-chloro-3-hydroxymethyl-indole-2-carboxamide scaffold with apoptotic antiproliferative activity. Bioorg. Chem. 2021, 112, 104960. [Google Scholar] [CrossRef] [PubMed]
- Hisham, M.; Youssif, B.G.; Osman, E.E.A.; Hayallah, A.M.; Abdel-Aziz, M. Synthesis and biological evaluation of novel xanthine derivatives as potential apoptotic antitumor agents. Eur. J. Med. Chem. 2019, 176, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Abou-Zied, H.A.; Youssif, B.G.; Mohamed, M.F.; Hayallah, A.M.; Abdel-Aziz, M. EGFR inhibitors and apoptotic inducers: Design, synthesis, anticancer activity and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorg. Chem. 2019, 89, 102997. [Google Scholar] [CrossRef] [PubMed]
- Hafliger, E.; Boccaccino, A.; Lapeyre-Prost, A.; Perret, A.; Gallois, C.; Antista, M.; Pilla, L.; Lecomte, T.; Scartozzi, M.; Soularue, E. Encorafenib plus cetuximab treatment in BRAF V600E-mutated metastatic colorectal cancer patients pre-treated with an anti-EGFR: An AGEO-GONO case series. Eur. J. Cancer 2022, 168, 34–40. [Google Scholar] [CrossRef]
- Bhat, M.A.; Tüzün, B.; Alsaif, N.A.; Khan, A.A.; Naglah, A.M. Synthesis, characterization, molecular modeling against EGFR target and ADME/T analysis of novel purine derivatives of sulfonamides. J. Mol. Struct. 2022, 1257, 132600. [Google Scholar] [CrossRef]
- Umar, A.B.; Uzairu, A.; Shallangwa, G.A.; Uba, S. QSAR modelling and molecular docking studies for anti-cancer compounds against melanoma cell line SK-MEL-2. Heliyon 2020, 6, e03640. [Google Scholar] [CrossRef]
- Gholivand, K.; Sabaghian, M.; Malekshah, R.E. Synthesis, characterization, cytotoxicity studies, theoretical approach of adsorptive removal and molecular calculations of four new phosphoramide derivatives and related graphene oxide. Bioorg. Chem. 2021, 115, 105193. [Google Scholar] [CrossRef]
- Chen, D.; Wang, H. HOMO-LUMO energy splitting in polycyclic aromatic hydrocarbons and their derivatives. Proc. Combust. Inst. 2019, 37, 953–959. [Google Scholar] [CrossRef]
- Kadam, P.R.; Bodke, Y.D.; Pushpavathi, I.; Satyanarayan, N.; Nippu, B. Synthesis, Characterization, DFT and Biological Study of New Methylene Thio-Linked Coumarin Derivatives. J. Mol. Struct. 2023, 1278, 134918. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef]
- Hunagund, U.; Shaikh, F.; Shastri, L.A.; Malimath, G.H.; Naikh, L.; Sunagar, V.S. Synthesis, characterization, photo physical and DFT studies of bicoumarin and 3-(3-benzofuranyl) coumarin derivatives. Chem. Data Collect. 2020, 30, 100537. [Google Scholar] [CrossRef]
- Rahuman, M.H.; Muthu, S.; Raajaraman, B.; Raja, M.; Umamahesvari, H. Investigations on 2-(4-Cyanophenylamino) acetic acid by FT-IR, FT-Raman, NMR and UV-Vis spectroscopy, DFT (NBO, HOMO-LUMO, MEP and Fukui function) and molecular docking studies. Heliyon 2020, 6, 4976. [Google Scholar] [CrossRef] [PubMed]
- Zia, M.; Muhammad, S.; Bibi, S.; Abbasi, S.W.; Al-Sehemi, A.G.; Chaudhary, A.R.; Bai, F.Q. Exploring the potential of novel phenolic compounds as potential therapeutic candidates against SARS-CoV-2, using quantum chemistry, molecular docking and dynamic studies. Bioorg. Med. Chem. Lett. 2021, 43, 128079. [Google Scholar] [CrossRef]
- Parmar, D.R.; Soni, J.Y.; Guduru, R.; Rayani, R.H.; Kusurkar, R.V.; Vala, A.G.; Talukdar, S.N.; Eissa, I.H.; Metwaly, A.M.; Khalil, A. Discovery of new anticancer thiourea-azetidine hybrids: Design, synthesis, in vitro antiproliferative, SAR, in silico molecular docking against VEGFR-2, ADMET, toxicity, and DFT studies. Bioorg. Chem. 2021, 115, 105206. [Google Scholar] [CrossRef] [PubMed]
- Hagar, M.; Ahmed, H.A.; Aljohani, G.; Alhaddad, O.A. Investigation of some antiviral N-heterocycles as COVID-19 drug: Molecular docking and DFT calculations. Int. J. Mol. Sci. 2020, 21, 3922. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lei, T.; Shen, C.; Wang, Z.; Cao, D.; Hou, T. ADMET evaluation in drug discovery. 19. Reliable prediction of human cytochrome P450 inhibition using artificial intelligence approaches. J. Chem. Inf. Model. 2019, 59, 4587–4601. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Sattar, N.E.; Badawy, E.H.; AbdEl-Hady, W.H.; Abo-Alkasem, M.I.; Mandour, A.A.; Ismail, N.S. Design and synthesis of new CDK2 inhibitors containing thiazolone and thiazolthione scafold with apoptotic activity. Chem. Pharm. Bull. 2021, 69, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Rai, H.; Barik, A.; Singh, Y.P.; Suresh, A.; Singh, L.; Singh, G.; Nayak, U.Y.; Dubey, V.K.; Modi, G. Molecular docking, binding mode analysis, molecular dynamics, and prediction of ADMET/toxicity properties of selective potential antiviral agents against SARS-CoV-2 main protease: An effort toward drug repurposing to combat COVID-19. Mol. Divers. 2021, 25, 1905–1927. [Google Scholar] [CrossRef]
- Roy, P.P.; Roy, K. QSAR Studies of CYP2D6 Inhibitor Aryloxypropanolamines Using 2D and 3D Descriptors. Chem. Biol. Drug Des. 2009, 73, 442–455. [Google Scholar] [CrossRef]
- Xia, X.; Maliski, E.G.; Gallant, P.; Rogers, D. Classification of Kinase Inhibitors Using a Bayesian Model. J. Med. Chem. 2004, 47, 4463–4470. [Google Scholar] [CrossRef]
- Goodrnan, G.; Wilson, R. Comparison of the Dependence of the TD50 on Maximum Tolerated Dose for Mutagens and Nonmutagens. Risk Anal. 1992, 12, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Gonella Diaza, R.; Manganelli, S.; Esposito, A.; Roncaglioni, A.; Manganaro, A.; Benfenati, E. Comparison of in silico tools for evaluating rat oral acute toxicity. SAR QSAR Environ. Res. 2015, 26, 523–555. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmus, K.R. The Draize Eye Test. Surv. Ophthalmol. 2001, 45, 493–515. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | Cell Viability % | Antiproliferative Activity IC50 ± SEM (µM) | ||||
|---|---|---|---|---|---|---|
| A-549 | MCF-7 | Panc-1 | HT-29 | Average IC50 (GI50) | ||
| 8 | 90 | 4.30 ± 0.40 | 4.10 ± 0.40 | 4.60 ± 0.40 | 4.50 ± 0.40 | 4.40 |
| 9 | 89 | 2.20 ± 0.20 | 1.90 ± 0.10 | 2.60 ± 0.20 | 2.50 ± 0.20 | 2.30 |
| 10 | 89 | 3.20 ± 0.30 | 2.90 ± 0.20 | 3.20 ± 0.30 | 3.10 ± 0.30 | 3.10 |
| 11 | 87 | 2.90 ± 0.20 | 2.70 ± 0.20 | 2.90 ± 0.20 | 3.00 ± 0.20 | 2.90 |
| 12 | 90 | 1.00 ± 0.10 | 0.90 ± 0.10 | 1.10 ± 0.10 | 1.20 ± 0.10 | 1.05 |
| 13 | 87 | 4.80 ± 0.40 | 4.60 ± 0.40 | 4.90 ± 0.40 | 5.00 ± 0.40 | 4.80 |
| 14 | 89 | 1.80 ± 0.10 | 1.60 ± 0.10 | 2.00 ± 0.10 | 2.00 ± 0.10 | 1.85 |
| 15 | 90 | 1.40 ± 0.10 | 1.20 ± 0.10 | 1.70 ± 0.10 | 1.70 ± 0.10 | 1.50 |
| 16 | 89 | 3.70 ± 0.30 | 3.50 ± 0.30 | 3.80 ± 0.30 | 3.90 ± 0.30 | 3.70 |
| 17 | 91 | 1.20 ± 0.10 | 1.00 ± 0.10 | 1.30 ± 0.10 | 1.30 ± 0.10 | 1.20 |
| Doxorubicin | -- | 1.40 ± 0.08 | 0.90 ± 0.02 | 1.00 ± 0.02 | 1.20 ± 0.03 | 1.10 |
| Compound | EGFR Inhibition IC50 ± SEM (nM) | BRAFV600E Inhibition IC50 ± SEM (nM) |
|---|---|---|
| 12 | 81 ± 5 | 93 ± 6 |
| 14 | 104 ± 9 | 125 ± 10 |
| 15 | 93 ± 7 | 98 ± 8 |
| 17 | 87 ± 6 | 107 ± 9 |
| Erlotinib | 80 ± 5 | 60 ± 5 |
| Compound Number | Caspase–3 | |
|---|---|---|
| Conc (pg/mL) | Fold Change | |
| 12 | 543.50 ± 4 | 8.5 |
| 17 | 469.50 ± 4 | 7.2 |
| Staurosporine | 503.00 ± 4 | 8.0 |
| Control | 65.50 | 1 |
| Compound Number | Caspase-8 | Bax | Bcl-2 | |||
|---|---|---|---|---|---|---|
| Conc (ng/mL) | Fold Change | Conc (pg/mL) | Fold Change | Conc (ng/mL) | Fold Reduction | |
| 12 | 1.87 | 21 | 296 | 37 | 1.05 | 5 |
| 17 | 1.77 | 20 | 285 | 35 | 1.15 | 4 |
| Staurosporine | 1.80 | 20 | 280 | 35 | 1.10 | 5 |
| Control | 0.09 | 1 | 8 | 1 | 5 | 1 |
| Comp. ID | 12 | 15 |
|---|---|---|
| EHOMO (eV) | −5.127 | −5.231 |
| ELUMO (eV) | −1.165 | −1.213 |
| ELUMO-EHOMO (eV) | 3.962 | 4.018 |
| Ionization potential (IP) | 5.127 | 5.231 |
| Electron affinity (EA) | 1.165 | 1.213 |
| Electronegativity (χ) | 3.146 | 3.222 |
| Chemical potential (μ) | −3.146 | −3.222 |
| Chemical hardness (η) | 1.981 | 2.009 |
| Chemical softness (S) | 0.252 | 0.249 |
| Comp. ID | PSA | PPB a | Absorption Level b | CYP2D6 Prediction c | BBB Level d | Solubility Level e | AlogP98 |
|---|---|---|---|---|---|---|---|
| 8 | 87.308 | TRUE | 2 | FALSE | 4 | 1 | 6 |
| 9 | 96.438 | TRUE | 2 | FALSE | 4 | 2 | 5.984 |
| 10 | 87.515 | TRUE | 2 | FALSE | 4 | 1 | 6.749 |
| 11 | 88.308 | TRUE | 2 | FALSE | 4 | 1 | 6.665 |
| 12 | 116.098 | FALSE | 2 | FALSE | 4 | 2 | 5.287 |
| 13 | 99.738 | FALSE | 2 | FALSE | 4 | 2 | 6.292 |
| 14 | 98.938 | FALSE | 2 | FALSE | 4 | 2 | 6.068 |
| 15 | 95.648 | TRUE | 2 | FALSE | 4 | 2 | 5.984 |
| 16 | 97.579 | TRUE | 2 | FALSE | 4 | 2 | 5.525 |
| 17 | 114.098 | FALSE | 2 | FALSE | 4 | 2 | 5.287 |
| Erlotinib | 71.052 | TRUE | 0 | FALSE | 1 | 2 | 4.309 |
| Comp. ID | FDA Rodent Carcinogenicity (Mouse–Female) | Rat Maximum Tolerated Dose (Feed) a | Rat Oral LD50 a | Ocular Irritancy | Skin Irritancy |
|---|---|---|---|---|---|
| 8 | Non-Carcinogenic | 0.271 | 0.871442 | Moderate | Non-Irritant |
| 9 | Non-Carcinogenic | 0.1722 | 1.35834 | Mild | Non-Irritant |
| 10 | Non-Carcinogenic | 0.1377 | 1.39385 | Moderate | Non-Irritant |
| 11 | Non-Carcinogenic | 0.2145 | 1.15043 | Moderate | Non-Irritant |
| 12 | Non-Carcinogenic | 0.1287 | 10.2483 | Mild | Non-Irritant |
| 13 | Non-Carcinogenic | 0.1108 | 2.4424 | Mild | Non-Irritant |
| 14 | Non-Carcinogenic | 0.1106 | 1.80353 | Mild | Non-Irritant |
| 15 | Non-Carcinogenic | 0.1722 | 1.35834 | Mild | Non-Irritant |
| 16 | Non-Carcinogenic | 0.1878 | 0.707581 | Mild | Non-Irritant |
| 17 | Non-Carcinogenic | 0.1287 | 10.2483 | Mild | Non-Irritant |
| Erlotinib | Non-Carcinogenic | 0.0828 | 0.662169 | Mild | Non-Irritant |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Wahaibi, L.H.; Abou-Zied, H.A.; Beshr, E.A.M.; Youssif, B.G.M.; Hayallah, A.M.; Abdel-Aziz, M. Design, Synthesis, Antiproliferative Actions, and DFT Studies of New Bis–Pyrazoline Derivatives as Dual EGFR/BRAFV600E Inhibitors. Int. J. Mol. Sci. 2023, 24, 9104. https://doi.org/10.3390/ijms24109104
Al-Wahaibi LH, Abou-Zied HA, Beshr EAM, Youssif BGM, Hayallah AM, Abdel-Aziz M. Design, Synthesis, Antiproliferative Actions, and DFT Studies of New Bis–Pyrazoline Derivatives as Dual EGFR/BRAFV600E Inhibitors. International Journal of Molecular Sciences. 2023; 24(10):9104. https://doi.org/10.3390/ijms24109104
Chicago/Turabian StyleAl-Wahaibi, Lamya H., Hesham A. Abou-Zied, Eman A. M. Beshr, Bahaa G. M. Youssif, Alaa M. Hayallah, and Mohamed Abdel-Aziz. 2023. "Design, Synthesis, Antiproliferative Actions, and DFT Studies of New Bis–Pyrazoline Derivatives as Dual EGFR/BRAFV600E Inhibitors" International Journal of Molecular Sciences 24, no. 10: 9104. https://doi.org/10.3390/ijms24109104