
Serum Vitamin D Metabolites by HPLC-MS/MS Combined with Differential Ion Mobility Spectrometry: Aspects of Sample Preparation without Derivatization

 ,
,
Abstract
:1. Introduction
2. Results
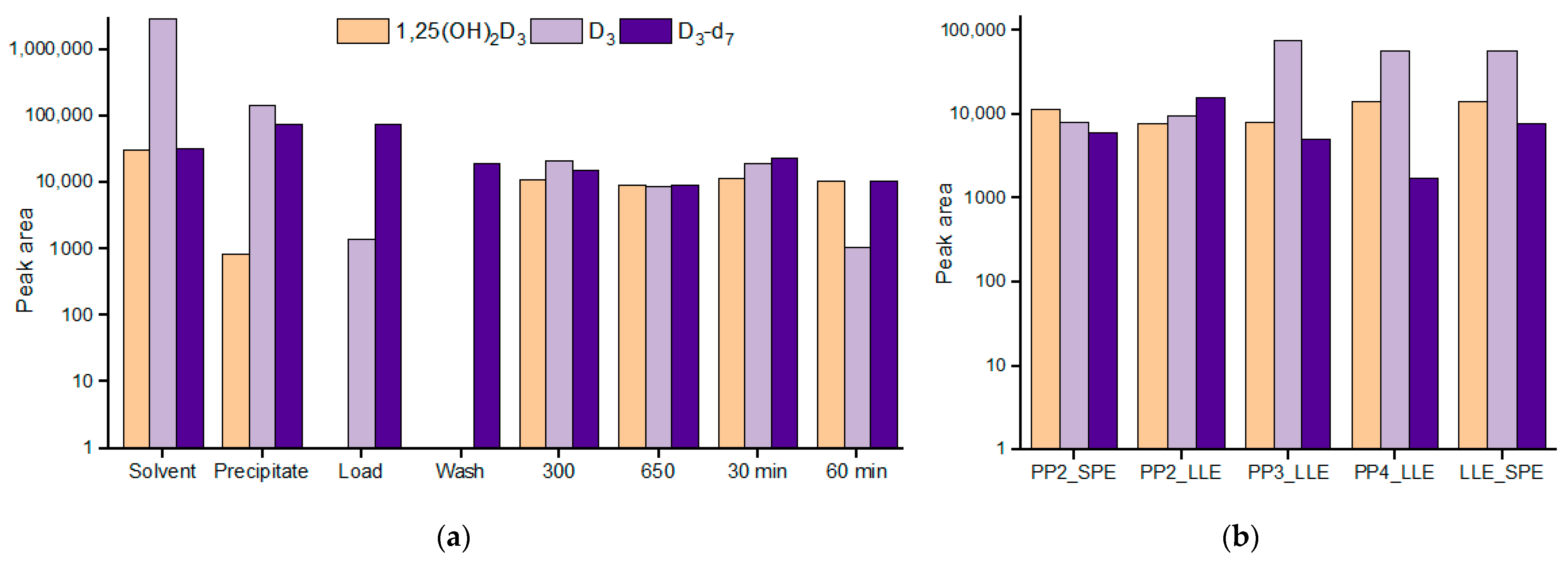
2.1. Principally Different Sample Preparation Approaches
2.2. Vitamin D3 Issue Sample Preparation Selection
2.3. LLE Followed by SPE Procedure
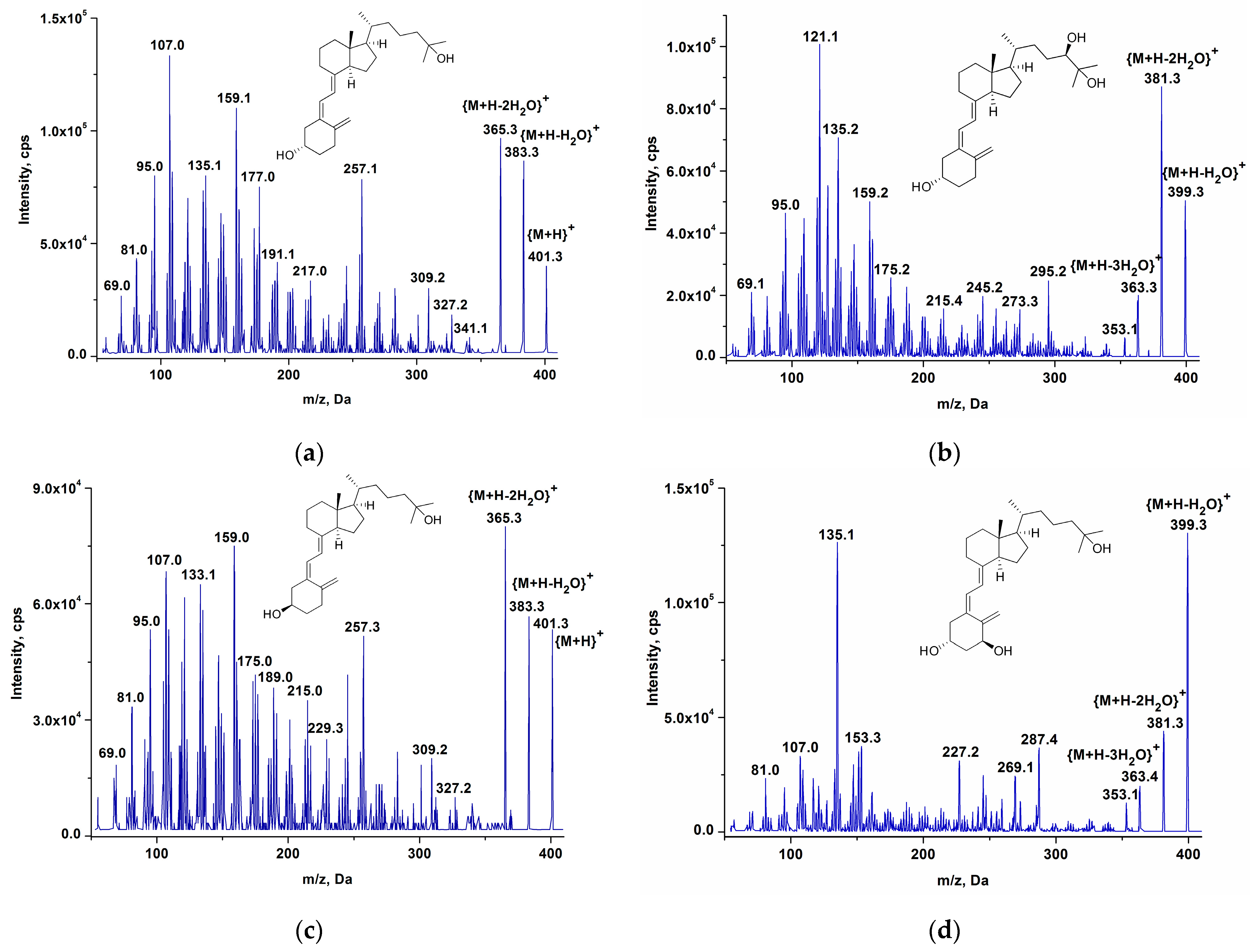
2.3.1. The Fragmentation Patterns
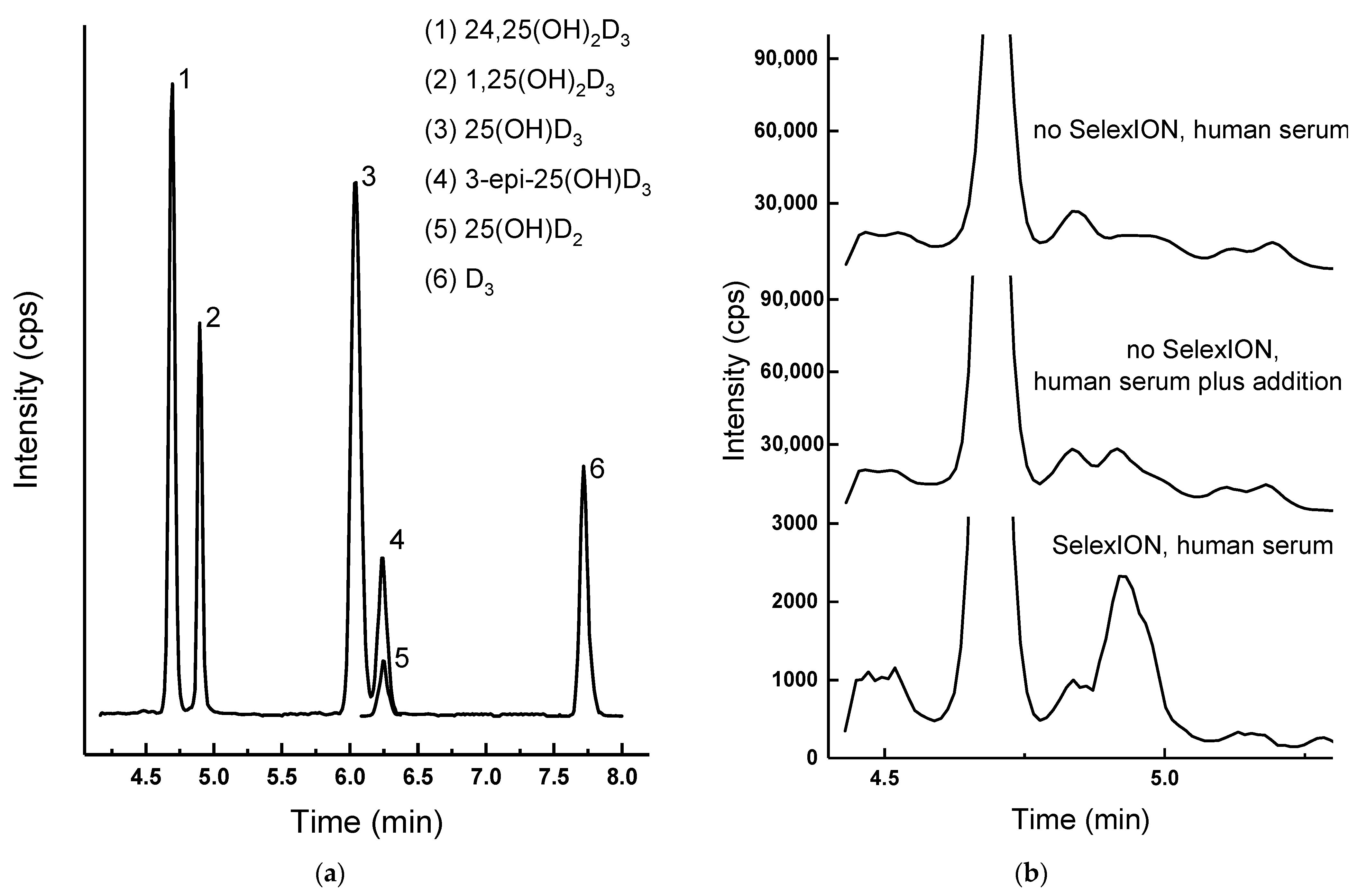
2.3.2. The SelexION Advantage
2.4. LLE Followed by SPE Procedure: Partial Validation, DEQAS Controls
3. Discussion
4. Materials and Methods
4.1. Reagents and Materials
4.2. LC–MS/MS Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time, Min | Acetonitrile, % | Methanol, % | Formic Acid in Water, % |
|---|---|---|---|
| 0 | 0 | 50 | 50 |
| 1 | 0 | 50 | 50 |
| 1.5 | 0 | 60 | 40 |
| 2 | 0 | 60 | 40 |
| 3.2 | 0 | 70 | 30 |
| 4.4 | 3 | 68 | 29 |
| 4.8 | 5 | 70 | 25 |
| 5.0 | 5 | 70 | 25 |
| 6.0 | 5 | 75 | 20 |
| 6.5 | 0 | 85 | 15 |
| 8.0 | 0 | 85 | 15 |
| 8.5 | 0 | 50 | 50 |
| 12 | 0 | 50 | 50 |
4.3. MALDI Mass Spectrometry
4.4. Sample Preparation Procedures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tuckey, R.C.; Cheng, C.Y.S.; Slominski, A.T. The Serum Vitamin D Metabolome: What We Know and What Is Still to Discover. J. Steroid Biochem. Mol. Biol. 2019, 186, 4–21. [Google Scholar] [CrossRef] [PubMed]
- Kowalówka, M.; Główka, A.K.; Karaźniewicz-łada, M.; Kosewski, G. Clinical Significance of Analysis of Vitamin d Status in Various Diseases. Nutrients 2020, 12, 2788. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, M.; Farrell, C.J.L.; Pusceddu, I.; Fabregat-Cabello, N.; Cavalier, E. Assessment of Vitamin D Status—A Changing Landscape. Clin. Chem. Lab. Med. 2017, 55, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Makin, H.L.J.; Jones, G.; Kaufmann, M.; Calverley, M.J. Analysis of Vitamins D, Their Metabolites and Analogues. In Steroid Analysis; Makin, H.L.J., Gower, D.B., Eds.; Springer: Dordrecht, The Netherlands, 2010; pp. 967–1096. ISBN 978-1-4020-9775-1. [Google Scholar] [CrossRef]
- Dirks, N.F.; Martens, F.; Vanderschueren, D.; Billen, J.; Pauwels, S.; Ackermans, M.T.; Endert, E.; den Heijer, M.; Blankenstein, M.A.; Heijboer, A.C. Determination of Human Reference Values for Serum Total 1,25-Dihydroxyvitamin D Using an Extensively Validated 2D ID-UPLC–MS/MS Method. J. Steroid Biochem. Mol. Biol. 2016, 164, 127–133. [Google Scholar] [CrossRef]
- Alonso, N.; Zelzer, S.; Eibinger, G.; Herrmann, M. Vitamin D Metabolites: Analytical Challenges and Clinical Relevance. Calcif. Tissue Int. 2023, 112, 158–177. [Google Scholar] [CrossRef]
- Volmer, D.A.; Mendes, L.R.B.C.; Stokes, C.S. Analysis of Vitamin D Metabolic Markers by Mass Spectrometry: Current Techniques, Limitations of the “Gold Standard” Method, and Anticipated Future Directions. Mass Spectrom. Rev. 2015, 34, 2–23. [Google Scholar] [CrossRef]
- White, P.; Cooke, N. The Multifunctional Properties and Characteristics of Vitamin D-Binding Protein. Trends Endocrinol. Metab. 2000, 11, 320–327. [Google Scholar] [CrossRef]
- Yin, S.; Yang, Y.; Wu, L.; Li, Y.; Sun, C. Recent Advances in Sample Preparation and Analysis Methods for Vitamin D and Its Analogues in Different Matrices. TrAC 2019, 110, 204–220. [Google Scholar] [CrossRef]
- Jenkinson, C.; Desai, R.; Slominski, A.T.; Tuckey, R.C.; Hewison, M.; Handelsman, D.J. Simultaneous Measurement of 13 Circulating Vitamin D3 and D2 Mono and Dihydroxy Metabolites Using Liquid Chromatography Mass Spectrometry. Clin. Chem. Lab. Med. 2021, 59, 1642–1652. [Google Scholar] [CrossRef]
- Gathungu, R.M.; Flarakos, C.C.; Satyanarayana Reddy, G.; Vouros, P. The Role of Mass Spectrometry in the Analysis of Vitamin D Compounds. Mass Spectrom. Rev. 2013, 32, 72–86. [Google Scholar] [CrossRef]
- Aronov, P.A.; Hall, L.M.; Dettmer, K.; Stephensen, C.B.; Hammock, B.D. Metabolic Profiling of Major Vitamin D Metabolites Using Diels–Alder Derivatization and Ultra-Performance Liquid Chromatography–Tandem Mass Spectrometry. Anal. Bioanal. Chem. 2008, 391, 1917–1930. [Google Scholar] [CrossRef] [PubMed]
- Hedman, C.J.; Wiebe, D.A.; Dey, S.; Plath, J.; Kemnitz, J.W.; Ziegler, T.E. Development of a Sensitive LC/MS/MS Method for Vitamin D Metabolites: 1,25 Dihydroxyvitamin D2&3 Measurement Using a Novel Derivatization Agent. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2014, 953–954, 62–67. [Google Scholar] [CrossRef]
- Casetta, B.; Jans, I.; Billen, J.; Vanderschueren, D.; Bouillon, R. Development of a Method for the Quantification of 1α,25(OH) 2-Vitamin D3 in Serum by Liquid Chromatography Tandem Mass Spectrometry without Derivatization. Eur. J. Mass Spectrom. 2010, 16, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Jenkinson, C.; Taylor, A.E.; Hassan-Smith, Z.K.; Adams, J.S.; Stewart, P.M.; Hewison, M.; Keevil, B.G. High Throughput LC-MS/MS Method for the Simultaneous Analysis of Multiple Vitamin D Analytes in Serum. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2016, 1014, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Abu Kassim, N.S.; Shaw, P.N.; Hewavitharana, A.K. Simultaneous Determination of 12 Vitamin D Compounds in Human Serum Using Online Sample Preparation and Liquid Chromatography-Tandem Mass Spectrometry. J. Chromatogr. A 2018, 1533, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Mena-Bravo, A.; Ferreiro-Vera, C.; Priego-Capote, F.; Maestro, M.A.; Mouriño, A.; Quesada-Gómez, J.M.; Luque de Castro, M.D. Quantitative Analytical Method to Evaluate the Metabolism of Vitamin D. Clin. Chim. Acta 2015, 442, 6–12. [Google Scholar] [CrossRef]
- Povaliaeva, A.; Pigarova, E.; Zhukov, A.; Bogdanov, V.; Dzeranova, L.; Mel’nikova, O.; Pekareva, E.; Malysheva, N.; Ioutsi, V.; Nikankina, L.; et al. Evaluation of Vitamin d Metabolism in Patients with Type 1 Diabetes Mellitus in the Setting of Cholecalciferol Treatment. Nutrients 2020, 12, 3873. [Google Scholar] [CrossRef]
- Hori, A.; Yamashita, M.; Yamaura, M.; Hongo, M.; Honda, T.; Hidaka, H. Rapid Quantitative Analysis of Human Serum Sphingomyelin Species Using MALDI-TOF Mass Spectrometry with Lipid Hydrolase Treatment. Clin. Chim. Acta 2016, 453, 95–99. [Google Scholar] [CrossRef]
- Valente, I.M.; Rodrigues, J.A. Recent Advances in Salt-Assisted LLE for Analyzing Biological Samples. Bioanalysis 2015, 7, 2187–2193. [Google Scholar] [CrossRef]
- Ioutsi, V.A.; Panov, Y.M.; Usol’tseva, L.O.; Smolin, E.S.; Antsupova, M.A.; Volchkov, P.Y.; Melnichenko, G.A.; Mokrysheva, N.G. Analysis of Serum Estrogens Using High-Performance Liquid Chromatography–Tandem Mass Spectrometry Coupled to Differential Ion Mobility Spectrometry. J. Anal. Chem. 2022, 77, 1760–1766. [Google Scholar] [CrossRef]
- Krylov, E.V.; Nazarov, E.G. Electric Field Dependence of the Ion Mobility. Int. J. Mass Spectrom. 2009, 285, 149–156. [Google Scholar] [CrossRef]
- Schneider, B.B.; Covey, T.R.; Coy, S.L.; Krylov, E.V.; Nazarov, E.G. Planar Differential Mobility Spectrometer as a Pre-Filter for Atmospheric Pressure Ionization Mass Spectrometry. Int. J. Mass Spectrom. 2010, 298, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Mahmoodani, F.; Perera, C.O.; Abernethy, G.; Fedrizzi, B.; Greenwood, D.; Chen, H. Identification of Vitamin D3 Oxidation Products Using High-Resolution and Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2018, 29, 1442–1455. [Google Scholar] [CrossRef] [PubMed]
- Coldwell, R.D.; Trafford, D.J.H.; Makin, H.L.J. Mass Fragmentographic Assay for 25-Hydroxyvitamin D in Plasma without Derivatization: Enhanced Sensitivity for Metabolites of Vitamins D2 and D3 after Pre-Column Dehydration. J. Mass Spectrom. 1995, 30, 348–356. [Google Scholar] [CrossRef]
- Huang, M.; Cadwallader, A.B.; Heltsley, R. Mechanism of Error Caused by Isotope-Labeled Internal Standard: Accurate Method for Simultaneous Measurement of Vitamin D and Pre-Vitamin D by Liquid Chromatography/Tandem Mass Spectrometry. Rapid Commun. Mass Spectrom. 2014, 28, 2101–2110. [Google Scholar] [CrossRef]
- Zhukov, A.; Povaliaeva, A.; Abilov, Z.; Kovaleva, E.; Usoltseva, L.; Eremkina, A.; Ioutsi, V.; Dzeranova, L.; Pigarova, E.; Rozhinskaya, L.; et al. Parameters of Vitamin D Metabolism in Patients with Hypoparathyroidism. Metabolites 2022, 12, 1279. [Google Scholar] [CrossRef]
- ICH Harmonised Guideline. Bioanalytical Method Validation and Study Sample Analysis M10; ICH Harmonised Guideline: Geneva, Switzerland, 2022. [Google Scholar]
- Zhang, Y.; Bala, V.; Mao, Z.; Chhonker, Y.S.; Murry, D.J. Quantification of Fat-Soluble Vitamins and Their Metabolites in Biological Matrices: An Updated Review. Bioanalysis 2020, 12, 625–640. [Google Scholar] [CrossRef]
- Sirajudeen, S.; Shah, I.; Ayoub, M.A.; Karam, S.M.; Menhali, A. Al Long-Term Vitamin D Deficiency Results in the Inhibition of Cell Proliferation and Alteration of Multiple Gastric Epithelial Cell Lineages in Mice. Int. J. Mol. Sci. 2022, 23, 6684. [Google Scholar] [CrossRef]
- Boelen, A.; Ruiter, A.F.C.; Claahsen-Van Der Grinten, H.L.; Endert, E.; Ackermans, M.T. Determination of a Steroid Profile in Heel Prick Blood Using LC-MS/MS. Bioanalysis 2016, 8, 375–384. [Google Scholar] [CrossRef]




| Analyte | Concentration, ng/mL 1 | Inter-Day Precision, % | Accuracy, % | IS Normalized MF 2 (% CV) | Analyte Recovery, % (% CV) | IS Recovery, % (% CV) |
|---|---|---|---|---|---|---|
| 1,25(OH)2D3 | 30 | 7.0 | 87.5 | 1.02 (9.7) | 95 (6.5) | 71 (2.3) |
| 225 | 3.6 | 95.9 | 0.90 (11.1) | 90 (7.7) | 73 (5.0) | |
| 24,25(OH)2D3 | 0.96 | 2.2 | 92.8 | 0.91 (2.0) | 79 (4.5) | 66 (4.0) |
| 7.2 | 3.7 | 92.2 | 0.99 (10.5) | 84 (3.8) | 70 (3.9) | |
| 25(OH)D3 | 7.5 | 3.5 | 95.4 | 0.89 (5.2) | 78 (5.9) | 67 (5.3) |
| 56.3 | 1.6 | 113.0 | 0.89 (10.2) | 82 (5.7) | 65 (5.2) | |
| 3-epi-25(OH)D3 | 2.25 | 3.7 | 89.6 | 0.94 (6.9) | 82 (7.2) | 68 (7.5) |
| 16.9 | 1.4 | 96.8 | 0.90 (10.4) | 76 (7.8) | 61 (8.8) | |
| 25(OH)D2 | 1.4 | 6.1 | 95.6 | 0.81 (3.8) | 77 (8.8) | 67 (5.3) 3 |
| 10.8 | 5.9 | 100.8 | 0.79 (10.0) | 81 (10.1) | 65 (5.2) 3 | |
| D3 | 37.5 | 5.5 | 90.2 | 0.98 (13.6) | 70 (12.4) | 62 (10.0) |
| 281 | 4.1 | 91.0 | 1.07 (9.3) | 72 (9.0) | 58 (9.2) |
| Analyte | Transition Type | Q1 | Q3 | tR, min | CE, V | DP, V | CXP, V |
|---|---|---|---|---|---|---|---|
| 1,25(OH)2D3 | quantifier | 399.3 | 135.1 | 4.9 | 28 | 89 | 16 |
| qualifier | 399.3 | 381.3 | 19 | 89 | 14 | ||
| 24,25(OH)2D3 | quantifier | 417.3 | 399.3 | 4.7 | 13 | 66 | 15 |
| qualifier | 417.3 | 381.3 | 15 | 66 | 14 | ||
| 25(OH)D3 | quantifier | 401.3 | 383.3 | 6.1 | 13 | 59 | 15 |
| qualifier | 401.3 | 365.4 | 17 | 59 | 13 | ||
| 3-epi-25(OH)D3 | quantifier | 401.29 | 383.2 | 6.3 | 14 | 110 | 9 |
| qualifier | 401.29 | 365.3 | 17 | 110 | 9 | ||
| 25(OH)D2 | quantifier | 413.3 | 355.3 | 6.3 | 15 | 110 | 7 |
| qualifier | 413.3 | 395.3 | 13 | 110 | 7 | ||
| D3 | quantifier | 385.4 | 259.3 | 7.7 | 20 | 100 | 30 |
| qualifier | 385.4 | 159.2 | 32 | 100 | 18 | ||
| 1,25(OH)2D3-d6 | IS | 405.3 | 135.0 | 4.9 | 30 | 170 | 12 |
| 24,25(OH)2D3-d6 | IS | 423.3 | 387.5 | 4.7 | 16 | 150 | 7 |
| 25(OH)D3-d6 | IS | 407.4 | 389.3 | 6.1 | 12 | 120 | 11 |
| 3-epi-25(OH)D3-d3 | IS | 404.4 | 368.3 | 6.3 | 18 | 150 | 6 |
| D3-d7 | IS | 392.4 | 266.3 | 7.7 | 20 | 90 | 30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usoltseva, L.; Ioutsi, V.; Panov, Y.; Antsupova, M.; Rozhinskaya, L.; Melnichenko, G.; Mokrysheva, N. Serum Vitamin D Metabolites by HPLC-MS/MS Combined with Differential Ion Mobility Spectrometry: Aspects of Sample Preparation without Derivatization. Int. J. Mol. Sci. 2023, 24, 8111. https://doi.org/10.3390/ijms24098111
Usoltseva L, Ioutsi V, Panov Y, Antsupova M, Rozhinskaya L, Melnichenko G, Mokrysheva N. Serum Vitamin D Metabolites by HPLC-MS/MS Combined with Differential Ion Mobility Spectrometry: Aspects of Sample Preparation without Derivatization. International Journal of Molecular Sciences. 2023; 24(9):8111. https://doi.org/10.3390/ijms24098111
Chicago/Turabian StyleUsoltseva, Liliia, Vitaliy Ioutsi, Yuriy Panov, Mariya Antsupova, Liudmila Rozhinskaya, Galina Melnichenko, and Natalia Mokrysheva. 2023. "Serum Vitamin D Metabolites by HPLC-MS/MS Combined with Differential Ion Mobility Spectrometry: Aspects of Sample Preparation without Derivatization" International Journal of Molecular Sciences 24, no. 9: 8111. https://doi.org/10.3390/ijms24098111