
The Triad Na+ Activated Na+ Channel (Nax)—Salt Inducible KINASE (SIK) and (Na+ + K+)-ATPase: Targeting the Villains to Treat Salt Resistant and Sensitive Hypertension
 ,
,
Abstract
:1. Introduction
2. Results
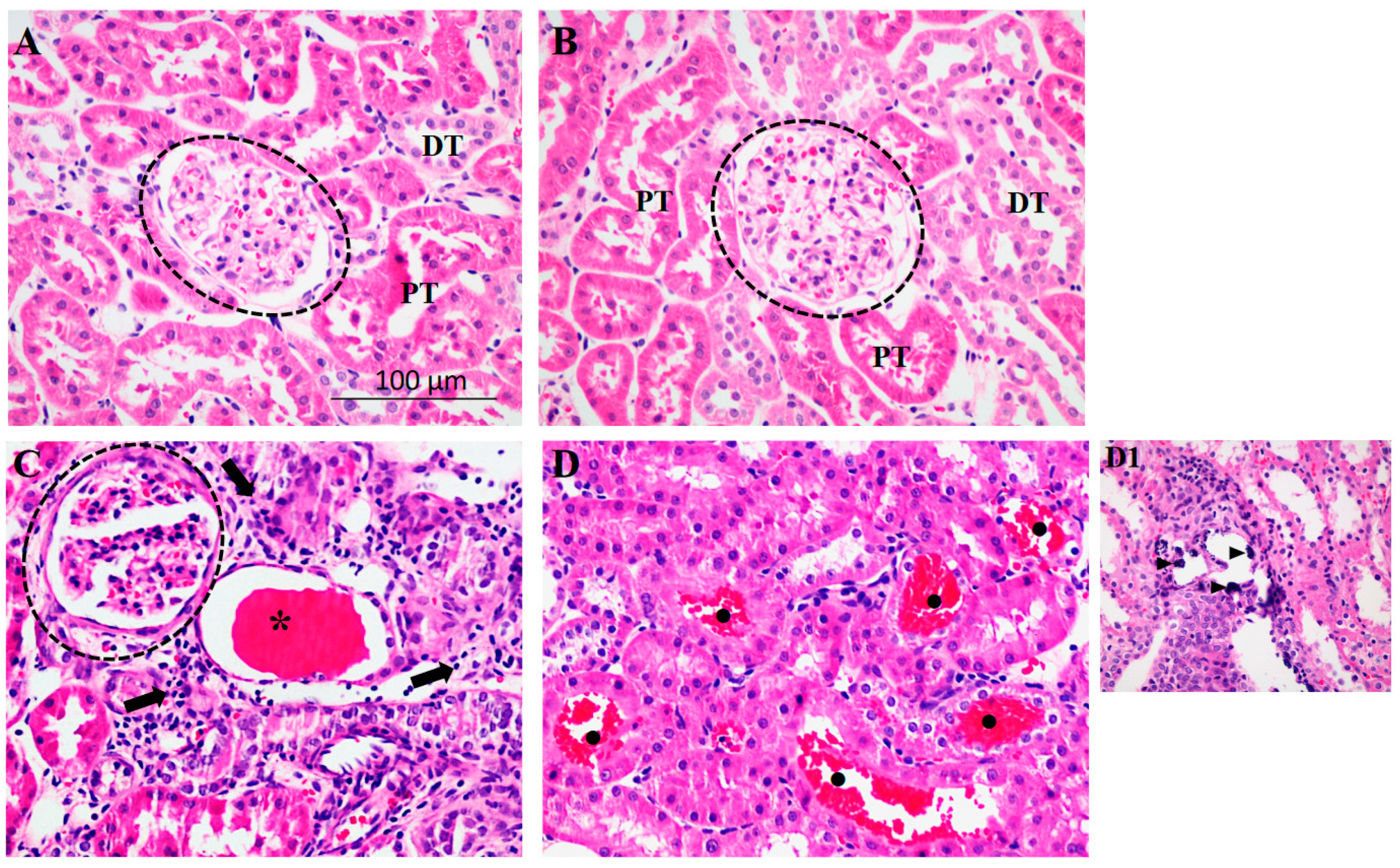
2.1. Physiological Parameters
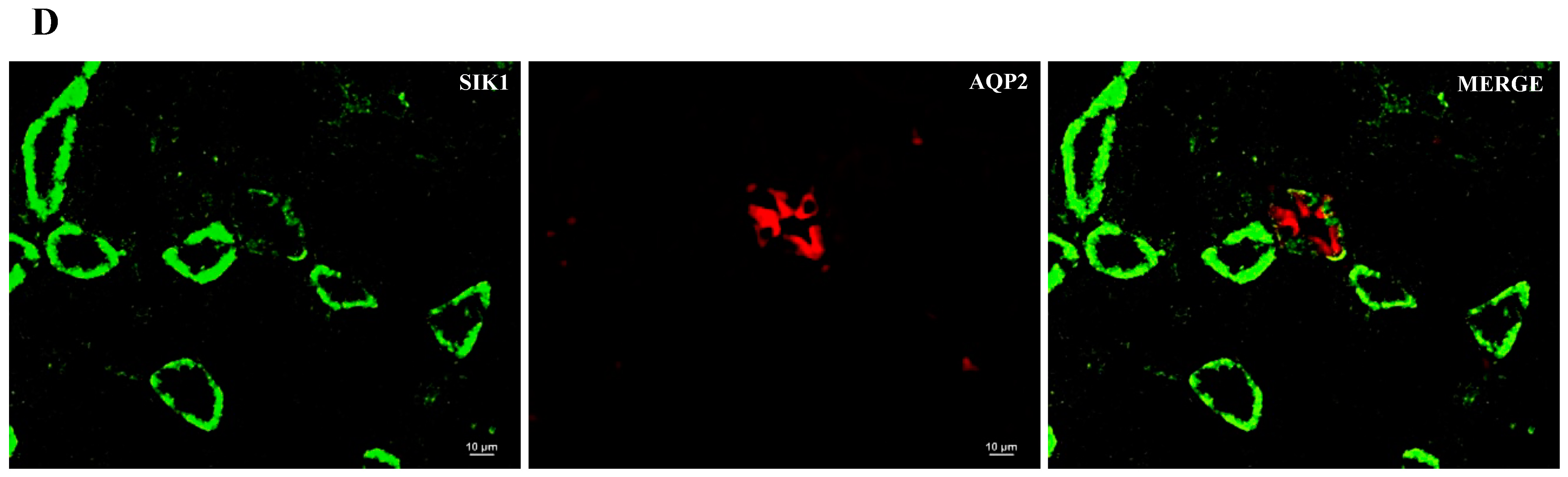
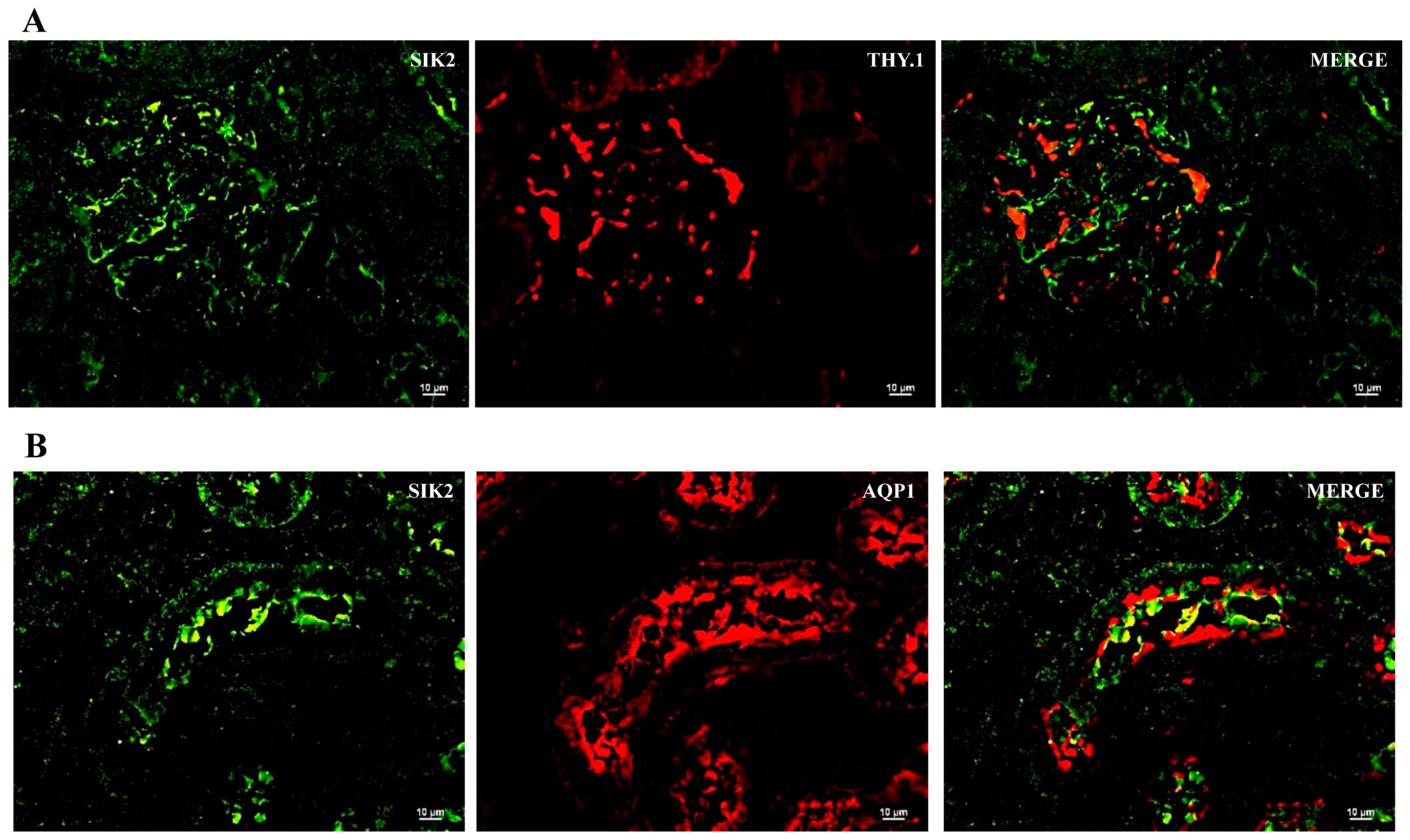
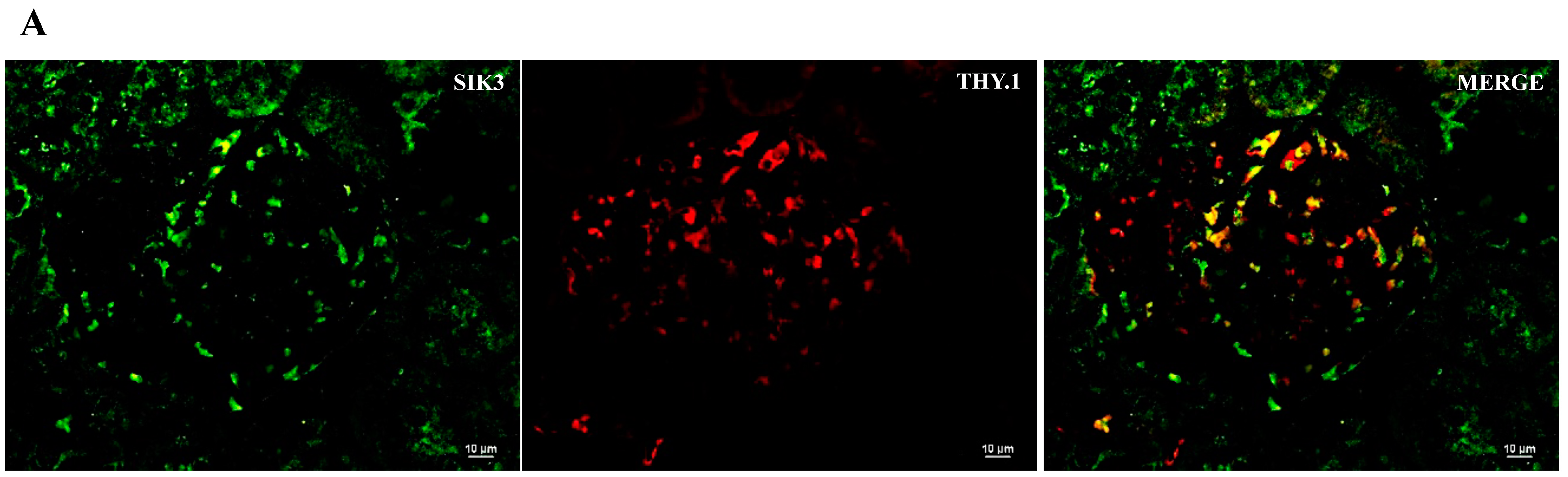
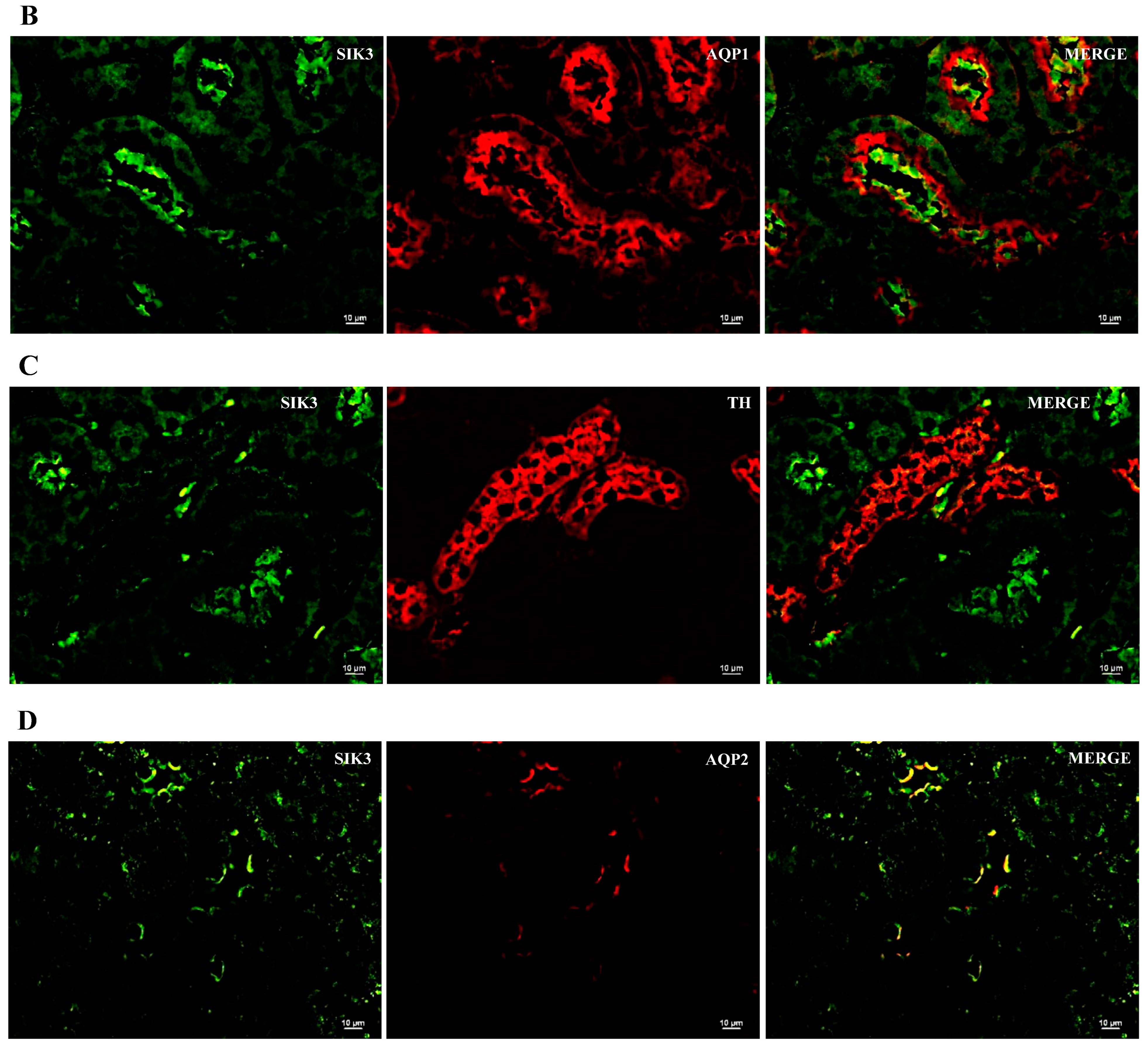
2.2. SIK Isoform Localization along the Nephron
2.3. Influence of HSD and HTN on SIK Content and Activity in the Kidney Cortex
2.4. The Impact of SIK Activity in Kidney Cortex (Na+ + K+)-ATPase Activity
3. Discussion
4. Materials and Methods
4.1. Experimental Design
4.2. Non-Invasive Systolic Blood Pressure (SBP) and Heart Rate (HR) Measurements
4.3. Urine and Blood Analysis
4.4. Renal Histology and SIK Immunolocalization
4.5. Western Blot Analysis
4.6. SIK Activity
4.7. Activity of Sodium Transporter
4.8. Statistical Analysis
5. Conclusions
- SIK isoforms localize throughout the nephron: (i) SIK1 is present in mesangial cells, the thick ascending limb and the collecting duct; (ii) SIK2 is present in glomeruli, the proximal tubule, the thick ascending limb, and the collecting duct; and (iii) SIK3 is present in mesangial cells, the proximal tubule, the thick ascending limb, and principal cells of the collecting duct.
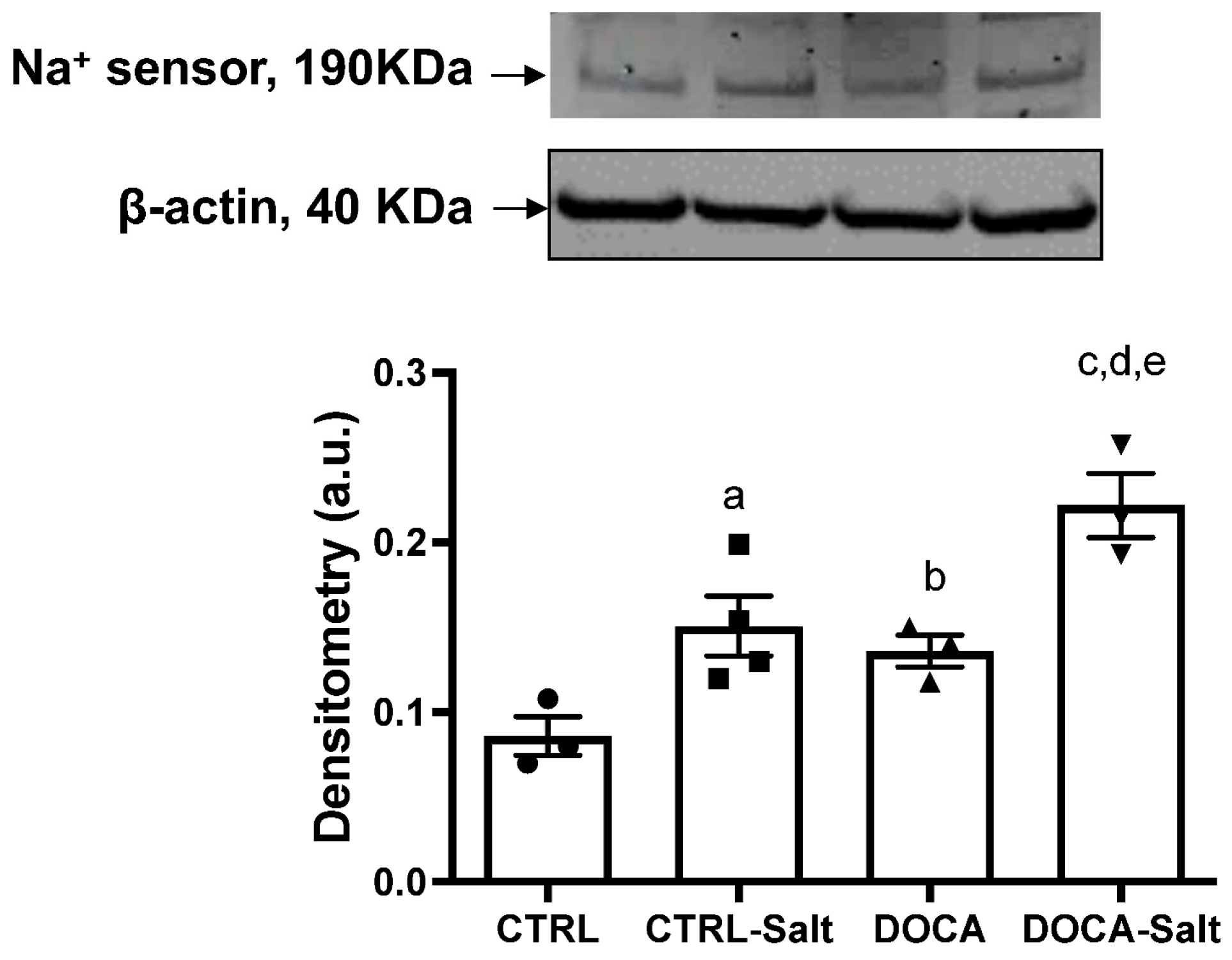
- Effect of HSD (CTRL-Salt vs. CTRL rats): CTRL-Salt rats elicit slight kidney function alteration, but not hypertension. In addition, these rats have augmented Nax content, total SIK activity, SIK1 content, and (Na+ + K+)-ATPase activity that is sensitive to SIK inhibition.
- Effect of HTN (DOCA vs. CTRL rats): DOCA rats exhibit moderate kidney function alteration and kidney injury along with augmented Nax and SIK1 contents, decreased total SIK activity, and unaltered (Na+ + K+)-ATPase activity that was insensitive to SIK inhibition.
- Effect of the combination of HSD and HTN:DOCA-Salt vs. DOCA rats: DOCA-Salt rats display exacerbation in SBP, severe kidney function decay, and kidney injury. In addition, these rats exhibit the greatest augmentation of Nax and SIK1 content, unaltered total SIK activity, and decreased (Na+ + K+)-ATPase activity sensitive to SIK inhibition.DOCA-Salt vs. CTRL-Salt rats: DOCA-Salt rats presented augmented SBP, HR, and a jeopardized tubular Na+ function, as FENa was decreased. This may be in part due to the diminished levels of SIK1, SIK activity, (Na+ + K+)-ATPase activity, and sensibility to SIK. However, compared to CTRL, Nax levels were increased in DOCA-Salt, which may contribute to the pathogenesis of salt sensitivity in this model.
- Fibrosis was attributed to augmented Nax content in CTRL-Salt, DOCA, and DOCA-Salt rats.
- We have identified the triad Nax/SIK/(Na+ + K+)-ATPase as the molecular key to preventing or treating the consequences of salt-sensitive HTN. HSD itself activates the triad Nax/SIK/(Na+ + K+)-ATPase, but on its own does not lead to HTN, although it initiates the progression of kidney function decay. In the present study, we demonstrated a disruption of the triad in the salt-sensitive HTN rats (DOCA-Salt rats). This is characterized by the intense expression of Nax and the attenuated response of SIK/(Na+ + K+)-ATPase in comparison to DOCA and CTRL-Salt rats. Thus, we conclude that Nax is associated with increased renal Na+ excretion and tissue fibrosis, which ultimately exacerbate HTN and the progression of chronic kidney disease.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef]
- LaMoia, T.E.; Shulman, G.I. Cellular and Molecular Mechanisms of Metformin Action. Endocr. Rev. 2020, 42, 77–96. [Google Scholar] [CrossRef]
- Katoh, Y.; Takemori, H.; Horike, N.; Doi, J.; Muraoka, M.; Min, L.; Okamoto, M. Salt-inducible kinase (SIK) isoforms: Their involvement in steroidogenesis and adipogenesis. Mol. Cell. Endocrinol. 2004, 217, 109–112. [Google Scholar] [CrossRef]
- Wang, Z.-N.; Takemori, H.; Halder, S.K.; Nonaka, Y.; Okamoto, M. Cloning of a novel kinase (SIK) of the SNF1/AMPK family from high salt diet-treated rat adrenal. FEBS Lett. 1999, 453, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Wein, M.N.; Foretz, M.; Fisher, D.E.; Xavier, R.J.; Kronenberg, H.M. Salt-Inducible Kinases: Physiology, Regulation by cAMP, and Therapeutic Potential. Trends Endocrinol. Metab. 2018, 29, 723–735. [Google Scholar] [CrossRef]
- Lin, X.-Z.; Takemori, H.; Katoh, Y.; Doi, J.; Horike, N.; Makino, A.; Nonaka, Y.; Okamoto, M. Salt-Inducible Kinase Is Involved in the ACTH/cAMP-Dependent Protein Kinase Signaling in Y1 Mouse Adrenocortical Tumor Cells. Mol. Endocrinol. 2001, 15, 1264–1276. [Google Scholar] [CrossRef]
- Koo, S.-H.; Flechner, L.; Qi, L.; Zhang, X.; Screaton, R.A.; Jeffries, S.; Hedrick, S.; Xu, W.; Boussouar, F.; Brindle, P.; et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 2005, 437, 1109–1114. [Google Scholar] [CrossRef]
- Liu, W.; Feldman, J.D.; Machado, H.B.; Vician, L.J.; Herschman, H.R. Expression of depolarization-induced immediate early gene proteins in PC12 cells. J. Neurosci. Res. 2003, 72, 670–678. [Google Scholar] [CrossRef]
- Jagannath, A.; Butler, R.; Godinho, S.I.H.; Couch, Y.; Brown, L.A.; Vasudevan, S.R.; Flanagan, K.C.; Anthony, D.; Churchill, G.C.; Wood, M.J.A.; et al. The CRTC1-SIK1 Pathway Regulates Entrainment of the Circadian Clock. Cell 2013, 154, 1100–1111. [Google Scholar] [CrossRef]
- Hashimoto, Y.K.; Satoh, T.; Okamoto, M.; Takemori, H. Importance of autophosphorylation at Ser186 in the A-loop of salt inducible kinase 1 for its sustained kinase activity. J. Cell. Biochem. 2008, 104, 1724–1739. [Google Scholar] [CrossRef]
- Bertorello, A.M.; Pires, N.; Igreja, B.; Pinho, M.J.; Vorkapic, E.; Wågsäter, D.; Wikström, J.; Behrendt, M.; Hamsten, A.; Eriksson, P.; et al. Increased Arterial Blood Pressure and Vascular Remodeling in Mice Lacking Salt-Inducible Kinase 1 (SIK1). Circ. Res. 2015, 116, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Hiyama, T.Y.; Watanabe, E.; Ono, K.; Inenaga, K.; Tamkun, M.M.; Yoshida, S.; Noda, M. Nax channel involved in CNS sodium-level sensing. Nat. Neurosci. 2002, 5, 511–512. [Google Scholar] [CrossRef]
- Noda, M. The Subfornical Organ, a Specialized Sodium Channel, and the Sensing of Sodium Levels in the Brain. Neuroscientist 2006, 12, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Dolivo, D.; Rodrigues, A.; Sun, L.; Li, Y.; Hou, C.; Galiano, R.; Hong, S.J.; Mustoe, T. The Nax (SCN7A) channel: An atypical regulator of tissue homeostasis and disease. Cell. Mol. Life Sci. 2021, 78, 5469–5488. [Google Scholar] [CrossRef] [PubMed]
- Noda, M.; Hiyama, T.Y. The Na(x) Channel: What It Is and What It Does. Neuroscientist 2015, 21, 399–412. [Google Scholar] [CrossRef]
- Lara, L.S.; Satou, R.; Bourgeois, C.R.T.; Gonzalez, A.A.; Zsombok, A.; Prieto, M.C.; Navar, L.G. The sodium-activated sodium channel is expressed in the rat kidney thick ascending limb and collecting duct cells and is upregulated during high salt intake. Am. J. Physiol. Ren. Physiol. 2012, 303, F105–F109. [Google Scholar] [CrossRef]
- Queiroz-Madeira, E.P.; Lara, L.S.; Wengert, M.; Landgraf, S.S.; Líbano-Soares, J.D.; Zapata-Sudo, G.; Sudo, R.T.; Takiya, C.M.; Gomes-Quintana, E.; Lopes, A.G.; et al. Na(+)-ATPase in spontaneous hypertensive rats: Possible AT(1) receptor target in the development of hypertension. Biochim. Biophys. Acta 2010, 1798, 360–366. [Google Scholar] [CrossRef]
- Taub, M.; Garimella, S.; Kim, D.; Rajkhowa, T.; Cutuli, F. Renal proximal tubule Na,K-ATPase is controlled by CREB-regulated transcriptional coactivators as well as salt-inducible kinase 1. Cell Signal. 2015, 27, 2568–2578. [Google Scholar] [CrossRef]
- Jaitovich, A.; Bertorello, A.M. Intracellular sodium sensing: SIK1 network, hormone action and high blood pressure. Biochim. Biophys. Acta 2010, 1802, 1140–1149. [Google Scholar] [CrossRef]
- Sjöström, M.; Stenström, K.; Eneling, K.; Zwiller, J.; Katz, A.I.; Takemori, H.; Bertorello, A.M. SIK1 is part of a cell sodium-sensing network that regulates active sodium transport through a calcium-dependent process. Proc. Natl. Acad. Sci. USA 2007, 104, 16922–16927. [Google Scholar] [CrossRef]
- Stenström, K.; Takemori, H.; Bianchi, G.; Katz, A.I.; Bertorello, A.M. Blocking the salt-inducible kinase 1 network prevents the increases in cell sodium transport caused by a hypertension-linked mutation in human α-adducin. J. Hypertens. 2009, 27, 2452–2457. [Google Scholar] [CrossRef] [PubMed]
- Berret, E.; Nehmé, B.; Henry, M.; Toth, K.; Drolet, G.; Mouginot, D. Regulation of Central Na+ Detection Requires the Cooperative Action of the NaX Channel and α1 Isoform of Na+/K+-ATPase in the Na+-Sensor Neuronal Population. J. Neurosci. 2013, 33, 3067–3078. [Google Scholar] [CrossRef] [PubMed]
- Pestana-Oliveira, N.; Nahey, D.B.; Johnson, T.; Collister, J.P. Development of the Deoxycorticosterone Acetate (DOCA)-salt Hypertensive Rat Model. Bio-protocol 2020, 10, e3708. [Google Scholar] [CrossRef] [PubMed]
- Lindoso, R.S.; Lopes, J.A.; Binato, R.; Abdelhay, E.; Takiya, C.M.; de Miranda, K.R.; Lara, L.S.; Viola, A.; Bussolati, B.; Vieyra, A.; et al. Adipose Mesenchymal Cells-Derived EVs Alleviate DOCA-Salt-Induced Hypertension by Promoting Cardio-Renal Protection. Mol. Ther. Methods Clin. Dev. 2019, 16, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Amara, V.R.; Surapaneni, S.K.; Tikoo, K. Dysregulation of microRNAs and renin-angiotensin system in high salt diet-induced cardiac dysfunction in uninephrectomized rats. PLoS ONE 2017, 12, e0180490. [Google Scholar] [CrossRef] [PubMed]
- Drenjančević-Perić, I.; Jelaković, B.; Lombard, J.H.; Kunert, M.P.; Kibel, A.; Gros, M. High-Salt Diet and Hypertension: Focus on the Renin-Angiotensin System. Kidney Blood Press. Res. 2011, 34, 1–11. [Google Scholar] [CrossRef]
- Drawz, P.; Rahman, M. Chronic Kidney Disease. Ann. Intern. Med. 2015, 162, ITC1–ITC16. [Google Scholar] [CrossRef]
- Popov, S.; Silveira, A.; Wågsäter, D.; Takemori, H.; Oguro, R.; Matsumoto, S.; Sugimoto, K.; Kamide, K.; Hirose, T.; Satoh, M.; et al. Salt-inducible kinase 1 influences Na(+),K(+)-ATPase activity in vascular smooth muscle cells and associates with variations in blood pressure. J. Hypertens. 2011, 29, 2395–2403. [Google Scholar] [CrossRef]
- Semplicini, A.; Ceolotto, G.; Sartori, M.; Maresca, A.M.; Baritono, E.; De Toni, R.; Paparella, I.; Calò, L. Regulation of glomerular filtration in essential hypertension: Role of abnormal Na+ transport and atrial natriuretic peptide. J. Nephrol. 2002, 15, 489–496. [Google Scholar]
- Kato, S.; Miyamoto, M.; Kato, M.; Kanamaru, T.; Takagi, M.; Nakanishi, N.; Sugino, N. Renal sodium metabolism in spontaneously hypertensive rats: Renal micropuncture study of the rate of 22Na recovery by the microinjection method. J. Hypertens. 1986, 4, S255–S256. [Google Scholar]
- Bova, S.; Goldman, W.F.; Yauan, X.J.; Blaustein, M.P. Influence of Na+ gradient on Ca2+ transients and contraction in vascular smooth muscle. Am. J. Physiol. 1990, 259, H409–H423. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, M.P. Physiological effects of endogenous ouabain: Control of intracellular Ca2+ stores and cell responsiveness. Am. J. Physiol. 1993, 264, C1367–C1387. [Google Scholar] [CrossRef] [PubMed]
- Bankir, L.; Figueres, L.; Prot-Bertoye, C.; Bouby, N.; Crambert, G.; Pratt, J.H.; Houillier, P. Medullary and cortical thick ascending limb: Similarities and differences. Am. J. Physiol. Ren. Physiol. 2020, 318, F422–F442. [Google Scholar] [CrossRef] [PubMed]
- Feraille, E.; Dizin, E. Coordinated Control of ENaC and Na+,K+-ATPase in Renal Collecting Duct. J. Am. Soc. Nephrol. 2016, 27, 2554–2563. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Armando, I.; Upadhyay, K.; Pascua, A.; Jose, P.A. The regulation of proximal tubular salt transport in hypertension: An update. Curr. Opin. Nephrol. Hypertens. 2009, 18, 412–420. [Google Scholar] [CrossRef]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Gu, Y.-Y.; Liu, X.-S.; Huang, X.-R.; Yu, X.-Q.; Lan, H.-Y. Diverse Role of TGF-β in Kidney Disease. Front. Cell Dev. Biol. 2020, 8, 123. [Google Scholar] [CrossRef]
- Kowanetz, M.; Lonn, P.; Vanlandewijck, M.; Kowanetz, K.; Heldin, C.-H.; Moustakas, A. TGFβ induces SIK to negatively regulate type I receptor kinase signaling. J. Cell Biol. 2008, 182, 655–662. [Google Scholar] [CrossRef]
- Singer, M.; Bulled, N.; Ostrach, B.; Mendenhall, E. Syndemics and the biosocial conception of health. Lancet 2017, 389, 941–950. [Google Scholar] [CrossRef]
- Rodriguez, V.J.; Chahine, A.; Parrish, M.S.; Alcaide, M.L.; Lee, T.K.; Hurwitz, B.; Sawhney, M.; Weiss, S.M.; Jones, D.L.; Kumar, M. The contribution of syndemic conditions to cardiovascular disease risk. AIDS Care 2021, 33, 585–593. [Google Scholar] [CrossRef]
- Basting, T.; Lazartigues, E. DOCA-Salt Hypertension: An Update. Curr. Hypertens. Rep. 2017, 19, 32. [Google Scholar] [CrossRef] [PubMed]
- NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in hypertension prevalence and progress in treatment and control from 1990 to 2019: A pooled analysis of 1201 population-representative studies with 104 million participants. Lancet 2021, 398, 957–980, Correction in Lancet 2022, 399, 520. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Mendes, L.V.P.; Gonsalez, S.R.; Oliveira-Pinto, L.M.; Pereira-Acácio, A.; Takiya, C.M.; Nascimento, J.H.M.; Einicker-Lamas, M.; Vieyra, A.; Cunha, V.M.N.; Lara, L.S. Long-term effect of a chronic low-protein multideficient diet on the heart: Hypertension and heart failure in chronically malnourished young adult rats. Int. J. Cardiol. 2017, 238, 43–56. [Google Scholar] [CrossRef]
- Cortes, A.L.; Gonsalez, S.R.; Rioja, L.S.; Oliveira, S.S.C.; Santos, A.L.S.; Prieto, M.C.; Melo, P.A.; Lara, L.S. Protective outcomes of low-dose doxycycline on renal function of Wistar rats subjected to acute ischemia/reperfusion injury. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Beiral, H.J.V.; Rodrigues-Ferreira, C.; Fernandes, A.M.; Gonsalez, S.R.; Mortari, N.C.; Takiya, C.M.; Sorenson, M.M.; Figueiredo-Freitas, C.; Galina, A.; Vieyra, A. The Impact of Stem Cells on Electron Fluxes, Proton Translocation, and ATP Synthesis in Kidney Mitochondria after Ischemia/Reperfusion. Cell Transplant. 2014, 23, 207–220. [Google Scholar] [CrossRef]
- Gonzalez, A.A.; Lara, L.S.; Luffman, C.; Seth, D.M.; Prieto, M.C. Soluble Form of the (Pro)Renin Receptor Is Augmented in the Collecting Duct and Urine of Chronic Angiotensin II–Dependent Hypertensive Rats. Hypertension 2011, 57, 859–864. [Google Scholar] [CrossRef]
- Muzi-Filho, H.; Bezerra, C.G.P.; Souza, A.M.; Boldrini, L.C.; Takiya, C.M.; Oliveira, F.L.; Nesi, R.T.; Valença, S.S.; Einicker-Lamas, M.; Vieyra, A.; et al. Undernutrition Affects Cell Survival, Oxidative Stress, Ca2+ Handling and Signaling Pathways in Vas Deferens, Crippling Reproductive Capacity. PLoS ONE 2013, 8, e69682. [Google Scholar] [CrossRef]
- Ingelfinger, J.R.; Jung, F.; Diamant, D.; Haveran, L.; Lee, E.; Brem, A.; Tang, S.S. Rat proximal tubule cell line transformed with origin-defective SV40 DNA: Autocrine ANG II feedback. Am. J. Physiol. 1999, 276, F218–F227. [Google Scholar] [CrossRef]
- Garagliano, J.M.; Katsurada, A.; Miyata, K.; Derbenev, A.V.; Zsombok, A.; Navar, L.G.; Satou, R. Advanced Glycation End Products Stimulate Angiotensinogen Production in Renal Proximal Tubular Cells. Am. J. Med. Sci. 2019, 357, 57–66. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | CTRL (n = 5) | CTRL-Salt (n = 5) | DOCA (n = 5) | DOCA-Salt (n = 5) |
|---|---|---|---|---|
| Body mass (g) | 305 ± 14 | 315 ± 0.5 (p = 0.9873) | 326 ± 21 (p = 0.7630) | 346 ± 4 (p = 0.1286; p = 0.7972; p = 0.3822) |
| Kidney mass (g) | 1.5 ± 0.1 | 1.9 ± 0.1 (p = 0.0320) | 2 ± 0.1 b (p = 0.0041) | 2.4 ± 0.1 c,e (p < 0.0001; p = 0.0320; p = 0.0041) |
| Kidney index (mg/g) | 4.8 ± 0.1 | 5.8 ± 0.2 (p = 0.0893) | 6.2 ± 0.2 (p = 0.0063) | 7.0 ± 0.5 c,e (p < 0.0001; p = 0.2558; p = 0.0256) |
| SBP (mmHg) | 104 ± 4 | 112 ± 1.2 (p = 0.8959) | 145 ± 3 b (p < 0.0001) | 160 ± 4 c,d,e (p < 0.0001; p = 0.0136; p < 0.0001) |
| Heart rate (bpm) | 280 ± 7 | 337 ± 10 a (p = 0.0008) | 452 ± 4 b (p < 0.0001) | 463 ± 5 c,e (p < 0.0001; p = 0.2385; p < 0.0001) |
| Water intake (mL/24 h) | 26 ± 2 | 49 ± 5 a (p = 0.0173) | 24 ± 2 (p = 0.9993) | 55 ± 6 c,d (p = 0.0007; p < 0.0001; p = 0.8677) |
| Urine volume (mL/24 h) | 9 ± 3 | 29 ± 3 a (p = 0.0022) | 7 ± 1 (p = 0.9964) | 36 ± 5 c,d (p < 0.0001; p < 0.0001; p = 0.4246) |
| Parameters | CTRL (n = 5) | CTRL-Salt (n = 5) | DOCA (n = 5) | DOCA-Salt (n = 5) |
|---|---|---|---|---|
| BUN (mg/dL) | 33 ± 1.2 | 43 ± 2.4 a (p = 0.0068) | 31 ± 1.6 (p = 0.8622) | 39 ± 1.5 c,d (p = 0.0868; p = 0.0114; p = 0.6234) |
| Proteinuria (mg/24 h) | 6.6 ± 1.3 | 21.5 ± 3.9 a (p = 0.0007) | 8.2 ± 0.7 (p = 0.9866) | 16 ± 1.7 c,d (p = 0.0243; p = 0.0497; p = 0.3452) |
| PCre (mg/dL) | 0.46 ± 0.03 | 0.49 ± 0.04 (p = 0.9971) | 0.56 ± 0.07 (p = 0.7118) | 0.75 ± 0.05 c,d,e (p = 0.0043; p = 0.0439; p = 0.0136) |
| UCre (mg/dL) | 178 ± 34 | 33 ± 4.6 a (p < 0.0001) | 128 ± 12 (p = 0.2449) | 35 ± 3.2 c,d (p < 0.0001; p = 0.0037; p > 0.9999) |
| UCre/PCre ratio | 402 ± 93 | 70 ± 14 a (p = 0.0007) | 207 ± 30 b (p = 0.0410) | 46 ± 4 c,d (p = 0.0002; p = 0.0116; p = 0.9981) |
| PNa (mEq/L) | 140 ± 4 | 155 ± 6 (p = 0.9407) | 153 ± 12 (p = 0.9415) | 131 ± 9 (p = 0.9787; p = 0.3905; p = 0.5485) |
| UNa (mEq/L) | 172 ± 0.9 | 476 ± 12 a (p < 0.0001) | 161 ± 17 (p = 0.9983) | 399 ± 26 c,d (p < 0.0001; p < 0.0001; p = 0.1852) |
| FLNa (mEq/min) | 0.17 ± 0.005 | 0.20 ± 0.008 (p = 0.4572) | 0.18 ± 0.014 (p = 0.9139) | 0.11 ± 0.008 c,d,e (p < 0.0243; p = 0.0003; p = 0.0004) |
| FENa (%) | 0.63 ± 0.14 | 4.8 ± 0.3 a (p = 0.0357) | 0.47 ± 0.08 (p > 0.9999) | 8.7 ± 1.07 c,d,e (p < 0.0001; p < 0.0001 p = 0.0351) |
| SIK Isoform | Glomerulus | Proximal Tubule | Thick Ascending Limb | Collecting Duct |
|---|---|---|---|---|
| SIK1 | Mesangial cells | − | + | + |
| SIK2 | + | + | + | + |
| SIK3 | Mesangial cells | Brush border | − | Principal cells |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonsalez, S.R.; Gomes, D.S.; de Souza, A.M.; Ferrão, F.M.; Vallotton, Z.; Gogulamudi, V.R.; Lowe, J.; Casarini, D.E.; Prieto, M.C.; Lara, L.S. The Triad Na+ Activated Na+ Channel (Nax)—Salt Inducible KINASE (SIK) and (Na+ + K+)-ATPase: Targeting the Villains to Treat Salt Resistant and Sensitive Hypertension. Int. J. Mol. Sci. 2023, 24, 7887. https://doi.org/10.3390/ijms24097887
Gonsalez SR, Gomes DS, de Souza AM, Ferrão FM, Vallotton Z, Gogulamudi VR, Lowe J, Casarini DE, Prieto MC, Lara LS. The Triad Na+ Activated Na+ Channel (Nax)—Salt Inducible KINASE (SIK) and (Na+ + K+)-ATPase: Targeting the Villains to Treat Salt Resistant and Sensitive Hypertension. International Journal of Molecular Sciences. 2023; 24(9):7887. https://doi.org/10.3390/ijms24097887
Chicago/Turabian StyleGonsalez, Sabrina R., Dayene S. Gomes, Alessandro M. de Souza, Fernanda M. Ferrão, Zoe Vallotton, Venkateswara R. Gogulamudi, Jennifer Lowe, Dulce E. Casarini, Minolfa C. Prieto, and Lucienne S. Lara. 2023. "The Triad Na+ Activated Na+ Channel (Nax)—Salt Inducible KINASE (SIK) and (Na+ + K+)-ATPase: Targeting the Villains to Treat Salt Resistant and Sensitive Hypertension" International Journal of Molecular Sciences 24, no. 9: 7887. https://doi.org/10.3390/ijms24097887