
Downregulation of Sirtuin 1 Does Not Account for the Impaired Long-Term Potentiation in the Prefrontal Cortex of Female APPswe/PS1dE9 Mice Modelling Alzheimer’s Disease
 , , , , , , ,
, , , , , , , 
Abstract
:1. Introduction
2. Results
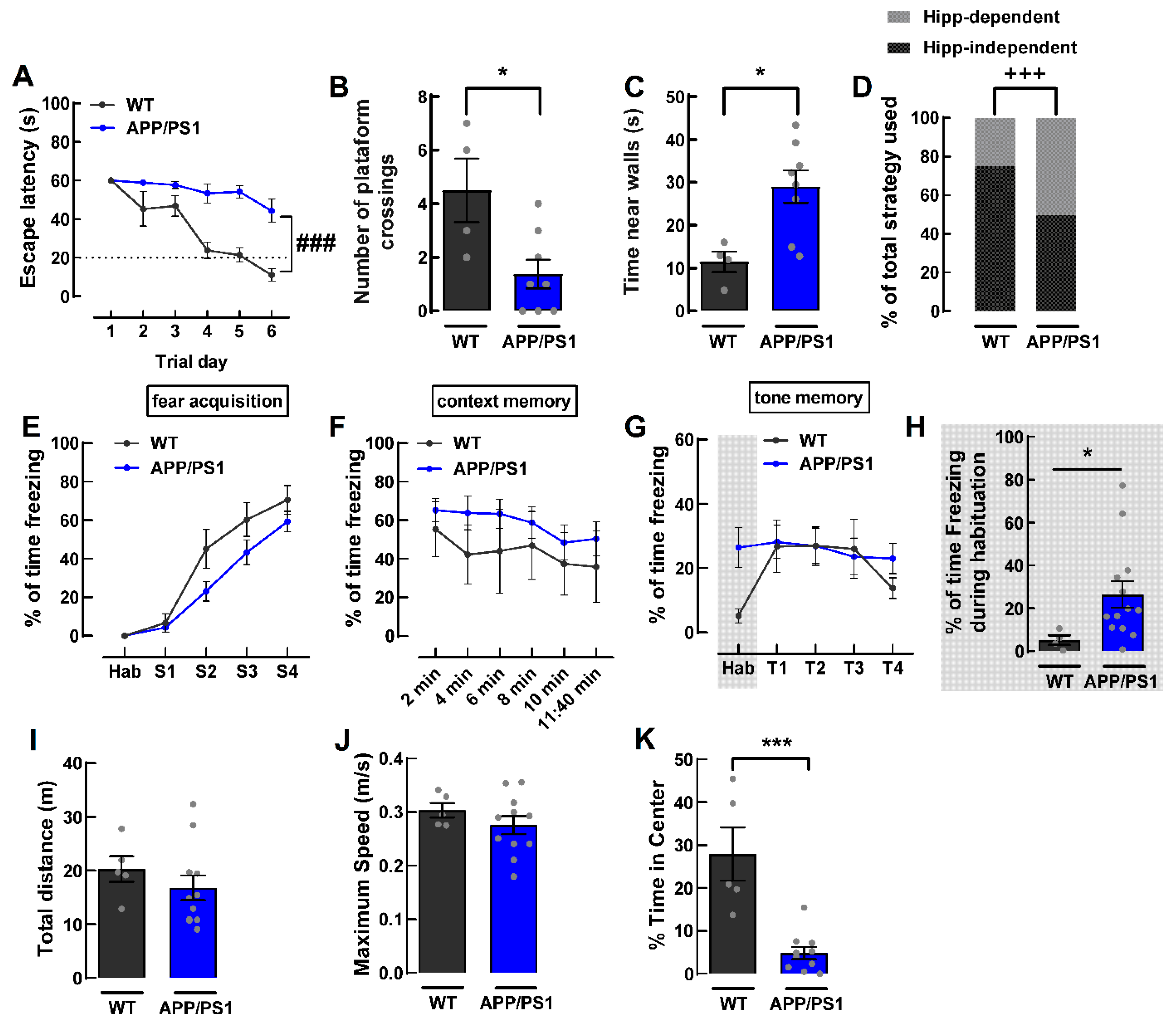
2.1. Female APP/PS1 Mice 9-Month-Old Display Mood and Memory Deficits
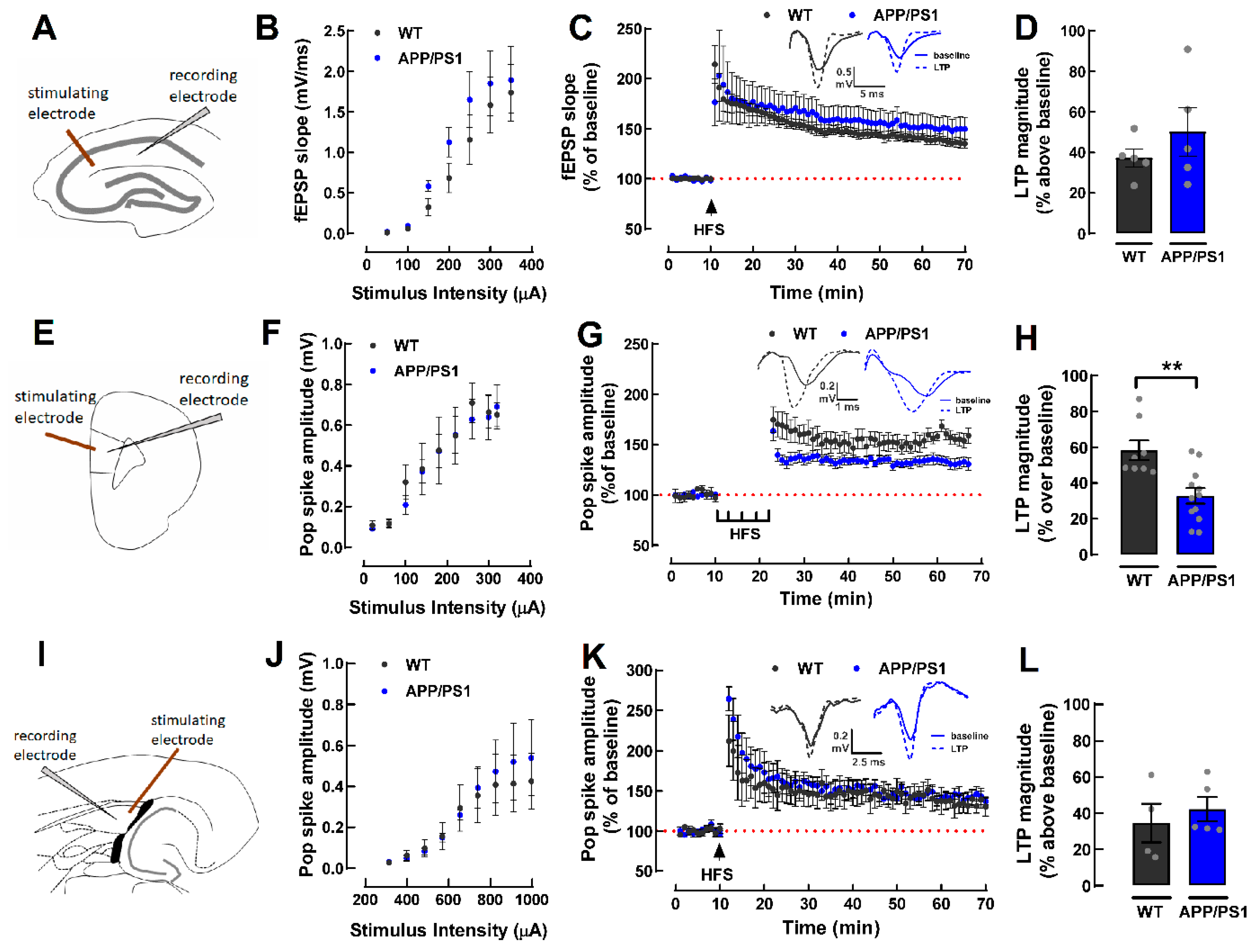
2.2. Lower Magnitude of LTP in the PFC of 9-Month-Old Female APP/PS1 Mice
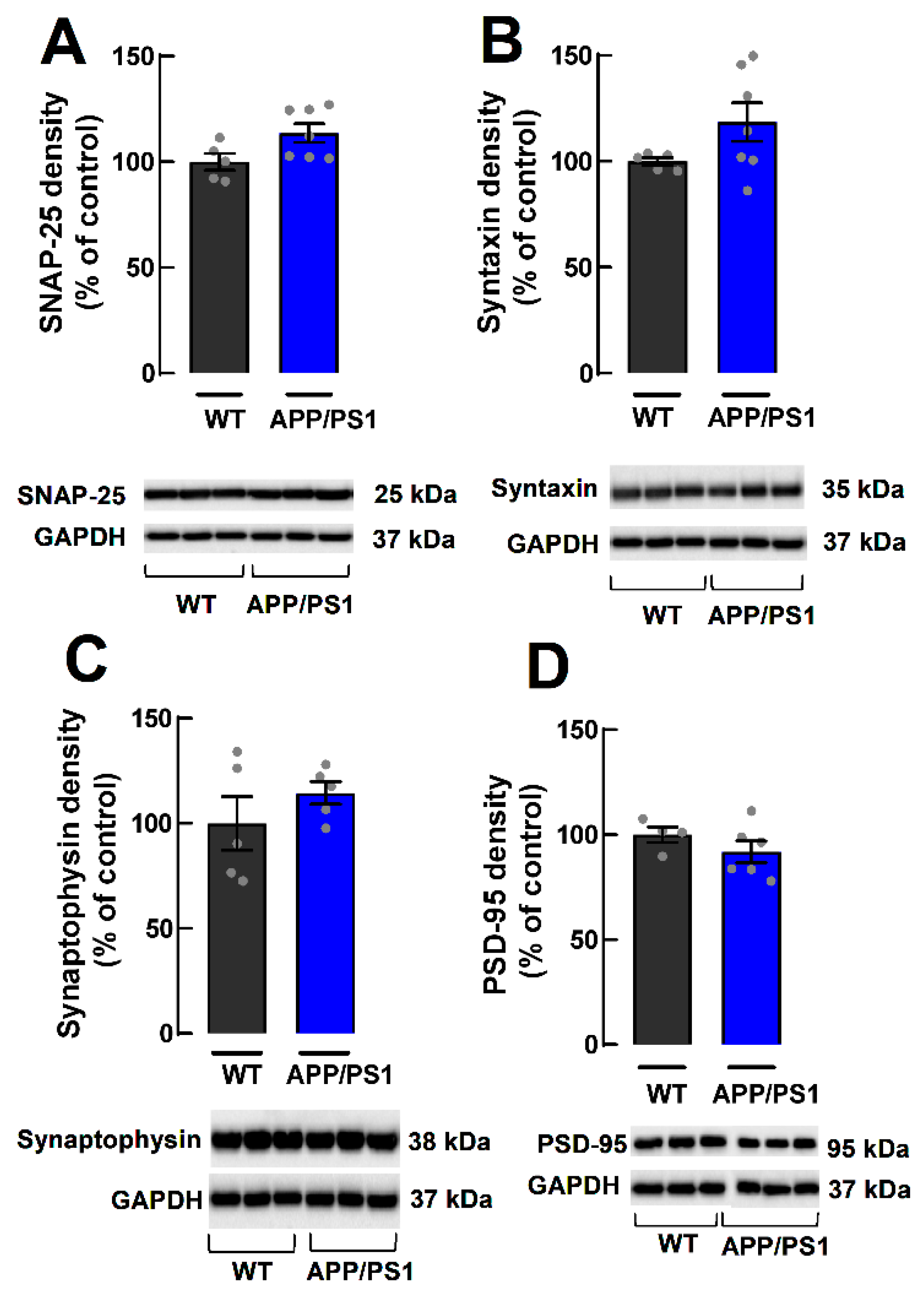
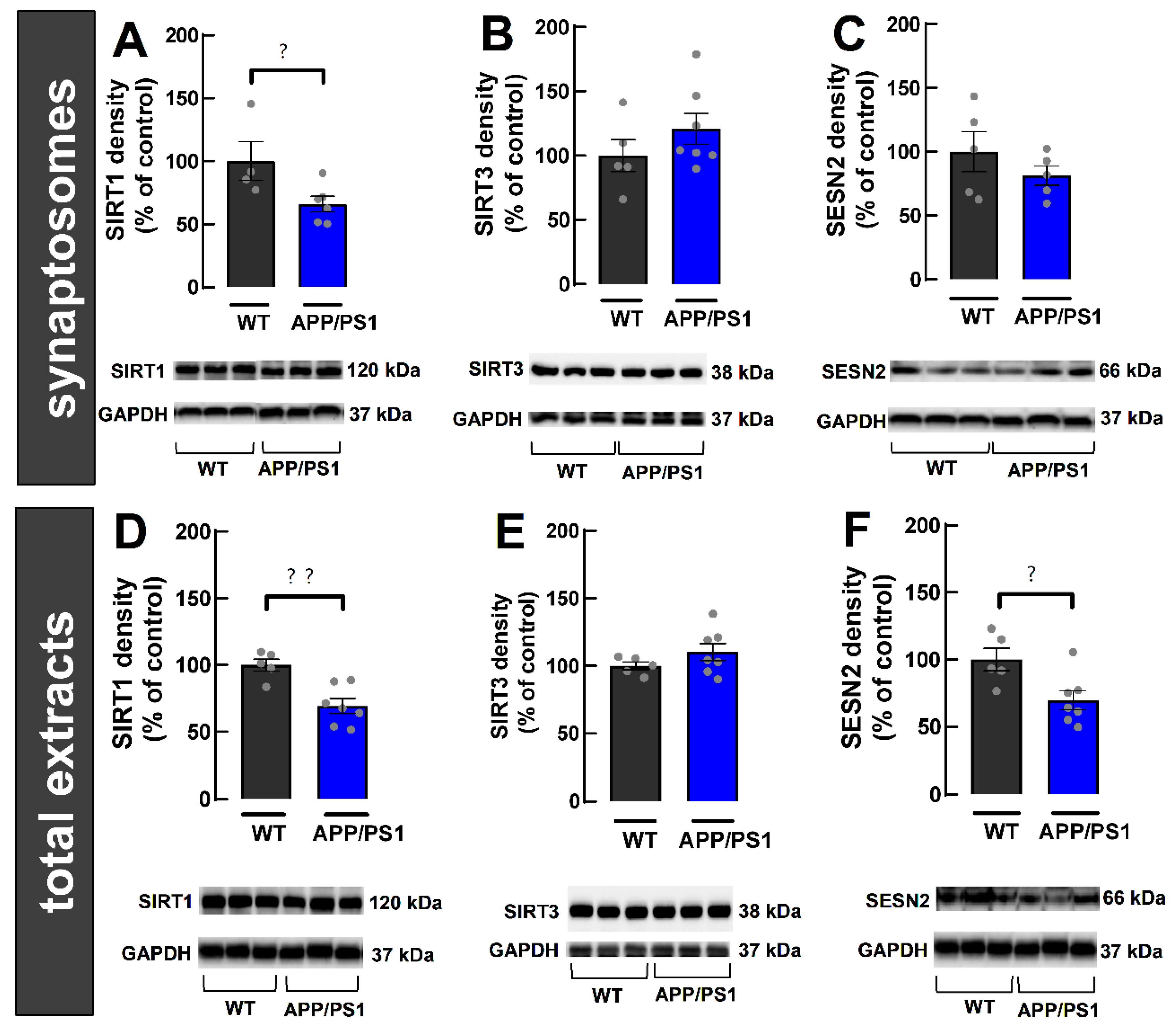
2.3. Lack of Alterations of Synaptic Markers in Cortical Synaptosomes of APP/PS1 Mice
2.4. Decreased SIRT1 in Cerebrocortical Synapses from 9-Month-Old Female APP/PS1 Mice
2.5. Decreased Density of SIRT1 and SESN2 in Total Extracts from the Cerebral Cortex of 9-Month-Old Female APP/PS1 Mice
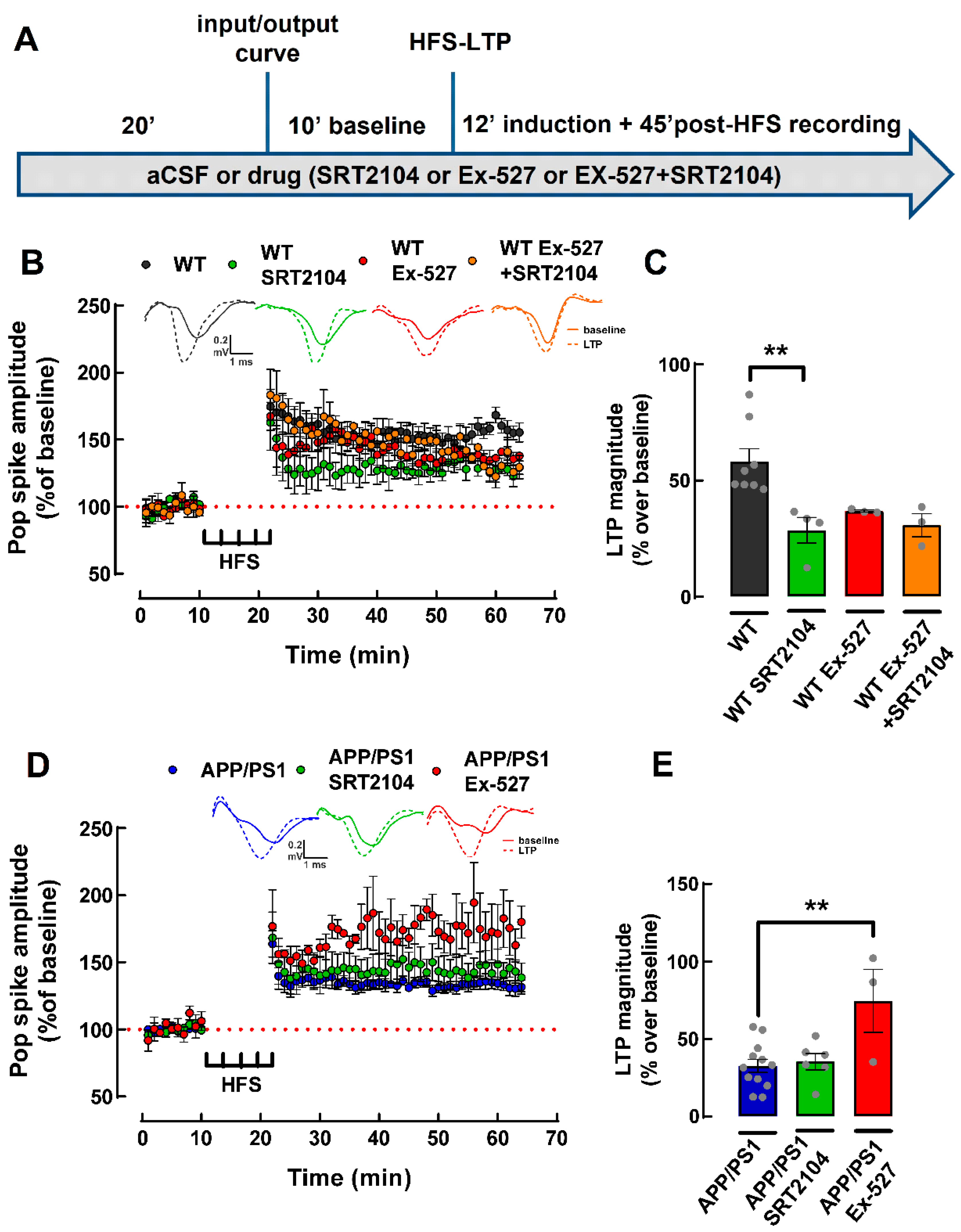
2.6. Pharmacological Activation of SIRT1 Does Not Recover LTP Deficit in PFC Slices from APP/PS1 Mice
3. Discussion
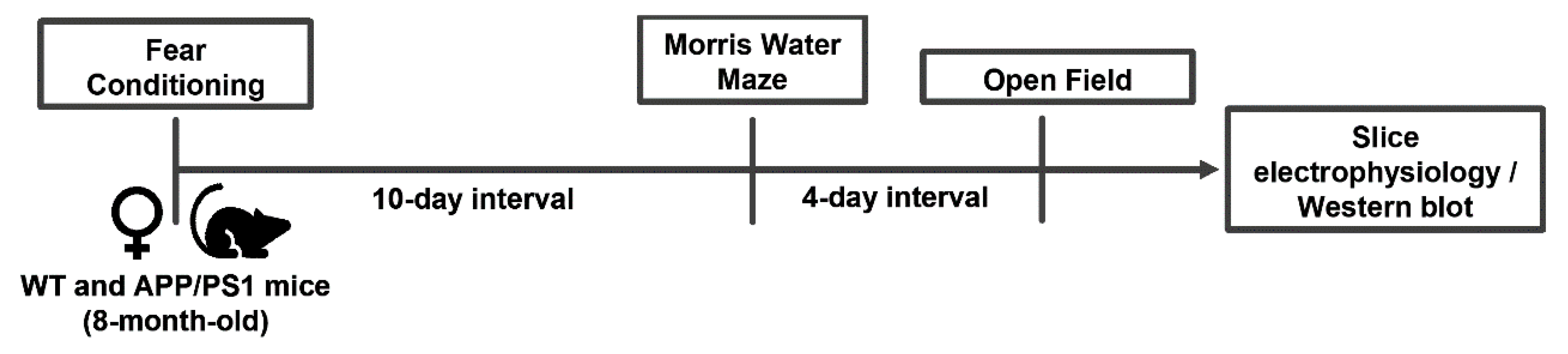
4. Materials and Methods
4.1. Animals
4.2. Drugs
4.3. Behavioral Analysis
4.4. Slice Electrophysiology
4.5. Preparation of Synaptosomes and Total Protein Extracts from the Cerebral Cortex
4.6. Western Blotting
4.7. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Oveisgharan, S.; Arvanitakis, Z.; Yu, L.; Farfel, J.; Schneider, J.A.; Bennett, D.A. Sex differences in Alzheimer’s disease and common neuropathologies of aging. Acta Neuropathol. 2018, 136, 887–900. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harbor Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [Green Version]
- García-Morales, V.; González-Acedo, A.; Melguizo-Rodríguez, L.; Pardo-Moreno, T.; Costela-Ruiz, V.J.; Montiel-Troya, M.; Ramos-Rodríguez, J.J. Current understanding of the physiopathology, diagnosis and therapeutic approach to Alzheimer’s disease. Biomedicines 2021, 9, 1910. [Google Scholar] [CrossRef]
- Kirvell, S.L.; Esiri, M.; Francis, P.T. Down-regulation of vesicular glutamate transporters precedes cell loss and pathology in Alzheimer’s disease. J. Neurochem. 2006, 98, 939–950. [Google Scholar] [CrossRef]
- Canas, P.M.; Simões, A.P.; Rodrigues, R.J.; Cunha, R.A. Predominant loss of glutamatergic terminal markers in a β-amyloid peptide model of Alzheimer’s disease. Neuropharmacology 2014, 76, 51–56. [Google Scholar] [CrossRef]
- John, A.; Reddy, P.H. Synaptic basis of Alzheimer’s disease: Focus on synaptic amyloid beta, P-tau and mitochondria. Ageing Res. Rev. 2021, 65, 101208. [Google Scholar] [CrossRef]
- Smailagic, N.; Lafortune, L.; Kelly, S.; Hyde, C.; Brayne, C. 18F-FDG PET for prediction of conversion to Alzheimer’s disease dementia in people with mild cognitive impairment: An updated systematic review of test accuracy. J. Alzheimer Dis. 2018, 64, 1175–1194. [Google Scholar] [CrossRef] [Green Version]
- Nogueiras, R.; Habegger, K.M.; Chaudhary, N.; Finan, B.; Banks, A.S.; Dietrich, M.O.; Horvath, T.L.; Sinclair, D.A.; Pfluger, P.T.; Tschöp, M.H. Sirtuin 1 and sirtuin 3: Physiological modulators of metabolism. Physiol. Rev. 2012, 92, 1479–1514. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Wang, W.Y.; Mao, Y.W.; Gräff, J.; Guan, J.S.; Pan, L.; Mak, G.; Kim, D.; Su, S.C.; Tsai, L.H. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 2010, 466, 1105–1109. [Google Scholar] [CrossRef] [Green Version]
- Michán, S.; Li, Y.; Chou, M.M.; Parrella, E.; Ge, H.; Long, J.M.; Allard, J.S.; Lewis, K.; Miller, M.; Xu, W.; et al. SIRT1 is essential for normal cognitive function and synaptic plasticity. J. Neurosci. 2010, 30, 9695–9707. [Google Scholar] [CrossRef] [Green Version]
- Demyanenko, S.; Gantsgorn, E.; Rodkin, S.; Sharifulina, S. Localization and expression of sirtuins 1, 2, 6 and plasticity-related proteins in the recovery period after a photothrombotic stroke in mice. J. Stroke Cerebrovasc. Dis. 2020, 29, 105152. [Google Scholar] [CrossRef]
- Hacioglu, C.; Kar, F.; Kanbak, G. Ex vivo investigation of bexarotene and nicotinamide function as a protectıve agent on rat synaptosomes treated with Aβ1-42. Neurochem. Res. 2021, 46, 804–818. [Google Scholar] [CrossRef]
- Zhang, Y.; Anoopkumar-Dukie, S.; Arora, D.; Davey, A.K. Review of the anti-inflammatory effect of SIRT1 and SIRT2 modulators on neurodegenerative diseases. Eur. J. Pharmacol. 2020, 867, 172847. [Google Scholar] [CrossRef]
- Fagerli, E.; Escobar, I.; Ferrier, F.J.; Jackson, C.W.; Perez-Lao, E.J.; Perez-Pinzon, M.A. Sirtuins and cognition: Implications for learning and memory in neurological disorders. Front. Physiol. 2022, 13, 908689. [Google Scholar] [CrossRef]
- Hohman, T.J.; Bush, W.S.; Jiang, L.; Brown-Gentry, K.D.; Torstenson, E.S.; Dudek, S.M.; Mukherjee, S.; Naj, A.; Kunkle, B.W.; Ritchie, M.D.; et al. Discovery of gene-gene interactions across multiple independent data sets of late onset Alzheimer disease from the Alzheimer Disease Genetics Consortium. Neurobiol. Aging 2016, 38, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Camporez, D.; Belcavello, L.; Almeida, J.F.F.; Silva-Sena, G.G.; Pimassoni, L.H.S.; Morelato, R.L.; do Carmo Pimentel Batitucci, M.; de Paula, F. Positive association of a Sirt1 variant and parameters of oxidative stress on Alzheimer’s disease. Neurol. Sci. 2021, 42, 1843–1851. [Google Scholar] [CrossRef]
- Lutz, M.I.; Milenkovic, I.; Regelsberger, G.; Kovacs, G.G. Distinct patterns of sirtuin expression during progression of Alzheimer’s disease. Neuromol. Med. 2014, 16, 405–414. [Google Scholar] [CrossRef]
- Song, S.; Li, B.; Jia, Z.; Guo, L. Sirtuin 3 mRNA expression is downregulated in the brain tissues of Alzheimer’s disease patients: A bioinformatic and data mining approach. Med. Sci. Monit. 2020, 26, e923547. [Google Scholar] [CrossRef]
- Yang, W.; Zou, Y.; Zhang, M.; Zhao, N.; Tian, Q.; Gu, M.; Liu, W.; Shi, R.; Lü, Y.; Yu, W. Mitochondrial Sirt3 expression is decreased in APP/PS1 double transgenic mouse model of Alzheimer’s disease. Neurochem. Res. 2015, 40, 1576–1582. [Google Scholar] [CrossRef]
- Wang, L.; Shi, F.X.; Li, N.; Cao, Y.; Lei, Y.; Wang, J.Z.; Tian, Q.; Zhou, X.W. AMPK ameliorates tau acetylation and memory impairment through Sirt1. Mol. Neurobiol. 2020, 57, 5011–5025. [Google Scholar] [CrossRef]
- Kim, D.; Nguyen, M.D.; Dobbin, M.M.; Fischer, A.; Sananbenesi, F.; Rodgers, J.T.; Delalle, I.; Baur, J.A.; Sui, G.; Armour, S.M.; et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007, 26, 3169–3179. [Google Scholar] [CrossRef] [Green Version]
- Corpas, R.; Revilla, S.; Ursulet, S.; Castro-Freire, M.; Kaliman, P.; Petegnief, V.; Giménez-Llort, L.; Sarkis, C.; Pallàs, M.; Sanfeliu, C. SIRT1 overexpression in mouse hippocampus induces cognitive enhancement through proteostatic and neurotrophic mechanisms. Mol. Neurobiol. 2017, 54, 5604–5619. [Google Scholar] [CrossRef]
- Liu, Y.; Cheng, A.; Li, Y.J.; Yang, Y.; Kishimoto, Y.; Zhang, S.; Wang, Y.; Wan, R.; Raefsky, S.M.; Lu, D.; et al. SIRT3 mediates hippocampal synaptic adaptations to intermittent fasting and ameliorates deficits in APP mutant mice. Nat. Comm. 2019, 10, 1886. [Google Scholar] [CrossRef] [Green Version]
- Julien, C.; Tremblay, C.; Emond, V.; Lebbadi, M.; Salem, N., Jr.; Bennett, D.A.; Calon, F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Torres-Lista, V.; Parrado-Fernández, C.; Alvarez-Montón, I.; Frontiñán-Rubio, J.; Durán-Prado, M.; Peinado, J.R.; Johansson, B.; Alcaín, F.J.; Giménez-Llort, L. Neophobia, NQO1 and SIRT1 as premorbid and prodromal indicators of AD in 3xTg-AD mice. Behav. Brain Res. 2014, 271, 140–146. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Ratovitski, T.; van Lare, J.; Lee, M.K.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Price, D.L.; Sisodia, S.S. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 1997, 19, 939–945. [Google Scholar] [CrossRef] [Green Version]
- Trinchese, F.; Liu, S.; Battaglia, F.; Walter, S.; Mathews, P.M.; Arancio, O. Progressive age-related development of Alzheimer-like pathology in APP/PS1 mice. Ann. Neurol. 2004, 55, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Kilgore, M.; Miller, C.A.; Fass, D.M.; Hennig, K.M.; Haggarty, S.J.; Sweatt, J.D.; Rumbaugh, G. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 2010, 35, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Viana da Silva, S.; Haberl, M.G.; Zhang, P.; Bethge, P.; Lemos, C.; Gonçalves, N.; Gorlewicz, A.; Malezieux, M.; Gonçalves, F.Q.; Grosjean, N.; et al. Early synaptic deficits in the APP/PS1 mouse model of Alzheimer’s disease involve neuronal adenosine A2A receptors. Nat. Comm. 2016, 7, 11915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treit, D.; Fundytus, M. Thigmotaxis as a test for anxiolytic activity in rats. Pharmacol. Biochem. Behav. 1988, 31, 959–962. [Google Scholar] [CrossRef] [PubMed]
- Garthe, A.; Kempermann, G. An old test for new neurons: Refining the Morris water maze to study the functional relevance of adult hippocampal neurogenesis. Front. Neurosci. 2013, 7, 63. [Google Scholar] [CrossRef] [Green Version]
- LeDoux, J.E. Emotion circuits in the brain. Ann. Rev. Neurosci. 2000, 23, 155–184. [Google Scholar] [CrossRef]
- Rusakov, D.A.; Harrison, E.; Stewart, M.G. Synapses in hippocampus occupy only 1–2% of cell membranes and are spaced less than half-micron apart: A quantitative ultrastructural analysis with discussion of physiological implications. Neuropharmacology 1998, 37, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Bonkowski, M.S.; Sinclair, D.A. Slowing ageing by design: The rise of NAD+ and sirtuin-activating compounds. Nature reviews. Mol. Cell. Biol. 2016, 17, 679–690. [Google Scholar] [CrossRef]
- Gertz, M.; Fischer, F.; Nguyen, G.T.; Lakshminarasimhan, M.; Schutkowski, M.; Weyand, M.; Steegborn, C. Ex-527 inhibits Sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 2772–2781. [Google Scholar] [CrossRef] [Green Version]
- Janus, C.; Flores, A.Y.; Xu, G.; Borchelt, D.R. Behavioral abnormalities in APPSwe/PS1dE9 mouse model of AD-like pathology: Comparative analysis across multiple behavioral domains. Neurobiol. Aging 2015, 36, 2519–2532. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Chang, C.P.; Lin, C.J.; Lai, H.L.; Kao, Y.H.; Cheng, S.J.; Chen, H.M.; Liao, Y.P.; Faivre, E.; Buée, L.; et al. Adenosine augmentation evoked by an ENT1 inhibitor improves memory impairment and neuronal plasticity in the APP/PS1 mouse model of Alzheimer’s disease. Mol. Neurobiol. 2018, 55, 8936–8952. [Google Scholar] [CrossRef]
- Gong, Z.; Huang, J.; Xu, B.; Ou, Z.; Zhang, L.; Lin, X.; Ye, X.; Kong, X.; Long, D.; Sun, X.; et al. Urolithin A attenuates memory impairment and neuroinflammation in APP/PS1 mice. J. Neuroinflamm. 2019, 16, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhao, J.; Guo, F.L.; Gao, X.; Xie, X.; Liu, S.; Yang, X.; Yang, X.; Zhang, L.; Ye, Y.; et al. Metformin ameliorates synaptic defects in a mouse model of AD by inhibiting Cdk5 activity. Front. Cell. Neurosci. 2020, 14, 170. [Google Scholar] [CrossRef] [PubMed]
- Knafo, S.; Venero, C.; Merino-Serrais, P.; Fernaud-Espinosa, I.; Gonzalez-Soriano, J.; Ferrer, I.; Santpere, G.; DeFelipe, J. Morphological alterations to neurons of the amygdala and impaired fear conditioning in a transgenic mouse model of Alzheimer’s disease. J. Pathol. 2009, 219, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Kommaddi, R.P.; Das, D.; Karunakaran, S.; Nanguneri, S.; Bapat, D.; Ray, A.; Shaw, E.; Bennett, D.A.; Nair, D.; Ravindranath, V. Aβ mediates F-actin disassembly in dendritic spines leading to cognitive deficits in Alzheimer’s disease. J. Neurosci. 2018, 38, 1085–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonardi, C.; de Pulford, F.; Jennings, D.; Pardon, M.C. A detailed analysis of the early context extinction deficits seen in APPswe/PS1dE9 female mice and their relevance to preclinical Alzheimer’s disease. Behav. Brain Res. 2011, 222, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Hulshof, L.A.; Frajmund, L.A.; van Nuijs, D.; van der Heijden, D.C.N.; Middeldorp, J.; Hol, E.M. Both male and female APPswe/PSEN1dE9 mice are impaired in spatial memory and cognitive flexibility at 9 months of age. Neurobiol. Aging 2022, 113, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Low, J.K.; Logge, W.; Garner, B.; Karl, T. Novel behavioural characteristics of female APPSwe/PS1ΔE9 double transgenic mice. Behav. Brain Res. 2014, 260, 111–118. [Google Scholar] [CrossRef]
- Dunsmoor, J.E.; Paz, R. Fear generalization and anxiety: Behavioral and neural mechanisms. Biol. Psychiatry 2015, 78, 336–343. [Google Scholar] [CrossRef]
- Zhao, Q.F.; Tan, L.; Wang, H.F.; Jiang, T.; Tan, M.S.; Tan, L.; Xu, W.; Li, J.Q.; Wang, J.; Lai, T.J.; et al. The prevalence of neuropsychiatric symptoms in Alzheimer’s disease: Systematic review and meta-analysis. J. Affect. Dis. 2016, 190, 264–271. [Google Scholar] [CrossRef]
- Hoefer, M.; Allison, S.C.; Schauer, G.F.; Neuhaus, J.M.; Hall, J.; Dang, J.N.; Weiner, M.W.; Miller, B.L.; Rosen, H.J. Fear conditioning in frontotemporal lobar degeneration and Alzheimer’s disease. Brain 2008, 131, 1646–1657. [Google Scholar] [CrossRef]
- Nasrouei, S.; Rattel, J.A.; Liedlgruber, M.; Marksteiner, J.; Wilhelm, F.H. Fear acquisition and extinction deficits in amnestic mild cognitive impairment and early Alzheimer’s disease. Neurobiol. Aging 2020, 87, 26–34. [Google Scholar] [CrossRef]
- Bannerman, D.M.; Sprengel, R.; Sanderson, D.J.; McHugh, S.B.; Rawlins, J.N.; Monyer, H.; Seeburg, P.H. Hippocampal synaptic plasticity, spatial memory and anxiety. Nat. Neurosci. Rev. 2014, 15, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Volianskis, A.; Køstner, R.; Mølgaard, M.; Hass, S.; Jensen, M.S. Episodic memory deficits are not related to altered glutamatergic synaptic transmission and plasticity in the CA1 hippocampus of the APPswe/PS1δE9-deleted transgenic mice model of ß-amyloidosis. Neurobiol. Aging 2010, 31, 1173–1187. [Google Scholar] [CrossRef]
- Xu, L.; Zhou, Y.; Hu, L.; Jiang, H.; Dong, Y.; Shen, H.; Lou, Z.; Yang, S.; Ji, Y.; Ruan, L.; et al. Deficits in N-methyl-D-aspartate receptor function and synaptic plasticity in hippocampal CA1 in APP/PS1 mouse model of Alzheimer’s disease. Front. Aging Neurosci. 2021, 13, 772980. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.T.; Zhang, S.J.; Witter, M.P.; Moser, E.I.; Moser, M.B. A prefrontal-thalamo-hippocampal circuit for goal-directed spatial navigation. Nature 2015, 522, 50–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negrón-Oyarzo, I.; Espinosa, N.; Aguilar-Rivera, M.; Fuenzalida, M.; Aboitiz, F.; Fuentealba, P. Coordinated prefrontal-hippocampal activity and navigation strategy-related prefrontal firing during spatial memory formation. Proc. Natl. Acad. Sci. USA 2018, 115, 7123–7128. [Google Scholar] [CrossRef] [Green Version]
- Maillet, D.; Rajah, M.N. Association between prefrontal activity and volume change in prefrontal and medial temporal lobes in aging and dementia: A review. Ageing Res. Rev. 2013, 12, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, F.; Motta, C.; Casula, E.P.; Bonnì, S.; Assogna, M.; Caltagirone, C.; Martorana, A.; Koch, G. LTP-like cortical plasticity predicts conversion to dementia in patients with memory impairment. Brain Stimul. 2020, 13, 1175–1182. [Google Scholar] [CrossRef]
- Battaglia, F.; Wang, H.Y.; Ghilardi, M.F.; Gashi, E.; Quartarone, A.; Friedman, E.; Nixon, R.A. Cortical plasticity in Alzheimer’s disease in humans and rodents. Biol. Psychiatry 2007, 62, 1405–1412. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, A.; Wang, Z.J.; Cao, Q.; Wang, W.; Lin, L.; Ma, K.; Zhang, F.; Wei, J.; Matas, E.; et al. Inhibition of EHMT1/2 rescues synaptic and cognitive functions for Alzheimer’s disease. Brain 2019, 142, 787–807. [Google Scholar] [CrossRef]
- Mu, L.; Cai, J.; Gu, B.; Yu, L.; Li, C.; Liu, Q.S.; Zhao, L. Treadmill exercise prevents decline in spatial learning and memory in 3×Tg-AD mice through enhancement of structural synaptic plasticity of the hippocampus and prefrontal cortex. Cells 2022, 11, 244. [Google Scholar] [CrossRef]
- Taxier, L.R.; Philippi, S.M.; Fleischer, A.W.; York, J.M.; LaDu, M.J.; Frick, K.M. APOE4 homozygote females are resistant to the beneficial effects of 17β-estradiol on memory and CA1 dendritic spine density in the EFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2022, 118, 13–24. [Google Scholar] [CrossRef]
- Canas, P.M.; Duarte, J.M.; Rodrigues, R.J.; Köfalvi, A.; Cunha, R.A. Modification upon aging of the density of presynaptic modulation systems in the hippocampus. Neurobiol. Aging 2009, 30, 1877–1884. [Google Scholar] [CrossRef] [Green Version]
- Kaster, M.P.; Machado, N.J.; Silva, H.B.; Nunes, A.; Ardais, A.P.; Santana, M.; Baqi, Y.; Müller, C.E.; Rodrigues, A.L.; Porciúncula, L.O.; et al. Caffeine acts through neuronal adenosine A2A receptors to prevent mood and memory dysfunction triggered by chronic stress. Proc. Natl. Acad. Sci. USA 2015, 112, 7833–7838. [Google Scholar] [CrossRef] [Green Version]
- Moreira-de-Sá, A.; Gonçalves, F.Q.; Lopes, J.P.; Silva, H.B.; Tomé, Â.R.; Cunha, R.A.; Canas, P.M. Adenosine A2A receptors format long-term depression and memory strategies in a mouse model of Angelman syndrome. Neurobiol. Dis. 2020, 146, 105137. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Kashani, A.; Lepicard, E.; Poirel, O.; Videau, C.; David, J.P.; Fallet-Bianco, C.; Simon, A.; Delacourte, A.; Giros, B.; Epelbaum, J.; et al. Loss of VGLUT1 and VGLUT2 in the prefrontal cortex is correlated with cognitive decline in Alzheimer disease. Neurobiol. Aging 2008, 29, 1619–1630. [Google Scholar] [CrossRef] [Green Version]
- Poirel, O.; Mella, S.; Videau, C.; Ramet, L.; Davoli, M.A.; Herzog, E.; Katsel, P.; Mechawar, N.; Haroutunian, V.; Epelbaum, J.; et al. Moderate decline in select synaptic markers in the prefrontal cortex (BA9) of patients with Alzheimer’s disease at various cognitive stages. Sci. Rep. 2018, 8, 938. [Google Scholar] [CrossRef] [Green Version]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; Mufson, E.J. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 1372–1384. [Google Scholar] [CrossRef] [PubMed]
- de Wilde, M.C.; Overk, C.R.; Sijben, J.W.; Masliah, E. Meta-analysis of synaptic pathology in Alzheimer’s disease reveals selective molecular vesicular machinery vulnerability. Alzheimers Dement. 2016, 12, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Cummings, D.M.; Liu, W.; Portelius, E.; Bayram, S.; Yasvoina, M.; Ho, S.H.; Smits, H.; Ali, S.S.; Steinberg, R.; Pegasiou, C.M.; et al. First effects of rising amyloid-β in transgenic mouse brain: Synaptic transmission and gene expression. Brain 2015, 138, 1992–2004. [Google Scholar] [CrossRef]
- Baglietto-Vargas, D.; Prieto, G.A.; Limon, A.; Forner, S.; Rodriguez-Ortiz, C.J.; Ikemura, K.; Ager, R.R.; Medeiros, R.; Trujillo-Estrada, L.; Martini, A.C.; et al. Impaired AMPA signaling and cytoskeletal alterations induce early synaptic dysfunction in a mouse model of Alzheimer’s disease. Aging Cell. 2018, 17, e12791. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.V.; Skotte, N.H.; Christensen, S.K.; Polli, F.S.; Shabani, M.; Markussen, K.H.; Haukedal, H.; Westi, E.W.; Diaz-delCastillo, M.; Sun, R.C.; et al. Hippocampal disruptions of synaptic and astrocyte metabolism are primary events of early amyloid pathology in the 5xFAD mouse model of Alzheimer’s disease. Cell. Death Dis. 2021, 12, 954. [Google Scholar] [CrossRef]
- Tang, J.; Oliveros, A.; Jang, M.H. Dysfunctional mitochondrial bioenergetics and synaptic degeneration in Alzheimer disease. Int. Neurourol. J. 2019, 23 (Suppl. S1), S5–S10. [Google Scholar] [CrossRef] [Green Version]
- Seo, N.Y.; Kim, G.H.; Noh, J.E.; Shin, J.W.; Lee, C.H.; Lee, K.J. Selective regional loss of cortical synapses lacking presynaptic mitochondria in the 5xFAD mouse model. Front. Neuroanat. 2021, 15, 690168. [Google Scholar] [CrossRef]
- Kumar, R.; Chaterjee, P.; Sharma, P.K.; Singh, A.K.; Gupta, A.; Gill, K.; Tripathi, M.; Dey, A.B.; Dey, S. Sirtuin1: A promising serum protein marker for early detection of Alzheimer’s disease. PLoS ONE 2013, 8, e61560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, H.J.; Murray, T.K.; Kehoe, P.G.; Love, S.; Verdin, E.M.; O’Neill, M.J.; Lane, J.D.; Balthasar, N. CNS SIRT3 expression is altered by reactive oxygen species and in Alzheimer’s disease. PLoS ONE 2012, 7, e48225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kim, Y.; Liu, T.; Hwang, Y.J.; Hyeon, S.J.; Im, H.; Lee, K.; Alvarez, V.E.; McKee, A.C.; Um, S.J.; et al. SIRT3 deregulation is linked to mitochondrial dysfunction in Alzheimer’s disease. Aging Cell. 2018, 17, e12679. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.D.; Yang, J.L.; Lin, T.K.; Yang, D.I. Emerging roles of sestrins in neurodegenerative diseases: Counteracting oxidative stress and beyond. J. Clin. Med. 2019, 8, 1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.S.; Chen, S.D.; Wu, C.L.; Huang, S.S.; Yang, D.I. Induction of sestrin2 as an endogenous protective mechanism against amyloid beta-peptide neurotoxicity in primary cortical culture. Exp. Neurol. 2014, 253, 63–71. [Google Scholar] [CrossRef]
- Rai, N.; Kumar, R.; Desai, G.R.; Venugopalan, G.; Shekhar, S.; Chatterjee, P.; Tripathi, M.; Upadhyay, A.D.; Dwivedi, S.; Dey, A.B.; et al. Relative alterations in blood-based levels of sestrin in Alzheimer’s disease and mild cognitive impairment patients. J. Alzheimer Dis. 2016, 54, 1147–1155. [Google Scholar] [CrossRef]
- Lei, Y.; Wang, J.; Wang, D.; Li, C.; Liu, B.; Fang, X.; You, J.; Guo, M.; Lu, X.Y. SIRT1 in forebrain excitatory neurons produces sexually dimorphic effects on depression-related behaviors and modulates neuronal excitability and synaptic transmission in the medial prefrontal cortex. Mol. Psychiatry 2020, 25, 1094–1111. [Google Scholar] [CrossRef] [Green Version]
- Cartas-Cejudo, P.; Lachén-Montes, M.; Ferrer, I.; Fernández-Irigoyen, J.; Santamaría, E. Sex-divergent effects on the NAD+-dependent deacetylase sirtuin signaling across the olfactory-entorhinal-amygdaloid axis in Alzheimer’s and Parkinson’s diseases. Biol. Sex. Diff. 2023, 14, 5. [Google Scholar] [CrossRef]
- Pearson-Smith, J.N.; Fulton, R.; Huynh, C.Q.; Figueroa, A.G.; Huynh, G.B.; Liang, L.P.; Gano, L.B.; Michel, C.R.; Reisdorph, N.; Reisdorph, R.; et al. Neuronal SIRT3 deletion predisposes to female-specific alterations in cellular metabolism, memory, and network excitability. J. Neurosci. 2023, 43, 1845–1857. [Google Scholar] [CrossRef]
- Simões, A.P.; Machado, N.J.; Gonçalves, N.; Kaster, M.P.; Simões, A.T.; Nunes, A.; Pereira de Almeida, L.; Goosens, K.A.; Rial, D.; Cunha, R.A. Adenosine A2A receptors in the amygdala control synaptic plasticity and contextual fear memory. Neuropsychopharmacology 2016, 41, 2862–2871. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, F.Q.; Pires, J.; Pliassova, A.; Beleza, R.; Lemos, C.; Marques, J.M.; Rodrigues, R.J.; Canas, P.M.; Köfalvi, A.; Cunha, R.A.; et al. Adenosine A2b receptors control A1 receptor-mediated inhibition of synaptic transmission in the mouse hippocampus. Eur. J. Neurosci. 2015, 41, 878–888. [Google Scholar] [CrossRef]
- Lopes, C.R.; Oliveira, A.; Gaspar, I.; Rodrigues, M.S.; Santos, J.; Szabó, E.; Silva, H.B.; Tomé, Â.R.; Canas, P.M.; Agostinho, P.; et al. Effects of chronic caffeine consumption on synaptic function, metabolism and adenosine modulation in different brain areas. Biomolecules 2023, 13, 106. [Google Scholar] [CrossRef]
- Anderson, W.W.; Collingridge, G.L. Capabilities of the WinLTP data acquisition program extending beyond basic LTP experimental functions. J. Neurosci. Meth. 2007, 162, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Canas, P.M.; Porciúncula, L.O.; Cunha, G.M.; Silva, C.G.; Machado, N.J.; Oliveira, J.M.; Oliveira, C.R.; Cunha, R.A. Adenosine A2A receptor blockade prevents synaptotoxicity and memory dysfunction caused by beta-amyloid peptides via p38 mitogen-activated protein kinase pathway. J. Neurosci. 2009, 29, 14741–14751. [Google Scholar] [CrossRef] [Green Version]
- Cunha, R.A.; Sebastião, A.M.; Ribeiro, J.A. Ecto-5’-nucleotidase is associated with cholinergic nerve terminals in the hippocampus but not in the cerebral cortex of the rat. J. Neurochem. 1992, 59, 657–666. [Google Scholar] [CrossRef]
- Rebola, N.; Pinheiro, P.C.; Oliveira, C.R.; Malva, J.O.; Cunha, R.A. Subcellular localization of adenosine A1 receptors in nerve terminals and synapses of the rat hippocampus. Brain Res. 2003, 987, 49–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Wu, M.; He, G.; Zhang, X.; Li, W.; Gao, Y.; Li, Z.; Wang, Z.; Zhang, C. Glyceraldehyde-3-phosphate dehydrogenase: A universal internal control for Western blots in prokaryotic and eukaryotic cells. Anal. Biochem. 2012, 423, 15–22. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Summary of Genotype Comparison | APPswe/PS1dE9 vs. WT | |
|---|---|---|
| Behavioral Test | ||
| Morris water maze | latency to find platform |  |
| platform crossings |  | |
| thigmotaxis |  | |
| search strategy |  hippocampal hippocampal | |
| Fear conditioning | acquisition—day 1 |  |
| context freezing—day 2 |  | |
| tone freezing—day 3 |  | |
| habituation freezing—day 4 |  | |
| Open field | total distance |  |
| maximum Speed |  | |
| % time in the center |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopes, C.R.; Silva, J.S.; Santos, J.; Rodrigues, M.S.; Madeira, D.; Oliveira, A.; Moreira-de-Sá, A.; Lourenço, V.S.; Gonçalves, F.Q.; Silva, H.B.; et al. Downregulation of Sirtuin 1 Does Not Account for the Impaired Long-Term Potentiation in the Prefrontal Cortex of Female APPswe/PS1dE9 Mice Modelling Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 6968. https://doi.org/10.3390/ijms24086968
Lopes CR, Silva JS, Santos J, Rodrigues MS, Madeira D, Oliveira A, Moreira-de-Sá A, Lourenço VS, Gonçalves FQ, Silva HB, et al. Downregulation of Sirtuin 1 Does Not Account for the Impaired Long-Term Potentiation in the Prefrontal Cortex of Female APPswe/PS1dE9 Mice Modelling Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(8):6968. https://doi.org/10.3390/ijms24086968
Chicago/Turabian StyleLopes, Cátia R., Joana S. Silva, Joana Santos, Matilde S. Rodrigues, Daniela Madeira, Andreia Oliveira, Ana Moreira-de-Sá, Vanessa S. Lourenço, Francisco Q. Gonçalves, Henrique B. Silva, and et al. 2023. "Downregulation of Sirtuin 1 Does Not Account for the Impaired Long-Term Potentiation in the Prefrontal Cortex of Female APPswe/PS1dE9 Mice Modelling Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 8: 6968. https://doi.org/10.3390/ijms24086968