
Inhibition of Prolyl Oligopeptidase Restores Prohibitin 2 Levels in Psychosis Models: Relationship to Cognitive Deficits in Schizophrenia
 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
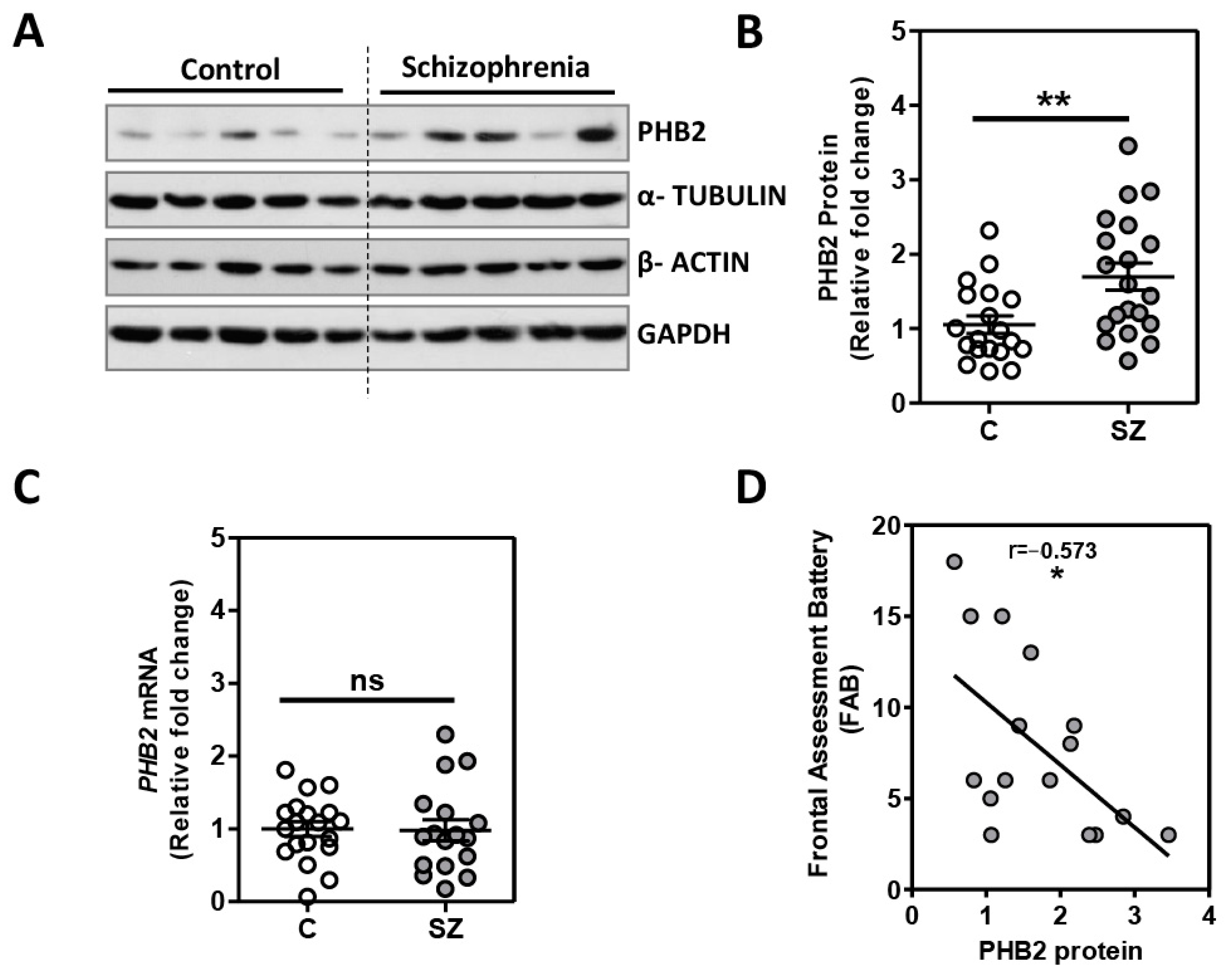
2.1. Analysis of PHB2 Levels in Postmortem Dorsolateral Prefrontal Cortex in Schizophrenia
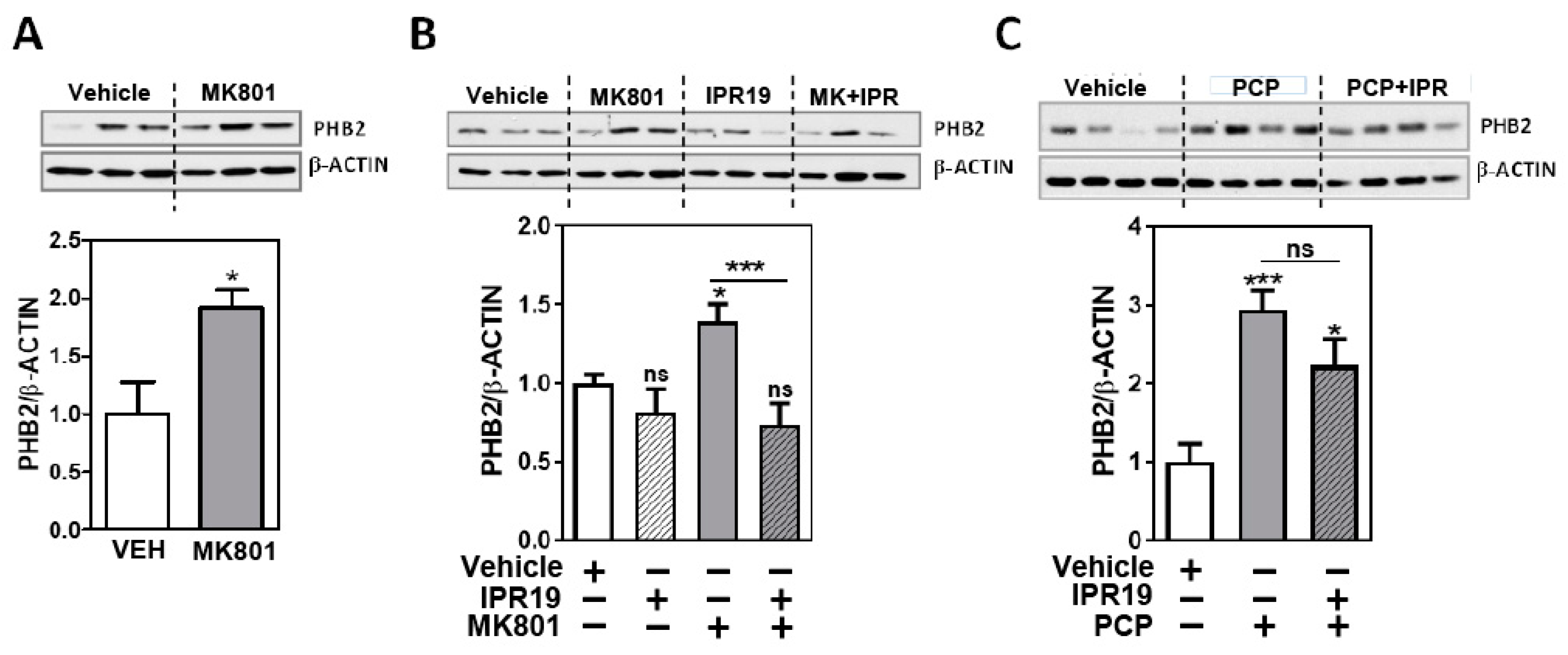
2.2. Modulation of PHB2 Protein Levels by NMDA Receptor Activity and the Prolyl Oligopeptidase Inhibitor IPR19 in Rodents
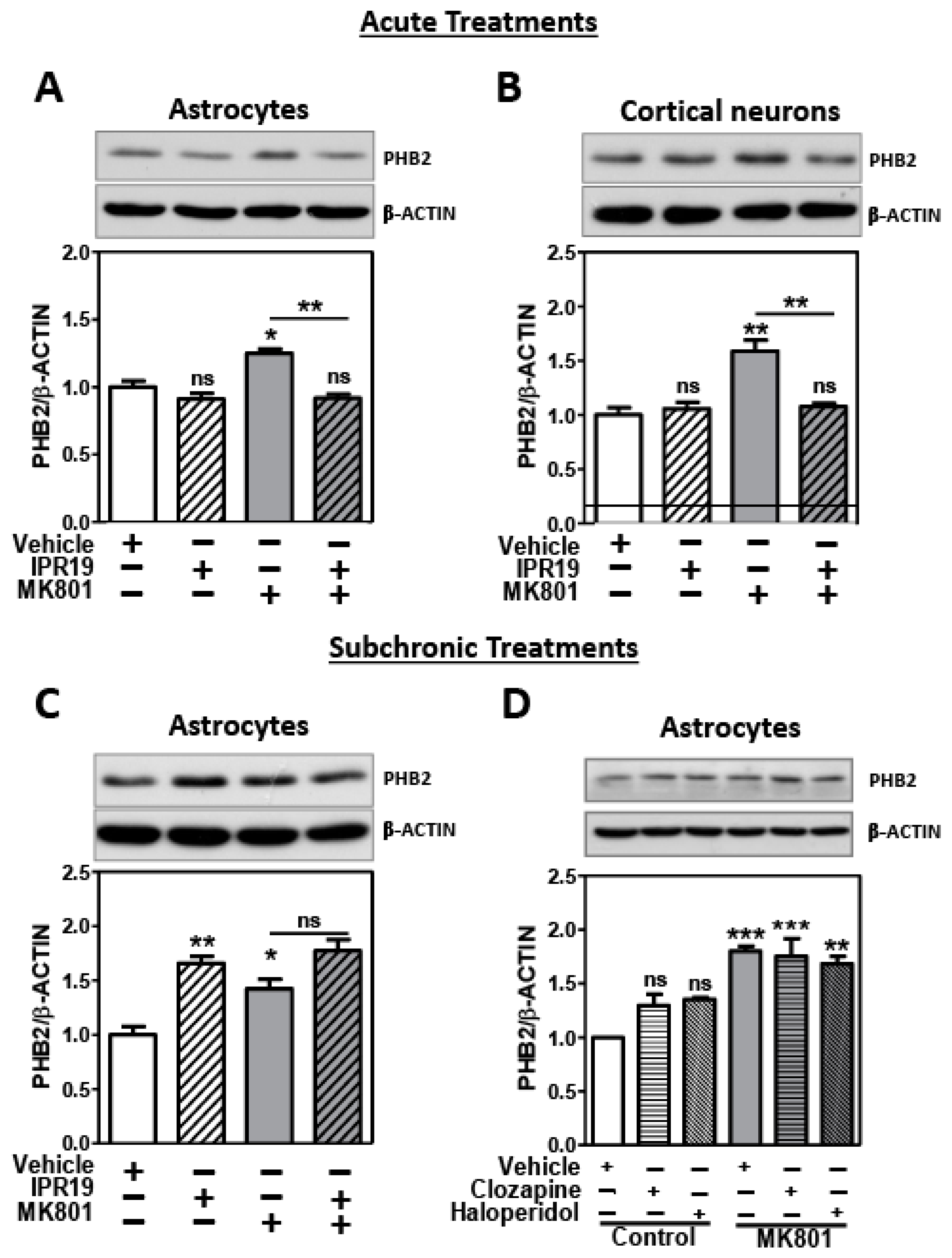
2.3. Modulation of PHB2 Protein Levels by NMDA Receptor Activity and the Prolyl Oligopeptidase Inhibitor IPR19 in Rat Cortical Astrocytes and Neurons
2.3.1. Acute Treatments
2.3.2. Subchronic Treatments
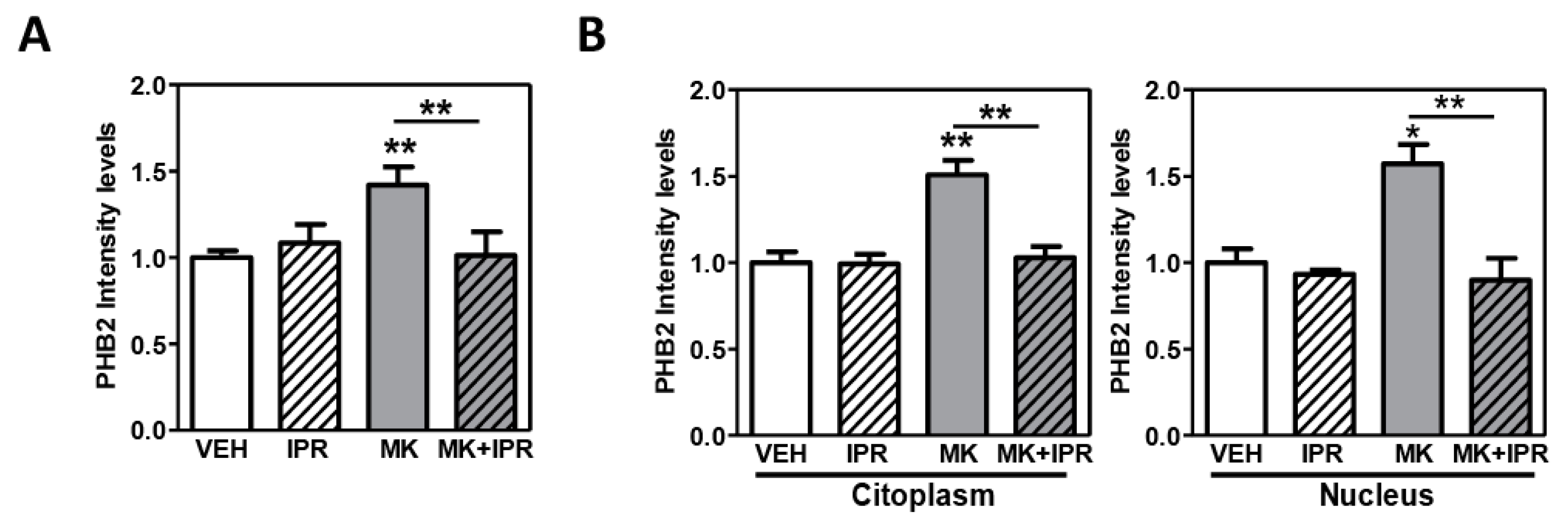
2.4. Characterization of PHB2 in Cortical Neuron Cultures Treated with NMDA Receptor Antagonist and the Inhibitor IPR19
3. Discussion
4. Materials and Methods
4.1. Brain Tissue Samples
4.2. Rat Cortical Cultures
4.3. Protein Extraction
4.4. Immunoblotting
4.5. Reverse Transcription Quantitative PCR (RT-qPCR)
4.6. Immunocytochemistry and Confocal Imaging
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Owen, M.J.; Sawa, A.; Mortensen, P.B. Schizophrenia. Lancet 2016, 388, 86–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatemi, S.H.; Folsom, T.D. The Neurodevelopmental Hypothesis of Schizophrenia, Revisited. Schizophr. Bull. 2009, 35, 528–548. [Google Scholar] [CrossRef] [PubMed]
- Huffman, R.E.; McGlashan, T.H. Synaptic Elimination, Neurodevelopment, and the Mechanism of Hallucinated “Voices” in Schizophrenia. Am. J. Psychiatry 1997, 154, 1683–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, J. How Synaptic Pruning Shapes Neural Wiring during Development and, Possibly, in Disease. Proc. Natl. Acad. Sci. USA 2020, 117, 16096–16099. [Google Scholar] [CrossRef]
- Bansal, V.; Chatterjee, I. Role of Neurotransmitters in Schizophrenia: A Comprehensive Study. Kuwait J. Sci. 2021, 48, 1–27. [Google Scholar] [CrossRef]
- Bubeníková-Valešová, V.; Horáček, J.; Vrajová, M.; Höschl, C. Models of Schizophrenia in Humans and Animals Based on Inhibition of NMDA Receptors. Neurosci. Biobehav. Rev. 2008, 32, 1014–1023. [Google Scholar] [CrossRef]
- Uno, Y.; Coyle, J.T. Glutamate Hypothesis in Schizophrenia. Psychiatry Clin. Neurosci. 2019, 73, 204–215. [Google Scholar] [CrossRef] [Green Version]
- McCutcheon, R.A.; Krystal, J.H.; Howes, O.D. Dopamine and Glutamate in Schizophrenia: Biology, Symptoms and Treatment. World Psychiatry 2020, 19, 15. [Google Scholar] [CrossRef] [Green Version]
- Bowie, C.R.; Harvey, P.D. Cognitive Deficits and Functional Outcome in Schizophrenia. Neuropsychiatr. Dis. Treat. 2006, 2, 531. [Google Scholar] [CrossRef] [Green Version]
- Krishnadas, R.; Moore, B.P.; Nayak, A.; Patel, R.R. Relationship of Cognitive Function in Patients with Schizophrenia in Remission to Disability: A Cross-Sectional Study in an Indian Sample. Ann. Gen. Psychiatry 2007, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Harvey, P.D.; Lombardi, J.; Leibman, M.; White, L.; Parrella, M.; Powchik, P.; Davidson, M. Cognitive Impairment and Negative Symptoms in Geriatric Chronic Schizophrenic Patients: A Follow-up Study. Schizophr. Res. 1996, 22, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.Y.; Lane, H.Y.; Lin, C.H. Medications Used for Cognitive Enhancement in Patients with Schizophrenia, Bipolar Disorder, Alzheimer’s Disease, and Parkinson’s Disease. Front. Psychiatry 2018, 9, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Śmiarowska, M.; Białecka, M.; Machoy-Mokrzyńska, A. Cannabis and Cannabinoids: Pharmacology and Therapeutic Potential. Neurol. Neurochir. Pol. 2022, 56, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Powchik, P.; Davidson, M.; Haroutunian, V.; Qabriel, S.M.; Purohit, D.P.; Perl, D.P.; Harvey, P.D.; Davis, K.L. Postmortem Studies in Schizophrenia. Schizophr. Bull. 1998, 24, 325–341. [Google Scholar] [CrossRef]
- Toru, M.; Watanabe, S.; Shibuya, H.; Nishikawa, T.; Noda, K.; Mitsushio, H.; Ichikawa, H.; Kurumaji, A.; Takashima, M.; Mataga, N.; et al. Neurotransmitters, Receptors and Neuropeptides in Post-mortem Brains of Chronic Schizophrenic Patients. Acta Psychiatr. Scand. 1988, 78, 121–137. [Google Scholar] [CrossRef]
- Arif, M.; Chikuma, T.; Ahmed, M.; Yoshida, S.; Kato, T. Suppressive Effect of Clozapine but Not Haloperidol on the Increases of Neuropeptide-Degrading Enzymes and Glial Cells in MK-801-Treated Rat Brain Regions. Neurosci. Res. 2007, 57, 248–258. [Google Scholar] [CrossRef]
- Albrecht, A.; Rahmoune, H.; Leedjärv, K.; Knorpp, T.; Joos, T.; Stocki, P.; Guest, P.C.; Bahn, S. Development of a Novel Assay for Proprotein Converting Enzyme Activity on a Multiplex Bead-Based Array System. Proteomics 2013, 13, 2976–2979. [Google Scholar] [CrossRef]
- Rodríguez, B.; Nani, J.V.; Almeida, P.G.C.; Brietzke, E.; Lee, R.S.; Hayashi, M.A.F. Neuropeptides and Oligopeptidases in Schizophrenia. Neurosci. Biobehav. Rev. 2020, 108, 679–693. [Google Scholar] [CrossRef]
- Babkova, K.; Korabecny, J.; Soukup, O.; Nepovimova, E.; Jun, D.; Kuca, K. Prolyl Oligopeptidase and Its Role in the Organism: Attention to the Most Promising and Clinically Relevant Inhibitors. Future Med. Chem. 2017, 9, 1015–1038. [Google Scholar] [CrossRef]
- Svarcbahs, R.; Julku, U.H.; Myöhänen, T.T. Inhibition of Prolyl Oligopeptidase Restores Spontaneous Motor Behavior in the ÷-Synuclein Virus Vector-Based Parkinson’s Disease Mouse Model by Decreasing α-Synuclein Oligomeric Species in Mouse Brain. J. Neurosci. 2016, 36, 12485–12497. [Google Scholar] [CrossRef] [Green Version]
- Prades, R.; Munarriz-Cuezva, E.; Urigüen, L.; Gil-Pisa, I.; Gómez, L.; Mendieta, L.; Royo, S.; Giralt, E.; Tarragó, T.; Meana, J.J. The Prolyl Oligopeptidase Inhibitor IPR19 Ameliorates Cognitive Deficits in Mouse Models of Schizophrenia. Eur. Neuropsychopharmacol. 2017, 27, 180–191. [Google Scholar] [CrossRef] [PubMed]
- López, A.; Mendieta, L.; Prades, R.; Royo, S.; Tarragó, T.; Giralt, E. Peptide POP Inhibitors for the Treatment of the Cognitive Symptoms of Schizophrenia. Future Med. Chem. 2013, 5, 1509–1523. [Google Scholar] [CrossRef]
- Woo, T.-U.W. Neurobiology of Schizophrenia Onset. Curr. Top. Behav. Neurosci. 2014, 16, 267–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wible, C.G.; Anderson, J.; Shenton, M.E.; Kricun, A.; Hirayasu, Y.; Tanaka, S.; Levitt, J.J.; O’donnell, B.F.; Kikinis, R.; Jolesz, F.A.; et al. Prefrontal Cortex, Negative Symptoms, and Schizophrenia: An MRI Study. Psychiatry Res. 2001, 108, 65–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Fan, L.; Qiu, C.; Jiang, T. Prefrontal Cortex and the Dysconnectivity Hypothesis of Schizophrenia. Neurosci. Bull. 2015, 31, 207–219. [Google Scholar] [CrossRef] [Green Version]
- Orellana, G.; Slachevsky, A.; Gaspar, P.A.; Alawieh, A.M. Executive Functioning in Schizophrenia. Front. Psychiatry 2013, 4, 35. [Google Scholar] [CrossRef] [Green Version]
- Giraldo-Chica, M.; Rogers, B.P.; Damon, S.M.; Landman, B.A.; Woodward, N.D. Prefrontal-Thalamic Anatomical Connectivity and Executive Cognitive Function in Schizophrenia. Biol. Psychiatry 2018, 83, 509–517. [Google Scholar] [CrossRef]
- Minzenberg, M.J.; Laird, A.R.; Thelen, S.; Carter, C.S.; Glahn, D.C. Meta-Analysis of 41 Functional Neuroimaging Studies of Executive Function in Schizophrenia. Arch. Gen. Psychiatry 2009, 66, 811–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinacho, R.; Villalmanzo, N.; Meana, J.J.; Ferrer, I.; Berengueras, A.; Haro, J.M.; Villén, J.; Ramos, B. Altered CSNK1E, FABP4 and NEFH Protein Levels in the Dorsolateral Prefrontal Cortex in Schizophrenia. Schizophr. Res. 2016, 177, 88–97. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.; Murphy, L.C.; Murphy, L.J. The Prohibitins: Emerging Roles in Diverse Functions. J. Cell. Mol. Med. 2006, 10, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Merkwirth, C.; Martinelli, P.; Korwitz, A.; Morbin, M.; Brönneke, H.S.; Jordan, S.D.; Rugarli, E.I.; Langer, T. Loss of Prohibitin Membrane Scaffolds Impairs Mitochondrial Architecture and Leads to Tau Hyperphosphorylation and Neurodegeneration. PLoS Genet. 2012, 8, e1003021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artal-Sanz, M.; Tavernarakis, N. Prohibitin and Mitochondrial Biology. Trends Endocrinol. Metab. 2009, 20, 394–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernando-Rodríguez, B.; Artal-Sanz, M. Cells Mitochondrial Quality Control Mechanisms and the PHB (Prohibitin) Complex. Cells 2018, 7, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Signorile, A.; Sgaramella, G.; Bellomo, F.; De Rasmo, D. Prohibitins: A Critical Role in Mitochondrial Functions and Implication in Diseases. Cells 2019, 8, 71. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Shen, J.; Ran, Z. Emerging Views of Mitophagy in Immunity and Autoimmune Diseases. Autophagy 2020, 16, 3–17. [Google Scholar] [CrossRef]
- Lahiri, V.; Klionsky, D.J. PHB2/Prohibitin 2: An Inner Membrane Mitophagy Receptor. Cell Res. 2017, 27, 311–312. [Google Scholar] [CrossRef]
- Wei, Y.; Chiang, W.C.; Sumpter, R.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238. [Google Scholar] [CrossRef] [Green Version]
- Bavelloni, A.; Piazzi, M.; Raffini, M.; Faenza, I.; Blalock, W.L.; Zhu, X.; Burger, M.; Doane, T.A.; Horwath, W.R. Prohibitin 2: At a Communications Crossroads. Proc. Natl. Acad. Sci. USA 2013, 67, 239–254. [Google Scholar] [CrossRef]
- Montano, M.M.; Ekena, K.; Delage-Mourroux, R.; Chang, W.; Martini, P.; Katzenellenbogen, B.S. An Estrogen Receptor-Selective Coregulator That Potentiates the Effectiveness of Antiestrogens and Represses the Activity of Estrogens. Proc. Natl. Acad. Sci. USA 1999, 96, 6947–6952. [Google Scholar] [CrossRef] [Green Version]
- Clark, D.; Dedova, I.; Cordwell, S.; Matsumoto, I. Proteome Analysis of the Anterior Cingulate Cortex Gray Matter in Schizophrenia A Proteome Analysis of the Anterior Cingulate Cortex Gray Matter in Schizophrenia. Mol. Psychiatry 2006, 11, 459–470. [Google Scholar] [CrossRef] [Green Version]
- Kasashima, K.; Ohta, E.; Kagawa, Y.; Endo, H. Mitochondrial Functions and Estrogen Receptor-Dependent Nuclear Translocation of Pleiotropic Human Prohibitin 2. J. Biol. Chem. 2006, 281, 36401–36410. [Google Scholar] [CrossRef] [Green Version]
- Balu, D.T. The NMDA Receptor and Schizophrenia. Adv. Pharmacol. 2016, 76, 351–382. [Google Scholar] [CrossRef] [Green Version]
- Howes, O.; Mccutcheon, R.; Stone, J. Glutamate and Dopamine in Schizophrenia: An Update for the 21st Century. J. Psychopharmacol. 2015, 29, 97–115. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.; Zhou, Y. NMDAR Hypofunction Animal Models of Schizophrenia. Front. Mol. Neurosci. 2019, 12, 185. [Google Scholar] [CrossRef] [Green Version]
- Pinacho, R.; Vila, E.; Prades, R.; Tarragó, T.; Castro, E.; Ferrer, I.; Ramos, B. The Glial Phosphorylase of Glycogen Isoform Is Reduced in the Dorsolateral Prefrontal Cortex in Chronic Schizophrenia. Schizophr. Res. 2016, 177, 37–43. [Google Scholar] [CrossRef]
- Tarasov, V.V.; Svistunov, A.A.; Chubarev, V.N.; Sologova, S.S.; Mukhortova, P.; Levushkin, D.; Somasundaram, S.G.; Kirkland, C.E.; Bachurin, S.O.; Aliev, G. Alterations of Astrocytes in the Context of Schizophrenic Dementia. Front. Pharmacol. 2019, 10, 1612. [Google Scholar] [CrossRef] [Green Version]
- Dietz, A.G.; Goldman, S.A.; Nedergaard, M. Glial Cells in Schizophrenia: A Unified Hypothesis. Lancet Psychiatry 2020, 7, 272–281. [Google Scholar] [CrossRef]
- Sullivan, C.R.; O’Donovan, S.M.; McCullumsmith, R.E.; Ramsey, A. Defects in Bioenergetic Coupling in Schizophrenia. Biol. Psychiatry 2018, 83, 739–750. [Google Scholar] [CrossRef]
- Smaili, S.S.; Ureshino, R.P.; Rodrigues, L.; Rocha, K.K.; Carvalho, J.T.; Oseki, K.T.; Bincoletto, C.; Lopes, G.S.; Hirata, H. The Role of Mitochondrial Function in Glutamate-Dependent Metabolism in Neuronal Cells. Curr. Pharm. Des. 2011, 17, 3865–3877. [Google Scholar] [CrossRef]
- Kolomeets, N.S.; Uranova, N. Ultrastructural Abnormalities of Astrocytes in the Hippocampus in Schizophrenia and Duration of Illness: A Postortem Morphometric Study. World J. Biol. Psychiatry 2010, 11, 282–292. [Google Scholar] [CrossRef]
- Yoon, J.H.; Minzenberg, M.J.; Ursu, S.; Walters, R.; Wendelken, C.; Ragland, J.D.; Carter, C.S. Association of Dorsolateral Prefrontal Cortex Dysfunction with Disrupted Coordinated Brain Activity in Schizophrenia: Relationship with Impaired Cognition, Behavioral Disorganization, and Global Function. Am. J. Psychiatry 2008, 165, 1006. [Google Scholar] [CrossRef] [Green Version]
- Smucny, J.; Dienel, S.J.; Lewis, D.A.; Carter, C.S. Mechanisms Underlying Dorsolateral Prefrontal Cortex Contributions to Cognitive Dysfunction in Schizophrenia. Neuropsychopharmacology 2021, 47, 292–308. [Google Scholar] [CrossRef]
- Lewis, D.A.; Moghaddam, B. Cognitive Dysfunction in Schizophrenia: Convergence of γ-Aminobutyric Acid and Glutamate Alterations. Arch. Neurol. 2006, 63, 1372–1376. [Google Scholar] [CrossRef]
- Snyder, M.A.; Gao, W.-J. NMDA Hypofunction as a Convergence Point for Progression and Symptoms of Schizophrenia. Front. Cell. Neurosci. 2013, 7, 31. [Google Scholar] [CrossRef] [Green Version]
- Picard, M.; McEwen, B.S. Mitochondria Impact Brain Function and Cognition. Proc. Natl. Acad. Sci. USA 2014, 111, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Vita, A.; De Peri, L.; Deste, G.; Sacchetti, E. Progressive Loss of Cortical Gray Matter in Schizophrenia: A Meta-Analysis and Meta-Regression of Longitudinal MRI Studies. Transl. Psychiatry 2012, 2, e190. [Google Scholar] [CrossRef] [Green Version]
- Jarskog, L.F.; Glantz, L.A.; Gilmore, J.H.; Lieberman, J.A. Apoptotic Mechanisms in the Pathophysiology of Schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2005, 29, 846–858. [Google Scholar] [CrossRef]
- Peng, Y.T.; Chen, P.; Ouyang, R.Y.; Song, L. Multifaceted Role of Prohibitin in Cell Survival and Apoptosis. Apoptosis 2015, 20, 1135–1149. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Xu, J.; Frolova, A.; Liao, L.; Malley, B.W.O.; Katzenellenbogen, B.S. Genetic Deletion of the Repressor of Estrogen Receptor Activity (REA) Enhances the Response to Estrogen in Target Tissues In Vivo Genetic Deletion of the Repressor of Estrogen Receptor Activity (REA) Enhances the Response to Estrogen in Target Tissues. Mol. Cell. Biol. 2005, 25, 1989–1999. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Gong, L.; Chen, L.; Xu, M.; Abou-Hamdan, H.; Tang, M.; Désaubry, L.; Song, Z. PHB2 (Prohibitin 2) Promotes PINK1-PRKN/Parkin-Dependent Mitophagy by the PARL-PGAM5-PINK1 Axis. Autophagy 2019, 16, 419–434. [Google Scholar] [CrossRef]
- Anderson, C.J.; Kahl, A.; Fruitman, H.; Qian, L.; Zhou, P.; Manfredi, G.; Costantino Iadecola, P. Levels Regulate OMA1 Activity and Turnover in Neurons. Cell Death Differ. 2019, 27, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Neill, J.C.; Barnes, S.; Cook, S.; Grayson, B.; Idris, N.F.; Mclean, S.L.; Snigdha, S.; Rajagopal, L.; Harte, M.K. Animal Models of Cognitive Dysfunction and Negative Symptoms of Schizophrenia: Focus on NMDA Receptor Antagonism. Pharmacol. Ther. 2010, 128, 419–432. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.; Watson, D.; Fone, K. Animal Models of Schizophrenia. Br. J. Pharmacol. 2011, 164, 1162–1194. [Google Scholar] [CrossRef] [PubMed]
- Roca Casasús, M.; Escanilla Casal, A.; Monje Hernández, A.; Baño Galindo, V.; Planchat Teruel, L.M.; Costa Escola, J.; Haro Abad, J.M. Banco de Tejidos Neurológicos de Sant Joan de Déu-Serveis de Salut Mental Para La Investigación de Las Enfermedades Mentales. La Importancia de Un Programa de Donación En Vida. Psiquiatr. Biol. 2008, 15, 73–79. [Google Scholar] [CrossRef]
- Gardner, D.M.; Murphy, A.L.; O’Donnell, H.; Centorrino, F.; Baldessarini, R.J. International Consensus Study of Antipsychotic Dosing. Am. J. Psychiatry 2010, 167, 686–693. [Google Scholar] [CrossRef] [Green Version]
- Cadinu, D.; Grayson, B.; Podda, G.; Harte, M.K.; Doostdar, N.; Neill, J.C. Invited Review NMDA Receptor Antagonist Rodent Models for Cognition in Schizophrenia and Identification of Novel Drug Treatments, an Update. Neuropharmacology 2017, 142, 41–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, K.; Fujita, Y.; Shimizu, E.; Iyo, M. Phencyclidine-Induced Cognitive Deficits in Mice Are Improved by Subsequent Subchronic Administration of Clozapine, but Not Haloperidol. Eur. J. Pharmacol. 2005, 519, 114–117. [Google Scholar] [CrossRef]
- Martins-De-Souza, D.; Lebar, M.; Turck, C.W. Proteome Analyses of Cultured Astrocytes Treated with MK-801 and Clozapine: Similarities with Schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 2011, 261, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Bolte, S.; Cordelières, F.P. A Guided Tour into Subcellular Colocalization Analysis in Light Microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Schizophrenia (n = 20) | Controls (n = 20) | Statistic | p Value | |
|---|---|---|---|---|
| Gender (male) | 100% (n = 20) | 100% (n = 20) | N/A | 1.000 a |
| Age (years) | 69 ± 11 | 74 ± 10 | 1.35; 38 b | 0.184 |
| PMD (hours) | 4.71 ± 2.51 | 5.45 ± 1.72 | 1.47; 38 b | 0.150 |
| pH | 6.74 ± 0.52 | 6.76 ± 0.35 | 195.5 c | 0.912 |
| SZ diagnosis | N/A | N/A | N/A | |
| Chronic residual | 70% (n = 14) | |||
| Chronic paranoid | 10% (n = 2) | |||
| Chronic disorganized | 10% (n = 2) | |||
| Chronic catatonic | 5% (n = 1) | |||
| Simple | 5% (n = 1) | |||
| Age of onset of illness (years) | 22 ± 7 | N/A | N/A | N/A |
| Duration of illness (years) | 51 ± 10 | N/A | N/A | N/A |
| Dosage of AP (mg/day) d | 584.60 ± 518.65 | N/A | N/A | N/A |
| AP treatment | N/A | N/A | N/A | |
| First-generation AP | 30% (n = 6) | |||
| Second-generation AP | 50% (n = 10) | |||
| None | 20% (n = 4) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vila, È.; Pinacho, R.; Prades, R.; Tarragó, T.; Castro, E.; Munarriz-Cuezva, E.; Meana, J.J.; Eugui-Anta, A.; Roldan, M.; Vera-Montecinos, A.; et al. Inhibition of Prolyl Oligopeptidase Restores Prohibitin 2 Levels in Psychosis Models: Relationship to Cognitive Deficits in Schizophrenia. Int. J. Mol. Sci. 2023, 24, 6016. https://doi.org/10.3390/ijms24076016
Vila È, Pinacho R, Prades R, Tarragó T, Castro E, Munarriz-Cuezva E, Meana JJ, Eugui-Anta A, Roldan M, Vera-Montecinos A, et al. Inhibition of Prolyl Oligopeptidase Restores Prohibitin 2 Levels in Psychosis Models: Relationship to Cognitive Deficits in Schizophrenia. International Journal of Molecular Sciences. 2023; 24(7):6016. https://doi.org/10.3390/ijms24076016
Chicago/Turabian StyleVila, Èlia, Raquel Pinacho, Roger Prades, Teresa Tarragó, Elena Castro, Eva Munarriz-Cuezva, J. Javier Meana, Ania Eugui-Anta, Mònica Roldan, América Vera-Montecinos, and et al. 2023. "Inhibition of Prolyl Oligopeptidase Restores Prohibitin 2 Levels in Psychosis Models: Relationship to Cognitive Deficits in Schizophrenia" International Journal of Molecular Sciences 24, no. 7: 6016. https://doi.org/10.3390/ijms24076016