
Hypophysitis: Defining Histopathologic Variants and a Review of Emerging Clinical Causative Entities
Abstract
:1. Introduction
2. Epidemiology
3. Subvariants
3.1. Primary Hyophysitis
3.1.1. Lymphocytic
3.1.2. Granulomatous
3.1.3. Xanthomatous
3.1.4. IgG4-Related Disease
3.1.5. Necrotizing
3.2. Secondary Causes
3.2.1. Immunotherapy
3.2.2. COVID-19
3.2.3. Paraneoplastic Syndrome
3.2.4. Autoimmune and Local Infiltrative Disease
4. Diagnosis
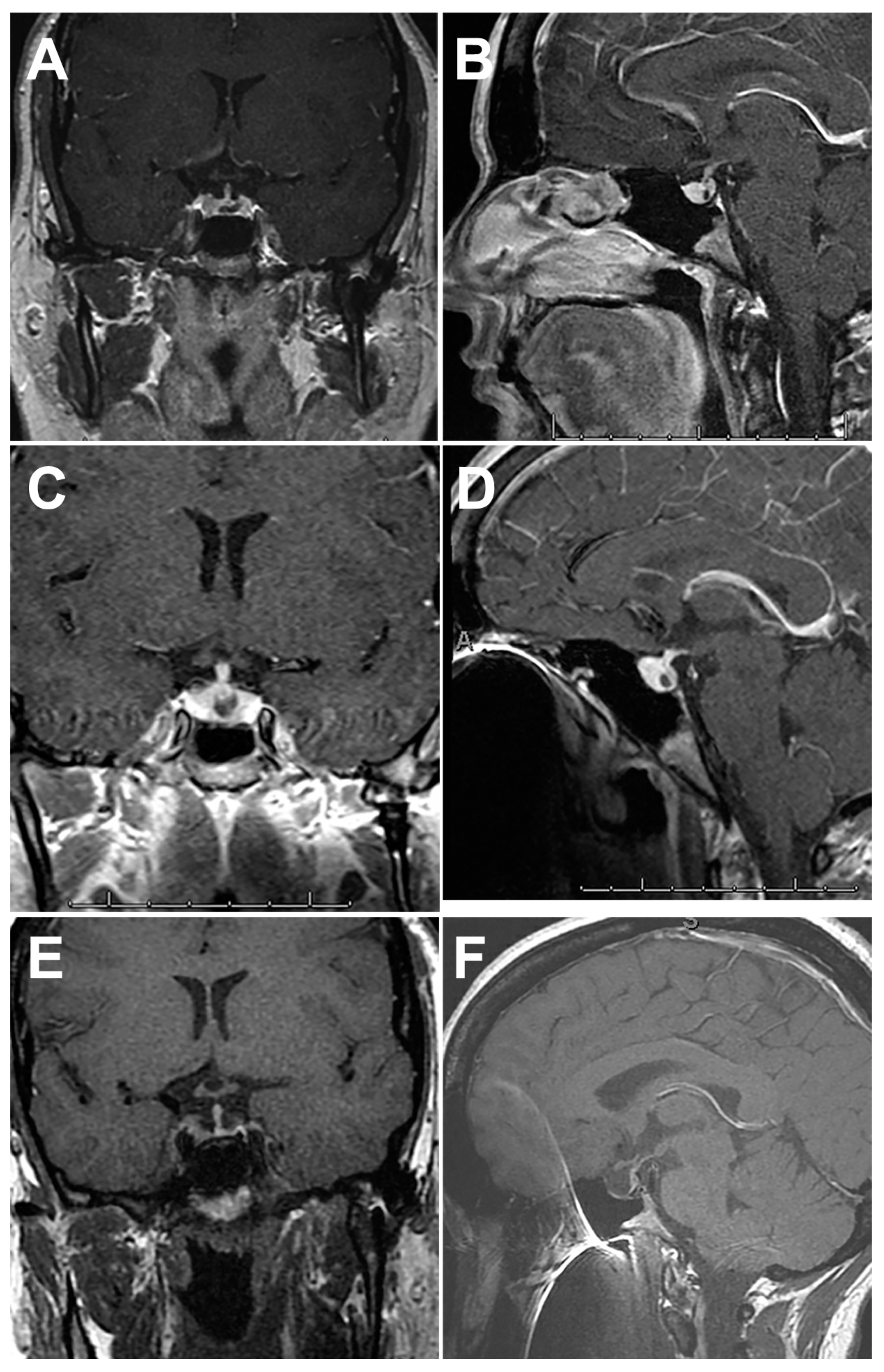
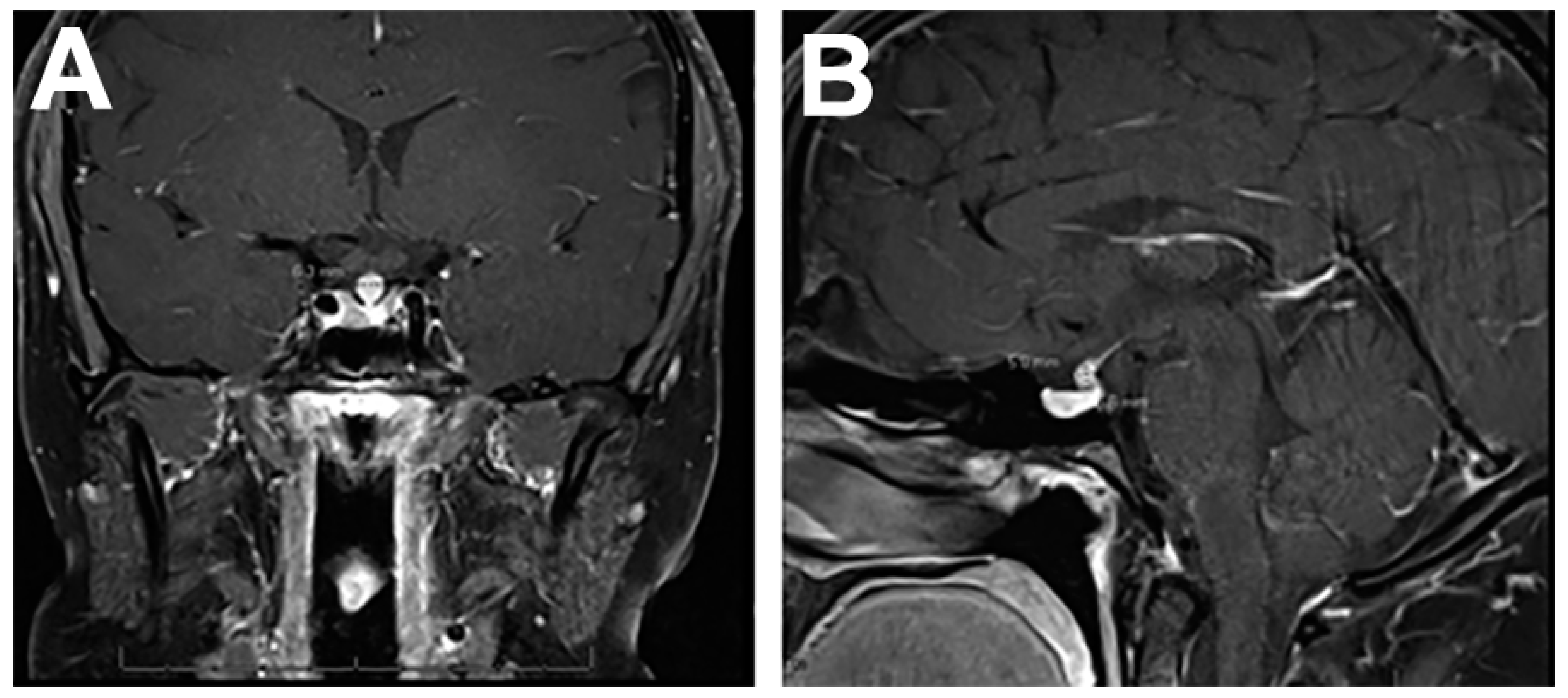
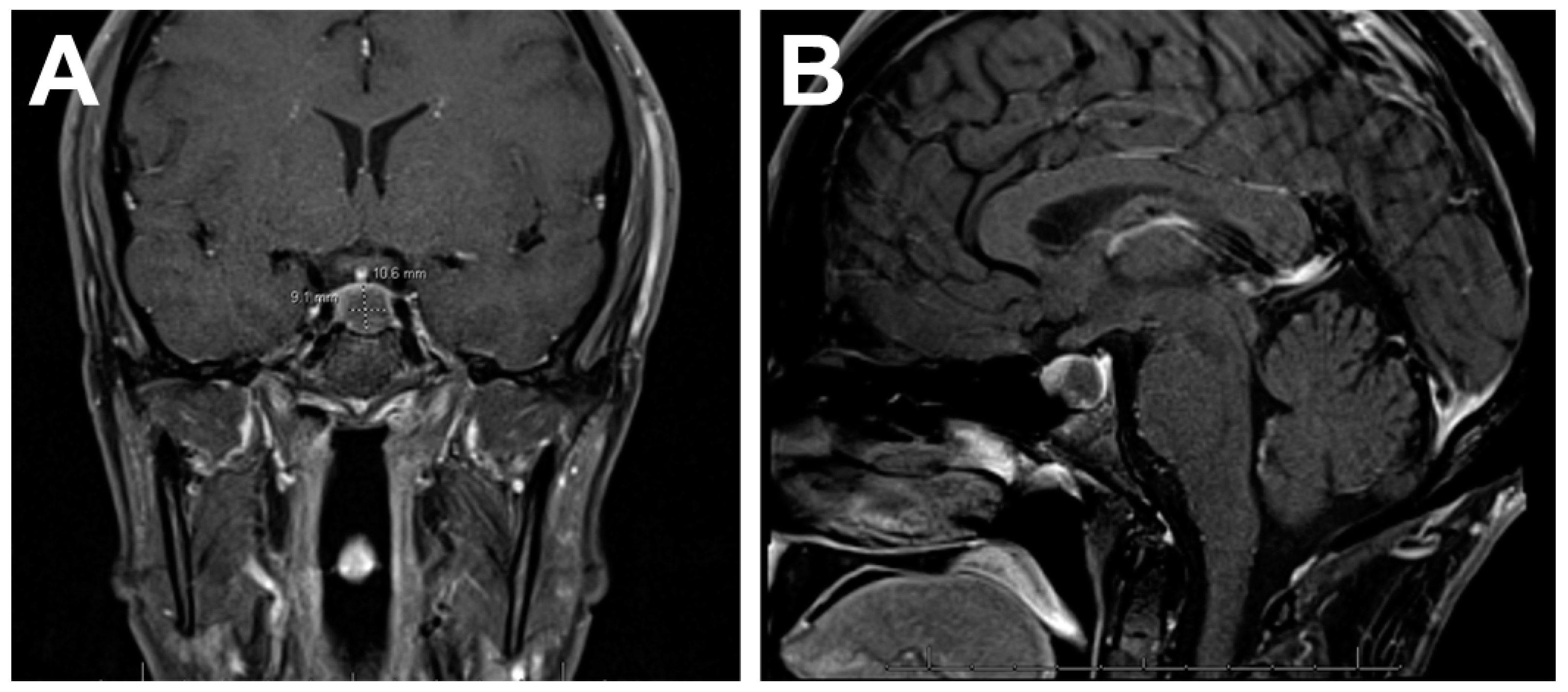
4.1. Imaging Features
4.2. Symptomatology
4.3. Management
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- El Sayed, S.A.; Fahmy, M.W.; Schwartz, J. Physiology, Pituitary Gland; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Joshi, M.N.; Whitelaw, B.C.; Carroll, P.V. Mechanisms in endocrinology: Hypophysitis: Diagnosis and treatment. Eur. J. Endocrinol. 2018, 179, R151–R163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langlois, F.; Varlamov, E.V.; Fleseriu, M. Hypophysitis, the growing spectrum of a rare pituitary disease. J. Clin. Endocrinol. Metab. 2022, 107, 10–28. [Google Scholar] [CrossRef] [PubMed]
- Prete, A.; Salvatori, R. Hypophysitis. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Gubbi, S.; Hannah-Shmouni, F.; Verbalis, J.G.; Koch, C.A. Hypophysitis: An update on the novel forms, diagnosis and management of disorders of pituitary inflammation. Best. Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101371. [Google Scholar] [CrossRef] [PubMed]
- Mathkour, M.; Zeoli, T.; Werner, C.; Scullen, T.; Garces, J.; Keen, J.; Ware, M. Recurring primary xanthomatous hypophysitis behaving like pituitary adenoma: Additional case and literature review. World Neurosurg. 2020, 138, 27–34. [Google Scholar] [CrossRef]
- Sharifi, G.; Mohajeri-Tehrani, M.R.; Navabakhsh, B.; Larijani, B.; Valeh, T. Idiopathic granulomatous hypophysitis presenting with galactorrhea, headache, and nausea in a woman: A case report and review of the literature. J. Med. Case Rep. 2019, 13, 334. [Google Scholar] [CrossRef]
- Caranci, F.; Leone, G.; Ponsiglione, A.; Muto, M.; Tortora, F.; Muto, M.; Cirillo, S.; Brunese, L.; Cerase, A. Imaging findings in hypophysitis: A review. Radiol. Med. 2020, 125, 319–328. [Google Scholar] [CrossRef]
- Glezer, A.; Bronstein, M.D. Pituitary autoimmune disease: Nuances in clinical presentation. Endocrine 2012, 42, 74–79. [Google Scholar] [CrossRef]
- Di Dalmazi, G.; Ippolito, S.; Lupi, I.; Caturegli, P. Hypophysitis induced by immune checkpoint inhibitors: A 10-year assessment. Expert. Rev. Endocrinol. Metab. 2019, 14, 381–398. [Google Scholar] [CrossRef]
- Carpinteri, R.; Patelli, I.; Casanueva, F.F.; Giustina, A. Pituitary tumours: Inflammatory and granulomatous expansive lesions of the pituitary. Best. Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 639–650. [Google Scholar] [CrossRef]
- Yuen, K.C.J.; Popovic, V.; Trainer, P.J. New causes of hypophysitis. Best. Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101276. [Google Scholar] [CrossRef]
- Romano, A.; Rigante, D.; Cipolla, C. Autoimmune phenomena involving the pituitary gland in children: New developing data about diagnosis and treatment. Autoimmun. Rev. 2019, 18, 102363. [Google Scholar] [CrossRef]
- Falorni, A.; Minarelli, V.; Bartoloni, E.; Alunno, A.; Gerli, R. Diagnosis and classification of autoimmune hypophysitis. Autoimmun. Rev. 2014, 13, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Force, B.K.; Vogel, T.P.; Nguyen, D.M.; Heck, K.A.; Sebastian, S.; Takashima, M.; Yoshor, D.; Samson, S.L. A remarkable response of granulomatous hypophysitis to infliximab in a patient with a background of Crohn’s disease—A case report. Front. Endocrinol. 2020, 11, 350. [Google Scholar] [CrossRef]
- Catala Bauset, M.; Gilsanz Peral, A.; Girbes Borras, J.; Zugasti Murillo, A.; Moreno Esteban, B.; Halperin Rabinovich, I.; Obiols Alfonso, G.; Pico Alfonso, A.; Del Pozo Pico, C.; Soto Moreno, A.; et al. Clinical practice guideline for the diagnosis and treatment of hypophysitis. Endocrinol. Nutr. 2008, 55, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.; Kim, H.; Hill, T.; Lee, M.; Orillac, C.; Mogar, N.; Pacione, D.; Agrawal, N. Preoperative differentiation of hypophysitis and pituitary adenomas using a novel clinicoradiologic scoring system. Pituitary 2022, 25, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Yoo, H.J.; Park, S.W.; Choi, M.G. A case of cystic lymphocytic hypophysitis with cacosmia and hypopituitarism. Endocr. J. 2004, 51, 375–380. [Google Scholar] [CrossRef] [Green Version]
- Joshi, M.; Gunawardena, S.; Goenka, A.; Ey, E.; Kumar, G. Post COVID-19 lymphocytic hypophysitis: A rare presentation. Child. Neurol. Open. 2022, 9, 2329048X221103051. [Google Scholar] [CrossRef]
- de Vries, F.; van Furth, W.R.; Biermasz, N.R.; Pereira, A.M. Hypophysitis: A comprehensive overview. Presse Med. 2021, 50, 104076. [Google Scholar] [CrossRef]
- Cemeroglu, A.P.; Blaivas, M.; Muraszko, K.M.; Robertson, P.L.; Vazquez, D.M. Lymphocytic hypophysitis presenting with diabetes insipidus in a 14-year-old girl: Case report and review of the literature. Eur. J. Pediatr. 1997, 156, 684–688. [Google Scholar] [CrossRef]
- Gutenberg, A.; Larsen, J.; Lupi, I.; Rohde, V.; Caturegli, P. A radiologic score to distinguish autoimmune hypophysitis from nonsecreting pituitary adenoma preoperatively. AJNR Am. J. Neuroradiol. 2009, 30, 1766–1772. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Wu, H.; Bao, X.; Wang, R. Lymphocytic hypophysitis secondary to ruptured Rathke cleft cyst: Case report and literature review. World Neurosurg. 2018, 114, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Hanna, B.; Li, Y.M.; Beutler, T.; Goyal, P.; Hall, W.A. Xanthomatous hypophysitis. J. Clin. Neurosci. 2015, 22, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Gezer, E.; Cabuk, B.; Bayrak, B.Y.; Canturk, Z.; Cetinarslan, B.; Selek, A.; Sozen, M.; Koksalan, D.; Ceylan, S. Xanthomatous hypophysitis secondary to a ruptured Rathke’s cleft cyst: A case report. Brain Tumor Res. Treat. 2022, 10, 48–54. [Google Scholar] [CrossRef] [PubMed]
- DeCou, S.; Recinos, P.F.; Prayson, R.A.; Karakasis, C.; Haider, A.; Patel, N. Successful immunomodulatory treatment for recurrent xanthogranulomatous hypophysitis in an adolescent: Illustrative case. J. Neurosurg. Case Lessons 2022, 4, CASE22191. [Google Scholar] [CrossRef]
- Balti, E.; Verhaeghe, S.; Kruse, V.; Roels, S.; Coremans, P. Exploring a new entity of single-agent pembrolizumab-associated hypophysitis. Cureus 2022, 14, e27763. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhu, C.Y.; Lin, J.; Chen, W.S.; Wang, Y.J.; Fu, H.Y.; Zhao, Q. Hypophysitis induced by anti-programmed cell death protein 1 immunotherapy in non-small cell lung cancer: Three case reports. World J. Clin. Cases 2022, 10, 11049–11058. [Google Scholar] [CrossRef]
- Bui, A.N.; Tyan, K.; Giobbie-Hurder, A.; Klein, I.A.; Manos, M.P.; Zubiri, L.; Reynolds, K.; Grover, S.; Weinhouse, G.L.; Ott, P.A.; et al. Impact of COVID-19 on patients with cancer receiving immune checkpoint inhibitors. J. Immunother. Precis. Oncol. 2021, 4, 35–44. [Google Scholar] [CrossRef]
- Levy, M.; Abeillon, J.; Dalle, S.; Assaad, S.; Borson-Chazot, F.; Disse, E.; Raverot, G.; Cugnet-Anceau, C. Anti-PD1 and anti-PDL1-induced hypophysitis: A cohort study of 17 patients with longitudinal follow-up. J. Clin. Med. 2020, 9, 3280. [Google Scholar] [CrossRef]
- Han, X.; Meng, M.; Zhang, T.; Wang, J.; Huang, G.; Ni, Y.; Li, W.; Dai, J.; Yang, X.; Ye, X. Hypophysitis: A rare but noteworthy immune-related adverse event secondary to camrelizumab therapy. J. Cancer Res. Ther. 2022, 18, 1440–1443. [Google Scholar] [CrossRef]
- Kotwal, A.; Rouleau, S.G.; Dasari, S.; Kottschade, L.; Ryder, M.; Kudva, Y.C.; Markovic, S.; Erickson, D. Immune checkpoint inhibitor-induced hypophysitis: Lessons learnt from a large cancer cohort. J. Investig. Med. 2022, 70, 939–946. [Google Scholar] [CrossRef]
- Geslot, A.; Chanson, P.; Caron, P. Covid-19, the thyroid and the pituitary-The real state of play. Ann. Endocrinol. 2022, 83, 103–108. [Google Scholar] [CrossRef]
- Ankireddypalli, A.R.; Chow, L.S.; Radulescu, A.; Kawakami, Y.; Araki, T. A case of hypophysitis associated with SARS-CoV-2 vaccination. AACE Clin. Case Rep. 2022, 8, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Gorbova, N.Y.; Vladimirova, V.P.; Rozhinskaya, L.Y.; Belaya, Z.Y. Hypophysitis and reversible hypopituitarism developed after COVID-19 infection-a clinical case report. Probl. Endokrinol. 2022, 68, 50–56. [Google Scholar] [CrossRef]
- Nonglait, P.L.; Naik, R.; Raizada, N. Hypophysitis after COVID-19 infection. Indian. J. Endocrinol. Metab. 2021, 25, 255–256. [Google Scholar] [CrossRef] [PubMed]
- Bando, H.; Kanie, K.; Takahashi, Y. Paraneoplastic autoimmune hypophysitis: An emerging concept. Best. Pract. Res. Clin. Endocrinol. Metab. 2022, 36, 101601. [Google Scholar] [CrossRef]
- Yamamoto, M.; Iguchi, G.; Bando, H.; Kanie, K.; Hidaka-Takeno, R.; Fukuoka, H.; Takahashi, Y. Autoimmune pituitary disease: New concepts with clinical implications. Endocr. Rev. 2020, 41, 261–272. [Google Scholar] [CrossRef]
- Amereller, F.; Kuppers, A.M.; Schilbach, K.; Schopohl, J.; Stormann, S. Clinical characteristics of primary hypophysitis—A single-centre series of 60 cases. Exp. Clin. Endocrinol. Diabetes 2021, 129, 234–240. [Google Scholar] [CrossRef]
- Perosevic, M.; Jones, P.S.; Tritos, N.A. Magnetic resonance imaging of the hypothalamo-pituitary region. Handb. Clin. Neurol. 2021, 179, 95–112. [Google Scholar] [CrossRef]
- Kurosaki, M.; Sakamoto, M.; Kambe, A.; Ogura, T. Up-to-date magnetic resonance imaging findings for the diagnosis of hypothalamic and pituitary tumors. Yonago Acta Med. 2021, 64, 155–161. [Google Scholar] [CrossRef]
- Gosangi, B.; McIntosh, L.; Keraliya, A.; Irugu, D.V.K.; Baheti, A.; Khandelwal, A.; Thomas, R.; Braschi-Amirfarzan, M. Imaging features of toxicities associated with immune checkpoint inhibitors. Eur. J. Radiol. Open. 2022, 9, 100434. [Google Scholar] [CrossRef]
- Lv, K.; Cao, X.; Geng, D.Y.; Zhang, J. Imaging findings of immunoglobin G4-related hypophysitis: A case report. World J. Clin. Cases 2022, 10, 9440–9446. [Google Scholar] [CrossRef] [PubMed]
- Nada, A.; Bhat, R.; Cousins, J. Magnetic resonance imaging criteria of immune checkpoint inhibitor-induced hypophysitis. Curr. Probl. Cancer 2021, 45, 100644. [Google Scholar] [CrossRef] [PubMed]
- Gamblin, A.S.; Twitchell, S.; Kundu, B.; Couldwell, W.T.; Menacho, S.T. Pembrolizumab-induced hypophysitis with acute progression of Rathke’s cyst. Clin. Exp. Neuroimmunol. 2021, 12, 276–280. [Google Scholar]
- Nikouline, A.; Carr, D. Postpartum headache: A broader differential. Am. J. Emerg. Med. 2021, 39, 258.e5–258.e6. [Google Scholar] [CrossRef]
- Amereller, F.; Deutschbein, T.; Joshi, M.; Schopohl, J.; Schilbach, K.; Detomas, M.; Duffy, L.; Carroll, P.; Papa, S.; Stormann, S. Differences between immunotherapy-induced and primary hypophysitis—A multicenter retrospective study. Pituitary 2022, 25, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Krishnappa, B.; Shah, R.; Memon, S.S.; Diwaker, C.; Lila, A.R.; Patil, V.A.; Shah, N.S.; Bandgar, T.R. Glucocorticoid therapy as first-line treatment in primary hypophysitis: A systematic review and individual patient data meta-analysis. Endocr. Connect. 2023, 12, e220311. [Google Scholar] [CrossRef] [PubMed]
- Kruse, M.; Olesen, T.B.; Markovic, L.; Glintborg, D.; Andersen, M.S. Recurrent autoimmune hypophysitis treated with rituximab: A case report. J. Med. Case Rep. 2021, 15, 591. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Subtype | Characteristics |
|---|---|
| Lymphocytic |
|
| Granulomatous |
|
| Xanthomatous |
|
| Ig-G4-related disease |
|
| Necrotizing |
|
| Subtypes | Characteristic |
|---|---|
| ICI-induced |
|
| COVID-19 |
|
| Paraneoplastic |
|
| Autoimmune |
|
| Granulomas and infection |
|
| Features | Adenoma | Hypophysitis |
|---|---|---|
| Clinical Symptoms |
|
|
| MRI |
|
|
| Management |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rawanduzy, C.A.; Winkler-Schwartz, A.; Couldwell, W.T. Hypophysitis: Defining Histopathologic Variants and a Review of Emerging Clinical Causative Entities. Int. J. Mol. Sci. 2023, 24, 5917. https://doi.org/10.3390/ijms24065917
Rawanduzy CA, Winkler-Schwartz A, Couldwell WT. Hypophysitis: Defining Histopathologic Variants and a Review of Emerging Clinical Causative Entities. International Journal of Molecular Sciences. 2023; 24(6):5917. https://doi.org/10.3390/ijms24065917
Chicago/Turabian StyleRawanduzy, Cameron A., Alexander Winkler-Schwartz, and William T. Couldwell. 2023. "Hypophysitis: Defining Histopathologic Variants and a Review of Emerging Clinical Causative Entities" International Journal of Molecular Sciences 24, no. 6: 5917. https://doi.org/10.3390/ijms24065917