
General Trends of the Camelidae Antibody VHHs Domain Dynamics
 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Dataset Description
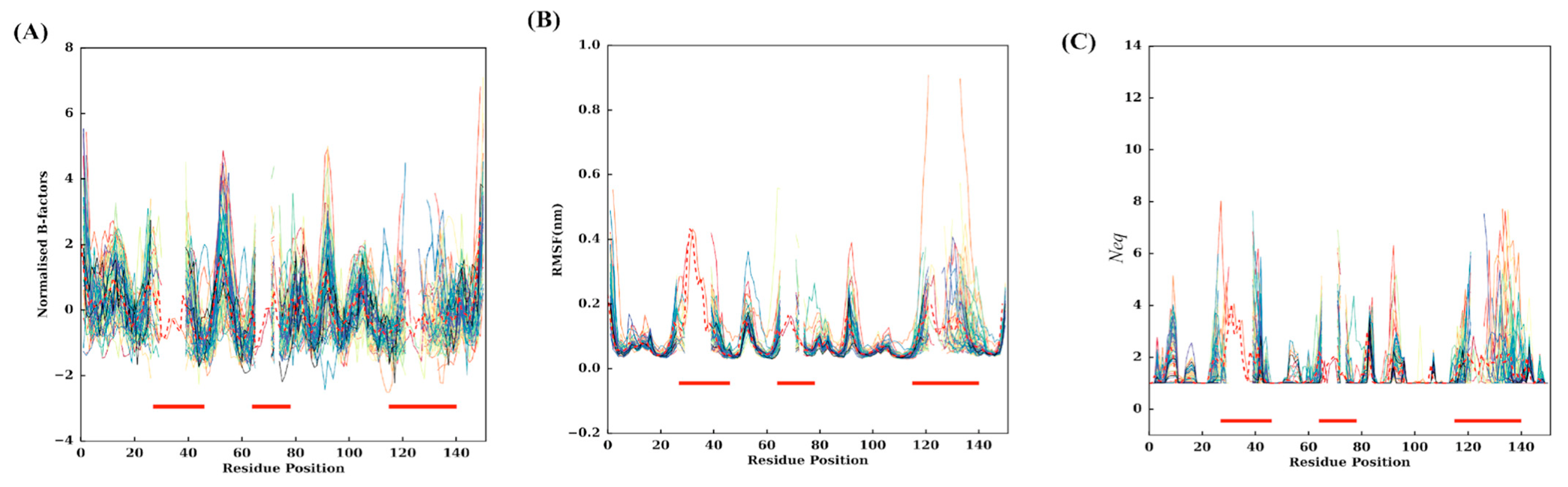
2.2. Assessment of Flexibility
2.3. Flexibility at Each Residue Position
2.4. Clustering of VHH Trajectories
2.5. Backbone Conformational Changes in Terms of Protein Blocks
2.6. Regions-Wise Correlation between Amino Acid Sequence and PB Sequences
2.6.1. Framework Region 1
2.6.2. Framework Region 2
2.6.3. Framework Region 3
2.6.4. Framework Region 4
2.6.5. Complementary Determining Regions
3. Discussion
4. Materials and Methods
4.1. Protein Structure Databank
4.2. Molecular Dynamics
4.3. Molecular Dynamics Analysis
4.4. Protein Structure and Trajectory Visualisation
4.5. Metric Normalisation
4.6. Hierarchical Clustering of VHH Dynamics
4.7. Scripting
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scully, M.; Cataland, S.R.; Peyvandi, F.; Coppo, P.; Knöbl, P.; Kremer Hovinga, J.A.; Metjian, A.; de la Rubia, J.; Pavenski, K.; Callewaert, F.; et al. Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N. Engl. J. Med. 2019, 380, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Jovčevska, I.; Muyldermans, S. The therapeutic potential of nanobodies. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2020, 34, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senolt, L. Emerging therapies in rheumatoid arthritis: Focus on monoclonal antibodies. F1000Research 2019, 8, F1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, J.; Le Bas, A.; Ruza, R.R.; Duyvesteyn, H.M.E.; Mikolajek, H.; Malinauskas, T.; Tan, T.K.; Rijal, P.; Dumoux, M.; Ward, P.N.; et al. Neutralizing nanobodies bind SARS-CoV-2 spike rbd and block interaction with ace2. Nat. Struct. Mol. Biol. 2020, 27, 846–854. [Google Scholar] [CrossRef]
- Wrapp, D.; De Vlieger, D.; Corbett, K.S.; Torres, G.M.; Wang, N.; Van Breedam, W.; Roose, K.; van Schie, L.; Hoffmann, M.; Pöhlmann, S.; et al. Structural basis for potent neutralization of betacoronaviruses by single-domain camelid antibodies. Cell 2020, 181, 1004–1015.e15. [Google Scholar] [CrossRef]
- Chen, F.; Liu, Z.; Jiang, F. Prospects of neutralizing nanobodies against SARS-CoV-2. Front. Immunol. 2021, 12, 690742. [Google Scholar] [CrossRef]
- Güttler, T.; Aksu, M.; Dickmanns, A.; Stegmann, K.M.; Gregor, K.; Rees, R.; Taxer, W.; Rymarenko, O.; Schünemann, J.; Dienemann, C.; et al. Neutralization of SARS-CoV-2 by highly potent, hyperthermostable, and mutation-tolerant nanobodies. EMBO J. 2021, 40, e107985. [Google Scholar] [CrossRef]
- Hanke, L.; Vidakovics Perez, L.; Sheward, D.J.; Das, H.; Schulte, T.; Moliner-Morro, A.; Corcoran, M.; Achour, A.; Karlsson Hedestam, G.B.; Hällberg, B.M.; et al. An alpaca nanobody neutralizes SARS-CoV-2 by blocking receptor interaction. Nat. Commun. 2020, 11, 4420. [Google Scholar] [CrossRef]
- Koenig, P.A.; Das, H.; Liu, H.; Kümmerer, B.M.; Gohr, F.N.; Jenster, L.M.; Schiffelers, L.D.J.; Tesfamariam, Y.M.; Uchima, M.; Wuerth, J.D.; et al. Structure-guided multivalent nanobodies block SARS-CoV-2 infection and suppress mutational escape. Science 2021, 371, eabe6230. [Google Scholar] [CrossRef]
- Schoof, M.; Faust, B.; Saunders, R.A.; Sangwan, S.; Rezelj, V.; Hoppe, N.; Boone, M.; Billesbølle, C.B.; Puchades, C.; Azumaya, C.M.; et al. An ultrapotent synthetic nanobody neutralizes SARS-CoV-2 by stabilizing inactive spike. Science 2020, 370, 1473–1479. [Google Scholar] [CrossRef]
- Wu, Y.; Li, C.; Xia, S.; Tian, X.; Kong, Y.; Wang, Z.; Gu, C.; Zhang, R.; Tu, C.; Xie, Y.; et al. Identification of human single-domain antibodies against SARS-CoV-2. Cell Host Microbe 2020, 27, 891–898.e895. [Google Scholar] [CrossRef]
- Xiang, Y.; Nambulli, S.; Xiao, Z.; Liu, H.; Sang, Z.; Duprex, W.P.; Schneidman-Duhovny, D.; Zhang, C.; Shi, Y. Versatile and multivalent nanobodies efficiently neutralize SARS-CoV-2. Science 2020, 370, 1479–1484. [Google Scholar] [CrossRef]
- Xu, J.; Xu, K.; Jung, S.; Conte, A.; Lieberman, J.; Muecksch, F.; Lorenzi, J.C.C.; Park, S.; Schmidt, F.; Wang, Z.; et al. Nanobodies from camelid mice and llamas neutralize SARS-CoV-2 variants. Nature 2021, 595, 278–282. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Agajanian, S.; Oztas, D.Y.; Gupta, G. Atomistic simulations and in silico mutational profiling of protein stability and binding in the SARS-CoV-2 spike protein complexes with nanobodies: Molecular determinants of mutational escape mechanisms. ACS Omega 2021, 6, 26354–26371. [Google Scholar] [CrossRef]
- Chothia, C.; Lesk, A.M. Canonical structures for the hypervariable regions of immunoglobulins. J. Mol. Biol. 1987, 196, 901–917. [Google Scholar] [CrossRef]
- Zuo, J.; Li, J.; Zhang, R.; Xu, L.; Chen, H.; Jia, X.; Su, Z.; Zhao, L.; Huang, X.; Xie, W. Institute collection and analysis of nanobodies (ican): A comprehensive database and analysis platform for nanobodies. BMC Genom. 2017, 18, 797. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Deszyński, P.; Młokosiewicz, J.; Volanakis, A.; Jaszczyszyn, I.; Castellana, N.; Bonissone, S.; Ganesan, R.; Krawczyk, K. Indi-integrated nanobody database for immunoinformatics. Nucleic Acids Res. 2022, 50, D1273–D1281. [Google Scholar] [CrossRef]
- Noël, F.; Malpertuy, A.; de Brevern, A.G. Global analysis of vhhs framework regions with a structural alphabet. Biochimie 2016, 131, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Melarkode Vattekatte, A.; Shinada, N.K.; Narwani, T.J.; Noël, F.; Bertrand, O.; Meyniel, J.P.; Malpertuy, A.; Gelly, J.C.; Cadet, F.; de Brevern, A.G. Discrete analysis of camelid variable domains: Sequences, structures, and in-silico structure prediction. PeerJ 2020, 8, e8408. [Google Scholar] [CrossRef]
- Melarkode Vattekatte, A.; Cadet, F.; Gelly, J.C.; de Brevern, A.G. Insights into comparative modeling of v(h)h domains. Int. J. Mol. Sci. 2021, 22, 9771. [Google Scholar]
- Wang, Y.T.; Liao, J.M.; Chen, C.L.; Su, Z.Y.; Chen, C.H.; Hu, J.J. Potential of mean force for human lysozyme–camelid vhh hl6 antibody interaction studies. Chem. Phys. Lett 2008, 455, 284–288. [Google Scholar] [CrossRef]
- Su, Z.Y.; Wang, Y.T. A molecular dynamics simulation of the human lysozyme—Camelid vhh hl6 antibody system. Int. J. Mol. Sci. 2009, 10, 1719–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velez-Vega, C.; Fenwick, M.K.; Escobedo, F.A. Simulated mutagenesis of the hypervariable loops of a llama vhh domain for the recovery of canonical conformations. J. Phys. Chem. B 2009, 113, 1785–1795. [Google Scholar] [CrossRef]
- Soler, M.A.; de Marco, A.; Fortuna, S. Molecular dynamics simulations and docking enable to explore the biophysical factors controlling the yields of engineered nanobodies. Sci. Rep. 2016, 6, 34869. [Google Scholar] [CrossRef] [Green Version]
- Mohseni, A.; Molakarimi, M.; Taghdir, M.; Sajedi, R.H.; Hasannia, S. Exploring single-domain antibody thermostability by molecular dynamics simulation. J. Biomol. Struct. Dyn. 2019, 37, 3686–3696. [Google Scholar] [CrossRef]
- Bekker, G.J.; Ma, B.; Kamiya, N. Thermal stability of single-domain antibodies estimated by molecular dynamics simulations. Protein Sci. Publ. Protein Soc. 2019, 28, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Zabetakis, D.; Shriver-Lake, L.C.; Olson, M.A.; Goldman, E.R.; Anderson, G.P. Experimental evaluation of single-domain antibodies predicted by molecular dynamics simulations to have elevated thermal stability. Protein Sci. Publ. Protein Soc. 2019, 28, 1909–1912. [Google Scholar] [CrossRef]
- Soler, M.A.; Fortuna, S.; de Marco, A.; Laio, A. Binding affinity prediction of nanobody-protein complexes by scoring of molecular dynamics trajectories. Phys. Chem. Chem. Phys. 2018, 20, 3438–3444. [Google Scholar] [CrossRef]
- Ikeuchi, E.; Kuroda, D.; Nakakido, M.; Murakami, A.; Tsumoto, K. Delicate balance among thermal stability, binding affinity, and conformational space explored by single-domain v(h)h antibodies. Sci. Rep. 2021, 11, 20624. [Google Scholar] [CrossRef]
- Fernández-Quintero, M.L.; DeRose, E.F.; Gabel, S.A.; Mueller, G.A.; Liedl, K.R. Nanobody paratope ensembles in solution characterized by md simulations and nmr. Int. J. Mol. Sci. 2022, 23, 5419. [Google Scholar] [CrossRef]
- Gray, E.R.; Brookes, J.C.; Caillat, C.; Turbé, V.; Webb, B.L.J.; Granger, L.A.; Miller, B.S.; McCoy, L.E.; El Khattabi, M.; Verrips, C.T.; et al. Unravelling the molecular basis of high affinity nanobodies against hiv p24: In vitro functional, structural, and in silico insights. ACS Infect. Dis. 2017, 3, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Kumachi, S.; Matsunaga, Y.; Sato, M.; Wakabayashi-Nakao, K.; Masaki, H.; Yonehara, R.; Motohashi, M.; Nemoto, N.; Tsuchiya, M. Construction of a humanized artificial vhh library reproducing structural features of camelid vhhs for therapeutics. Antibodies 2022, 11, 10. [Google Scholar] [CrossRef]
- Lesne, J.; Chang, H.J.; De Visch, A.; Paloni, M.; Barthe, P.; Guichou, J.F.; Mayonove, P.; Barducci, A.; Labesse, G.; Bonnet, J.; et al. Structural basis for chemically-induced homodimerization of a single domain antibody. Sci. Rep. 2019, 9, 1840. [Google Scholar] [CrossRef] [Green Version]
- Offmann, B.; Tyagi, M.; de Brevern, A.G. Local protein structures. Curr. Bioinform. 2007, 3, 165–202. [Google Scholar] [CrossRef] [Green Version]
- Craveur, P.; Joseph, A.P.; Esque, J.; Narwani, T.J.; Noël, F.; Shinada, N.; Goguet, M.; Leonard, S.; Poulain, P.; Bertrand, O.; et al. Protein flexibility in the light of structural alphabets. Front. Mol. Biosci. 2015, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Melarkode Vattekatte, A.; Narwani, T.J.; Floch, A.; Maljković, M.; Bisoo, S.; Shinada, N.K.; Kranjc, A.; Gelly, J.C.; Srinivasan, N.; Mitić, N.; et al. A structural entropy index to analyse local conformations in intrinsically disordered proteins. J. Struct. Biol. 2020, 210, 107464. [Google Scholar]
- De Brevern, A.G. Analysis of protein disorder predictions in the light of a protein structural alphabet. Biomolecules 2020, 10, 1080. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Mitchell, L.S.; Colwell, L.J. Comparative analysis of nanobody sequence and structure data. Proteins 2018, 86, 697–706. [Google Scholar] [CrossRef]
- Kelow, S.P.; Adolf-Bryfogle, J.; Dunbrack, R.L. Hiding in plain sight: Structure and sequence analysis reveals the importance of the antibody de loop for antibody-antigen binding. mAbs 2020, 12, 1840005. [Google Scholar] [CrossRef] [PubMed]
- North, B.; Lehmann, A.; Dunbrack, R.L., Jr. A new clustering of antibody cdr loop conformations. J. Mol. Biol. 2011, 406, 228–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Brevern, A.G.; Etchebest, C.; Hazout, S. Bayesian probabilistic approach for predicting backbone structures in terms of protein blocks. Proteins 2000, 41, 271–287. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.P.; Agarwal, G.; Mahajan, S.; Gelly, J.C.; Swapna, L.S.; Offmann, B.; Cadet, F.; Bornot, A.; Tyagi, M.; Valadié, H.; et al. A short survey on protein blocks. Biophys. Rev. 2010, 2, 137–147. [Google Scholar] [CrossRef]
- Bornot, A.; Etchebest, C.; de Brevern, A.G. A new prediction strategy for long local protein structures using an original description. Proteins 2009, 76, 570–587. [Google Scholar] [CrossRef] [Green Version]
- Narwani, T.J.; Craveur, P.; Shinada, N.K.; Floch, A.; Santuz, H.; Vattekatte, A.M.; Srinivasan, N.; Rebehmed, J.; Gelly, J.C.; Etchebest, C.; et al. Discrete analyses of protein dynamics. J. Biomol. Struct. Dyn. 2020, 38, 2988–3002. [Google Scholar] [CrossRef]
- Goguet, M.; Narwani, T.J.; Petermann, R.; Jallu, V.; de Brevern, A.G. In silico analysis of glanzmann variants of calf-1 domain of α(iib)β(3) integrin revealed dynamic allosteric effect. Sci. Rep. 2017, 7, 8001. [Google Scholar] [CrossRef] [Green Version]
- Anies, S.; Jallu, V.; Diharce, J.; Narwani, T.J.; de Brevern, A.G. Analysis of integrin α(iib) subunit dynamics reveals long-range effects of missense mutations on calf domains. Int. J. Mol. Sci. 2022, 23, 858. [Google Scholar] [CrossRef]
- Wesolowski, J.; Alzogaray, V.; Reyelt, J.; Unger, M.; Juarez, K.; Urrutia, M.; Cauerhff, A.; Danquah, W.; Rissiek, B.; Scheuplein, F.; et al. Single domain antibodies: Promising experimental and therapeutic tools in infection and immunity. Med. Microbiol. Immunol. 2009, 198, 157–174. [Google Scholar] [CrossRef] [Green Version]
- Bornot, A.; Etchebest, C.; de Brevern, A.G. Predicting protein flexibility through the prediction of local structures. Proteins 2011, 79, 839–852. [Google Scholar] [CrossRef] [Green Version]
- De Brevern, A.G.; Bornot, A.; Craveur, P.; Etchebest, C.; Gelly, J.C. Predyflexy: Flexibility and local structure prediction from sequence. Nucleic Acids Res. 2012, 40, W317–W322. [Google Scholar] [CrossRef] [Green Version]
- Narwani, T.J.; Etchebest, C.; Craveur, P.; Léonard, S.; Rebehmed, J.; Srinivasan, N.; Bornot, A.; Gelly, J.C.; de Brevern, A.G. In silico prediction of protein flexibility with local structure approach. Biochimie 2019, 165, 150–155. [Google Scholar] [CrossRef] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. Gromacs: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Rakhshani, H.; Dehghanian, E.; Rahati, A. Enhanced gromacs: Toward a better numerical simulation framework. J. Mol. Model. 2019, 25, 355. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the amber ff99sb protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Barnoud, J.; Santuz, H.; Craveur, P.; Joseph, A.P.; Jallu, V.; de Brevern, A.G.; Poulain, P. Pbxplore: A tool to analyze local protein structure and deformability with protein blocks. PeerJ 2017, 5, e4013. [Google Scholar] [CrossRef] [Green Version]
- Faure, G.; Joseph, A.P.; Craveur, P.; Narwani, T.J.; Srinivasan, N.; Gelly, J.C.; Rebehmed, J.; de Brevern, A.G. Ipbavizu: A pymol plugin for an efficient 3d protein structure superimposition approach. Source Code Biol. Med. 2019, 14, 5. [Google Scholar] [CrossRef]
- Joseph, A.P.; Srinivasan, N.; de Brevern, A.G. Improvement of protein structure comparison using a structural alphabet. Biochimie 2011, 93, 1434–1445. [Google Scholar] [CrossRef] [Green Version]
- Léonard, S.; Joseph, A.P.; Srinivasan, N.; Gelly, J.C.; de Brevern, A.G. Mulpba: An efficient multiple protein structure alignment method based on a structural alphabet. J. Biomol. Struct. Dyn. 2014, 32, 661–668. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol; Delano Scientific: San Carlos, CA, USA, 2002; 700p. [Google Scholar]
- Delano, W.L. The Pymol Molecular Graphics System on World Wide Web. 2013. Available online: http://www.pymol.org (accessed on 8 January 2023).
- Humphrey, W.; Dalke, A.; Schulten, K. Vmd: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 27–38. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.K.; Radivojac, P.; Obradovic, Z.; Dunker, A.K.; Zhu, G. Improved amino acid flexibility parameters. Protein Sci. Publ. Protein Soc. 2003, 12, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celton, M.; Malpertuy, A.; Lelandais, G.; de Brevern, A.G. Comparative analysis of missing value imputation methods to improve clustering and interpretation of microarray experiments. BMC Genom. 2010, 11, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Brevern, A.G.; Hazout, S.; Malpertuy, A. Influence of microarrays experiments missing values on the stability of gene groups by hierarchical clustering. BMC Bioinform. 2004, 5, 114. [Google Scholar] [CrossRef] [Green Version]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with numpy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- Team, R.D.C. R: A Language and Environment for Statistical Computing; R Foundation for Statisical Computing: Vienna, Austria, 2011. [Google Scholar]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vattekatte, A.M.; Diharce, J.; Rebehmed, J.; Cadet, F.; Gardebien, F.; Etchebest, C.; de Brevern, A.G. General Trends of the Camelidae Antibody VHHs Domain Dynamics. Int. J. Mol. Sci. 2023, 24, 4511. https://doi.org/10.3390/ijms24054511
Vattekatte AM, Diharce J, Rebehmed J, Cadet F, Gardebien F, Etchebest C, de Brevern AG. General Trends of the Camelidae Antibody VHHs Domain Dynamics. International Journal of Molecular Sciences. 2023; 24(5):4511. https://doi.org/10.3390/ijms24054511
Chicago/Turabian StyleVattekatte, Akhila Melarkode, Julien Diharce, Joseph Rebehmed, Frédéric Cadet, Fabrice Gardebien, Catherine Etchebest, and Alexandre G. de Brevern. 2023. "General Trends of the Camelidae Antibody VHHs Domain Dynamics" International Journal of Molecular Sciences 24, no. 5: 4511. https://doi.org/10.3390/ijms24054511