
The Interleukin-11/IL-11 Receptor Promotes Glioblastoma Survival and Invasion under Glucose-Starved Conditions through Enhanced Glutaminolysis
 , ,
, ,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
2.1. IL-11/IL-11Rα Expression Is Elevated in Glioblastoma Tumors and Primary Cell Lines
2.2. IL-11Rα Expression Promotes Cancer Cell Survival in Glucose-Starved Conditions
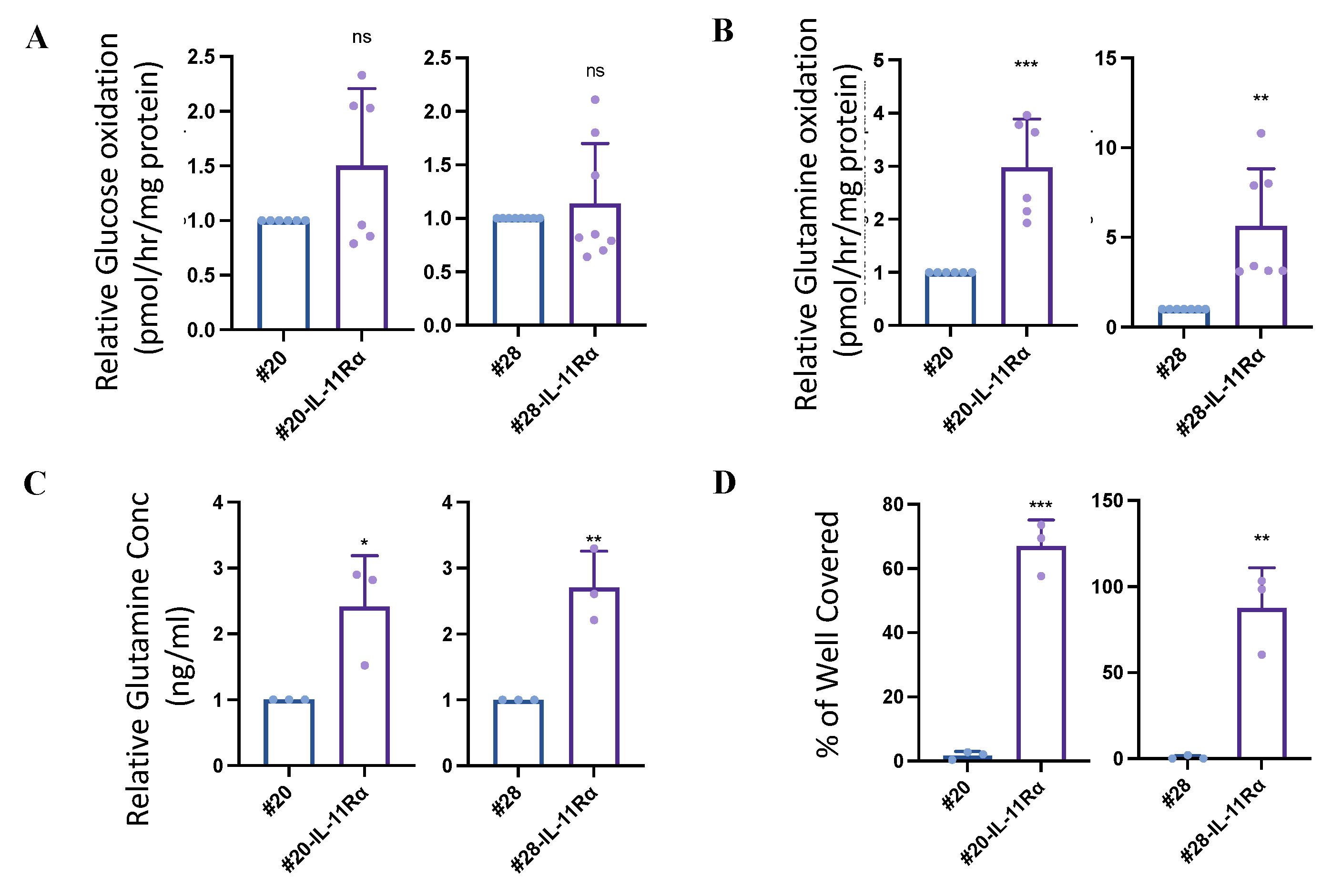
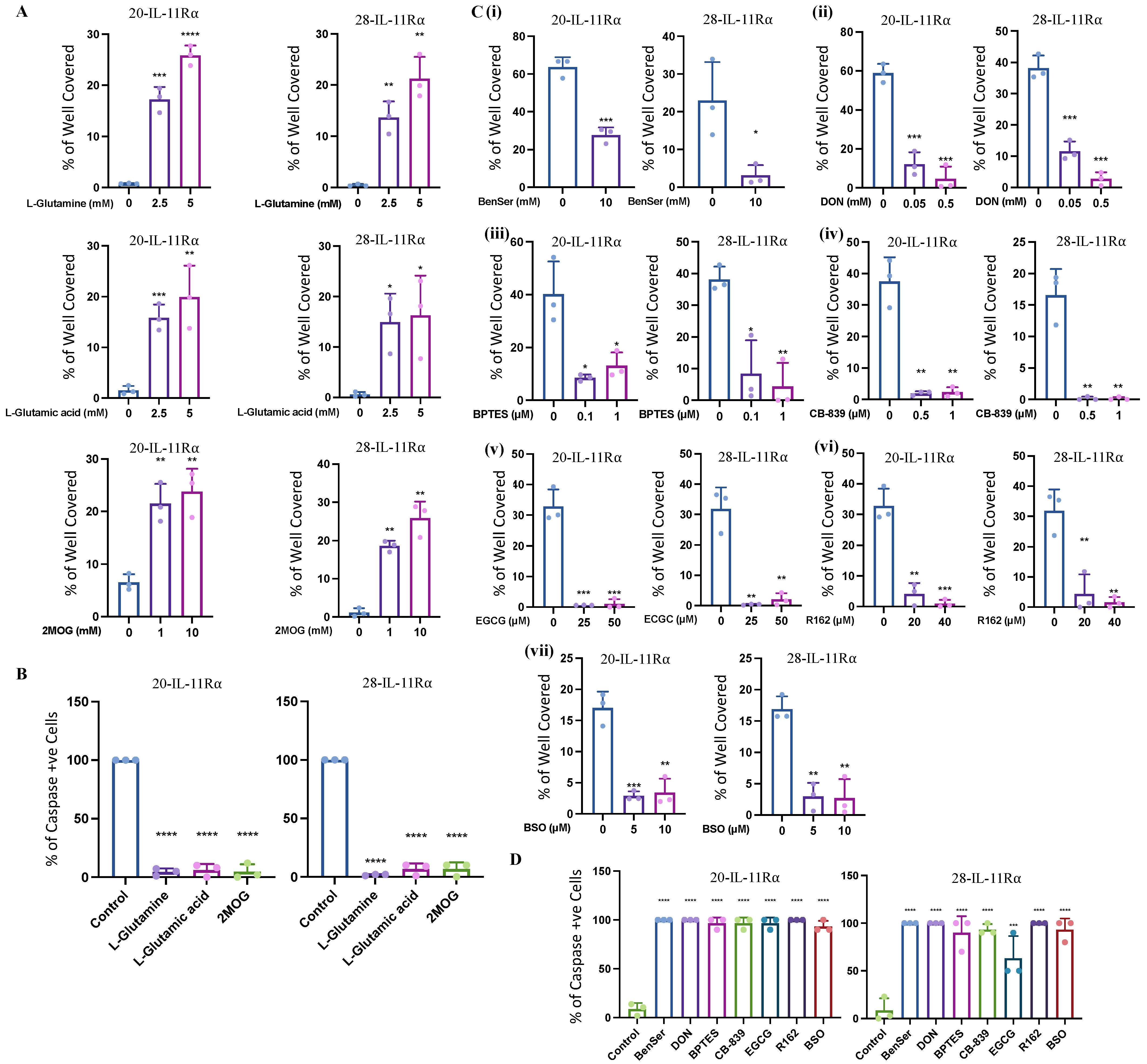
2.3. IL-11Rα Expression Promotes Survival through Glutaminolysis
2.4. IL-11Rα-Driven Migration and Invasion Is Glutamine-Dependent but Glucose-Independent
2.5. IL-11Rα Expression Correlates with Glutamine–Glutamate-Related Genes in Glioblastoma Patient Samples
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. Inhibitors and Reagents
4.3. Cell Culture
4.4. Cell Viability Assays
4.5. Immunohistochemistry
4.6. RNA Extraction and RT-PCR
4.7. Scratch/Wound-Healing Assay
4.8. Transwell Migration and Invasion Assays
4.9. Apoptosis Assay
4.10. Glucose and Glutamine Oxidation Assay
4.11. Glutamate-Detection Assay
4.12. OncoLnc (TCGA)
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef]
- Natarajan, S.K.; Venneti, S. Glutamine Metabolism in Brain Tumors. Cancers 2019, 11, 1628. [Google Scholar] [CrossRef]
- Marin-Valencia, I.; Yang, C.; Mashimo, T.; Cho, S.; Baek, H.; Yang, X.L.; Rajagopalan, K.N.; Maddie, M.; Vemireddy, V.; Zhao, Z.; et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012, 15, 827–837. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Mukherjee, P. Targeting energy metabolism in brain cancer: Review and hypothesis. Nutr. Metab. 2005, 2, 30. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Wu, Q.; Hitomi, M.; Rahim, N.; Kim, Y.; Sloan, A.E.; Weil, R.J.; Nakano, I.; Sarkaria, J.N.; Stringer, B.W.; et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat. Neurosci. 2013, 16, 1373–1382. [Google Scholar] [CrossRef]
- Aftab, S.; Shakoori, A.R. Low glucose availability alters the expression of genes involved in initial adhesion of human glioblastoma cancer cell line SF767. J. Cell. Biochem. 2019, 120, 16824–16839. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Mischel, P.S. Acyl-CoA-Binding Protein Fuels Gliomagenesis. Cell Metab. 2019, 30, 229–230. [Google Scholar] [CrossRef]
- Zaugg, K.; Yao, Y.; Reilly, P.T.; Kannan, K.; Kiarash, R.; Mason, J.; Huang, P.; Sawyer, S.K.; Fuerth, B.; Faubert, B.; et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011, 25, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Azzalin, A.; Brambilla, F.; Arbustini, E.; Basello, K.; Speciani, A.; Mauri, P.; Bezzi, P.; Magrassi, L. A New Pathway Promotes Adaptation of Human Glioblastoma Cells to Glucose Starvation. Cells 2020, 9, 1249. [Google Scholar] [CrossRef]
- Duman, C.; Yaqubi, K.; Hoffmann, A.; Acikgoz, A.A.; Korshunov, A.; Bendszus, M.; Herold-Mende, C.; Liu, H.K.; Alfonso, J. Acyl-CoA-Binding Protein Drives Glioblastoma Tumorigenesis by Sustaining Fatty Acid Oxidation. Cell Metab. 2019, 30, 274–289.e5. [Google Scholar] [CrossRef]
- Yang, C.; Ko, B.; Hensley, C.T.; Jiang, L.; Wasti, A.T.; Kim, J.; Sudderth, J.; Calvaruso, M.A.; Lumata, L.; Mitsche, M.; et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol. Cell 2014, 56, 414–424. [Google Scholar] [CrossRef]
- Yang, C.; Sudderth, J.; Dang, T.; Bachoo, R.M.; McDonald, J.G.; DeBerardinis, R.J. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res. 2009, 69, 7986–7993. [Google Scholar] [CrossRef]
- Chen, L.; Cui, H. Targeting Glutamine Induces Apoptosis: A Cancer Therapy Approach. Int. J. Mol. Sci. 2015, 16, 22830–22855. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef]
- van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef] [PubMed]
- Mates, J.M.; Segura, J.A.; Martin-Rufian, M.; Campos-Sandoval, J.A.; Alonso, F.J.; Marquez, J. Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr. Mol. Med. 2013, 13, 514–534. [Google Scholar] [CrossRef]
- Wang, J.B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.; Dias, S.M.; Dang, C.V.; et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.; Antoniewicz, M.R.; Stephanopoulos, G.; Kelleher, J.K. Quantifying reductive carboxylation flux of glutamine to lipid in a brown adipocyte cell line. J. Biol. Chem. 2008, 283, 20621–20627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, K.; Hartmann, R.; Suwala, A.K.; Rios, D.H.; Kamp, M.A.; Sabel, M.; Steiger, H.J.; Willbold, D.; Sharma, A.; Kahlert, U.D.; et al. Overexpression of Cystine/Glutamate Antiporter xCT Correlates with Nutrient Flexibility and ZEB1 Expression in Highly Clonogenic Glioblastoma Stem-like Cells (GSCs). Cancers 2021, 13, 6001. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed]
- Zuo, H.; Wan, Y. Metabolic Reprogramming in Mitochondria of Myeloid Cells. Cells 2019, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Carow, B.; Rottenberg, M.E. SOCS3, a Major Regulator of Infection and Inflammation. Front. Immunol. 2014, 5, 58. [Google Scholar] [CrossRef]
- Putoczki, T.L.; Thiem, S.; Loving, A.; Busuttil, R.A.; Wilson, N.J.; Ziegler, P.K.; Nguyen, P.M.; Preaudet, A.; Farid, R.; Edwards, K.M.; et al. Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer Cell 2013, 24, 257–271. [Google Scholar] [CrossRef]
- West, A.J.; Tsui, V.; Stylli, S.S.; Nguyen, H.P.T.; Morokoff, A.P.; Kaye, A.H.; Luwor, R.B. The role of interleukin-6-STAT3 signalling in glioblastoma. Oncol. Lett. 2018, 16, 4095–4104. [Google Scholar] [CrossRef] [PubMed]
- Lokau, J.; Agthe, M.; Flynn, C.M.; Garbers, C. Proteolytic control of Interleukin-11 and Interleukin-6 biology. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2105–2117. [Google Scholar] [CrossRef]
- Metcalfe, R.D.; Aizel, K.; Zlatic, C.O.; Nguyen, P.M.; Morton, C.J.; Lio, D.S.; Cheng, H.C.; Dobson, R.C.J.; Parker, M.W.; Gooley, P.R.; et al. The structure of the extracellular domains of human interleukin 11alpha receptor reveals mechanisms of cytokine engagement. J. Biol. Chem. 2020, 295, 8285–8301. [Google Scholar] [CrossRef]
- Dams-Kozlowska, H.; Gryska, K.; Kwiatkowska-Borowczyk, E.; Izycki, D.; Rose-John, S.; Mackiewicz, A. A designer hyper interleukin 11 (H11) is a biologically active cytokine. BMC Biotechnol. 2012, 12, 8. [Google Scholar] [CrossRef]
- Wang, X.; Che, X.; Liu, C.; Fan, Y.; Bai, M.; Hou, K.; Shi, X.; Zhang, X.; Liu, B.; Zheng, C.; et al. Cancer-associated fibroblasts-stimulated interleukin-11 promotes metastasis of gastric cancer cells mediated by upregulation of MUC1. Exp. Cell. Res. 2018, 368, 184–193. [Google Scholar] [CrossRef]
- Wei, J.; Ma, L.; Lai, Y.H.; Zhang, R.; Li, H.; Li, C.; Lin, J. Bazedoxifene as a novel GP130 inhibitor for Colon Cancer therapy. J. Exp. Clin. Cancer Res. 2019, 38, 63. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, C.N.; Chand, A.; Putoczki, T.L.; Ernst, M. Emerging roles for IL-11 signaling in cancer development and progression: Focus on breast cancer. Cytokine Growth Factor Rev. 2015, 26, 489–498. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin. Immunol. 2014, 26, 54–74. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Cao, Y.; Xiao, H.; Li, C.; Lin, J. Bazedoxifene as a Novel GP130 Inhibitor for Pancreatic Cancer Therapy. Mol. Cancer Ther. 2016, 15, 2609–2619. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, S.; Lin, X.; Lu, Y.; Gu, P. MicroRNA-124a inhibits cell proliferation and migration in liver cancer by regulating interleukin-11. Mol. Med. Rep. 2018, 17, 3972–3978. [Google Scholar] [CrossRef]
- Tan, F.H.; Putoczki, T.L.; Lou, J.; Hinde, E.; Hollande, F.; Giraud, J.; Stylli, S.S.; Paradiso, L.; Zhu, H.J.; Sieber, O.M.; et al. Ponatinib Inhibits Multiple Signaling Pathways Involved in STAT3 Signaling and Attenuates Colorectal Tumor Growth. Cancers 2018, 10, 526. [Google Scholar] [CrossRef]
- Thilakasiri, P.; Huynh, J.; Poh, A.R.; Tan, C.W.; Nero, T.L.; Tran, K.; Parslow, A.C.; Afshar-Sterle, S.; Baloyan, D.; Hannan, N.J.; et al. Repurposing the selective estrogen receptor modulator bazedoxifene to suppress gastrointestinal cancer growth. EMBO Mol. Med. 2019, 11, e9539. [Google Scholar] [CrossRef]
- Belanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed]
- Cannino, G.; Ciscato, F.; Masgras, I.; Sanchez-Martin, C.; Rasola, A. Metabolic Plasticity of Tumor Cell Mitochondria. Front. Oncol. 2018, 8, 333. [Google Scholar] [CrossRef] [PubMed]
- Oizel, K.; Chauvin, C.; Oliver, L.; Gratas, C.; Geraldo, F.; Jarry, U.; Scotet, E.; Rabe, M.; Alves-Guerra, M.C.; Teusan, R.; et al. Efficient Mitochondrial Glutamine Targeting Prevails Over Glioblastoma Metabolic Plasticity. Clin. Cancer Res. 2017, 23, 6292–6304. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Sasayama, T.; Irino, Y.; Takata, K.; Nagashima, H.; Satoh, N.; Kyotani, K.; Mizowaki, T.; Imahori, T.; Ejima, Y.; et al. Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J. Clin. Investig. 2015, 125, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Li, X.; Wu, Y.; Zhang, G.; Liu, X.; Li, Y.; Bao, Y.; Yang, W.; Cui, H. EGFR activates GDH1 transcription to promote glutamine metabolism through MEK/ERK/ELK1 pathway in glioblastoma. Oncogene 2020, 39, 2975–2986. [Google Scholar] [CrossRef] [PubMed]
- Plaitakis, A.; Kalef-Ezra, E.; Kotzamani, D.; Zaganas, I.; Spanaki, C. The Glutamate Dehydrogenase Pathway and Its Roles in Cell and Tissue Biology in Health and Disease. Biology 2017, 6, 11. [Google Scholar] [CrossRef]
- Zhang, J.; Fan, J.; Venneti, S.; Cross, J.R.; Takagi, T.; Bhinder, B.; Djaballah, H.; Kanai, M.; Cheng, E.H.; Judkins, A.R.; et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol. Cell 2014, 56, 205–218. [Google Scholar] [CrossRef]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.O.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lin, J.; Feng, S.; Chen, X.; Huang, H.; Wang, C.; Yu, Y.; He, Y.; Han, S.; Zheng, L.; et al. SIRT4 inhibits the proliferation, migration, and invasion abilities of thyroid cancer cells by inhibiting glutamine metabolism. OncoTargets Ther. 2019, 12, 2397–2408. [Google Scholar] [CrossRef]
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014, 10, 728. [Google Scholar] [CrossRef]
- Ahluwalia, M.S.; Patton, C.; Stevens, G.; Tekautz, T.; Angelov, L.; Vogelbaum, M.A.; Weil, R.J.; Chao, S.; Elson, P.; Suh, J.H.; et al. Phase II trial of ritonavir/lopinavir in patients with progressive or recurrent high-grade gliomas. J. Neurooncol. 2011, 102, 317–321. [Google Scholar] [CrossRef]
- Tateishi, K.; Iafrate, A.J.; Ho, Q.; Curry, W.T.; Batchelor, T.T.; Flaherty, K.T.; Onozato, M.L.; Lelic, N.; Sundaram, S.; Cahill, D.P.; et al. Myc-Driven Glycolysis Is a Therapeutic Target in Glioblastoma. Clin. Cancer Res. 2016, 22, 4452–4465. [Google Scholar] [CrossRef]
- Dang, C.V. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.; Najdovska, M.; Grail, D.; Lundgren-May, T.; Buchert, M.; Tye, H.; Matthews, V.B.; Armes, J.; Bhathal, P.S.; Hughes, N.R.; et al. STAT3 and STAT1 mediate IL-11-dependent and inflammation-associated gastric tumorigenesis in gp130 receptor mutant mice. J. Clin. Investig. 2008, 118, 1727–1738. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Ma, Q.; Ding, N.; Luo, F.; Bai, Y.; Kang, F.; Gong, X.; Dong, R.; Dai, J.; Dai, Q.; et al. IL-11 is essential in promoting osteolysis in breast cancer bone metastasis via RANKL-independent activation of osteoclastogenesis. Cell Death Dis. 2019, 10, 353. [Google Scholar] [CrossRef]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef]
- Nobusawa, S.; Watanabe, T.; Kleihues, P.; Ohgaki, H. IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin. Cancer Res. 2009, 15, 6002–6007. [Google Scholar] [CrossRef]
- Liu, F.M.; Gao, Y.F.; Kong, Y.; Guan, Y.; Zhang, J.; Li, S.H.; Ye, D.; Wen, W.; Zuo, C.; Hua, W. The diagnostic value of lower glucose consumption for IDH1 mutated gliomas on FDG-PET. BMC Cancer 2021, 21, 83. [Google Scholar] [CrossRef]
- Guzman, C.; Bagga, M.; Kaur, A.; Westermarck, J.; Abankwa, D. ColonyArea: An ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS ONE 2014, 9, e92444. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| <6 Month Survival; n (%) | 6–18 Month Survival; n (%) | >18 Month Survival; n (%) | |
|---|---|---|---|
| Negative Staining | 5 (29) | 30 (51) | 17 (78) |
| Positive Staining | 12 (71) | 29 (49) | 5 (22) |
| Total Samples | 17 | 59 | 22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stuart, S.F.; Bezawork-Geleta, A.; Areeb, Z.; Gomez, J.; Tsui, V.; Zulkifli, A.; Paradiso, L.; Jones, J.; Nguyen, H.P.T.; Putoczki, T.L.; et al. The Interleukin-11/IL-11 Receptor Promotes Glioblastoma Survival and Invasion under Glucose-Starved Conditions through Enhanced Glutaminolysis. Int. J. Mol. Sci. 2023, 24, 3356. https://doi.org/10.3390/ijms24043356
Stuart SF, Bezawork-Geleta A, Areeb Z, Gomez J, Tsui V, Zulkifli A, Paradiso L, Jones J, Nguyen HPT, Putoczki TL, et al. The Interleukin-11/IL-11 Receptor Promotes Glioblastoma Survival and Invasion under Glucose-Starved Conditions through Enhanced Glutaminolysis. International Journal of Molecular Sciences. 2023; 24(4):3356. https://doi.org/10.3390/ijms24043356
Chicago/Turabian StyleStuart, Sarah F., Ayenachew Bezawork-Geleta, Zammam Areeb, Juliana Gomez, Vanessa Tsui, Ahmad Zulkifli, Lucia Paradiso, Jordan Jones, Hong P. T. Nguyen, Tracy L. Putoczki, and et al. 2023. "The Interleukin-11/IL-11 Receptor Promotes Glioblastoma Survival and Invasion under Glucose-Starved Conditions through Enhanced Glutaminolysis" International Journal of Molecular Sciences 24, no. 4: 3356. https://doi.org/10.3390/ijms24043356