
Localization of Pyranose 2-Oxidase from Kitasatospora aureofaciens: A Step Closer to Elucidate a Biological Role


1
Food Biotechnology Laboratory, Department of Food Science and Technology, BOKU—University of Natural Resources and Life Sciences, Muthgasse 11, 1190 Vienna, Austria
2
Doctoral Programme BioToP—Biomolecular Technology of Proteins, BOKU—University of Natural Resources and Life Sciences, Muthgasse 11, 1190 Vienna, Austria
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2023, 24(3), 1975; https://doi.org/10.3390/ijms24031975
Submission received: 22 December 2022
/
Revised: 11 January 2023
/
Accepted: 16 January 2023
/
Published: 19 January 2023
(This article belongs to the Special Issue Biotechnological Applications of Oxidoreductases)
Abstract
:Lignin degradation in fungal systems is well characterized. Recently, a potential for lignin depolymerization and modification employing similar enzymatic activities by bacteria is increasingly recognized. The presence of genes annotated as peroxidases in Actinobacteria genomes suggests that these bacteria should contain auxiliary enzymes such as flavin-dependent carbohydrate oxidoreductases. The only auxiliary activity subfamily with significantly similar representatives in bacteria is pyranose oxidase (POx). A biological role of providing H2O2 for peroxidase activation and reduction of radical degradation products suggests an extracellular localization, which has not been established. Analysis of the genomic locus of POX from Kitasatospora aureofaciens (KaPOx), which is similar to fungal POx, revealed a start codon upstream of the originally annotated one, and the additional sequence was considered a putative Tat-signal peptide by computational analysis. We expressed KaPOx including this additional upstream sequence as well as fusion constructs consisting of the additional sequence, the KaPOx mature domain and the fluorescent protein mRFP1 in Streptomyces lividans. The putative signal peptide facilitated secretion of KaPOx and the fusion protein, suggesting a natural extracellular localization and supporting a potential role in providing H2O2 and reducing radical compounds derived from lignin degradation.
1. Introduction
Lignocellulose, a composite of the polymers lignin, cellulose, hemicellulose and pectin, provides structural integrity and resistance to pathogens and herbivores to plants. The paracristallinity of cellulose, the complexity of the hemicellulose matrix and the linkage to lignin are major barriers against the enzymatic degradation of lignocellulose [1]. The lignin fraction is a heterogenous alkyl-aromatic polymer formed from three aromatic alcohols with different degrees of methoxylation. Lignification happens through monomer and polymer crosslinking via radicals produced by oxidases and is characterized by a large number of different interunit linkages, resulting in a remarkable recalcitrance to degradation [1,2].In nature, degradation of lignocellulose is feasible at physiological conditions by a number of organisms. The process of lignin degradation is mostly described in white-rot and brown-rot fungi [3,4], which mineralize lignin through a cocktail of oxidative enzymes, namely polyphenol oxidases (laccases) and peroxidases (lignin, manganese and versatile peroxidases), as well as auxiliary enzymes such as H2O2-generating oxidases. These peroxide-providing enzymes can belong to the Glucose-Methanol-Choline (GMC) oxidoreductase family of flavoenzymes (Auxiliary Activities Family 3), such as aryl alcohol oxidases, pyranose oxidases or cellobiose dehydrogenases, or to the copper radical oxidases (Auxiliary Activities Family 5), such as glyoxal oxidase [2]. Additionally, peroxide providing enzymes may be involved in the oxidative depolymerization of cellulose and hemicellulose through lytic polysaccharide monoxygenases (LPMOs), for which hydrogen peroxide is also an essential co-substrate [5]. Most flavoprotein oxidases, such as aryl alcohol oxidases and pyranose oxidases, also have a significant dehydrogenase activity using (substituted) quinones as electron acceptors, often with a higher efficiency than molecular oxygen [6,7,8]. This dehydrogenase activity may also be involved in lignin depolymerization, as has been shown in vitro for combinations of fungal laccase and pyranose oxidase [9] as well as lignin peroxidase and pyranose oxidase [6,10], where addition of pyranose oxidase increased the degree of depolymerization markedly, presumably by reducing the resulting lignin breakdown products and preventing their immediate repolymerization.
While the enzymatic system(s) for depolymerization of lignin in fungi are well investigated, this is not the case for bacteria, although a potential for lignin depolymerization and modification using similar enzymatic activities such as laccases and peroxidases is increasingly recognized [1,11,12,13]. To date, there are only few reports on flavoprotein oxidoreductases that can be considered Auxiliary Activities for lignocellulose degradation. Pyranose oxidases were characterized from Pseudarthrobacter siccitolerans [14], Kitasatospora aureofaciens [6] and Streptomyces canus [15]. The sequence of POx derived from K. aureofaciens (KaPOx) is closely related to fungal POx [16] and shows both oxidase and dehydrogenase activity with comparable substrate preferences to fungal enzymes. The oxidase activity of POx is suitable for providing H2O2 for peroxidase activation for lignin depolymerization, while the dehydrogenase activity can play a role in preventing re-polymerization of lignin-derived radicals as well as protecting the cells from damage by those radicals. Both putative biological roles are only plausible if POx is located extracellularly, otherwise the produced hydrogen peroxide would have to be transported out of the cells for activation of peroxidases, and free radicals derived from lignin depolymerization would have to be imported into the cells. Both bacterial genes were identified by sequence similarities and were expressed in E. coli for characterization, but their natural subcellular localization has not been established. It should be noted that a copper radical oxidase with catalytic similarities to glyoxal oxidase (for which similar biological functions are discussed) has been described as a secretory enzyme in Streptomyces coelicolor and Streptomyces lividans [17]. We investigated the subcellular location of KaPOx by heterologous expression using Gram-positive bacterial expression systems and included additional sequences upstream of the annotated ATG that may constitute a functional signal peptide. To accommodate the detection of secreted enzyme, fusion constructs with a fluorescent protein (mono-Red Fluorescent Protein, mRFP) were used as reporter.
2. Results
2.1. Signal Peptide Prediction
The K. aureofaciens genomic sequence upstream of the coding region of KaPOx was analyzed using ORF Finder: https://www.ncbi.nlm.nih.gov/orffinder/ (accessed on 11 November 2019), and two Met residues were discovered upstream and in frame with the previously annotated start codon (Figure 1). These additional sequences amount to 63 amino acids starting from the first and 44 amino acids starting from the second encoded Met residue. These sequences were analyzed using TatP-1.0 [18]: https://services.healthtech.dtu.dk/service.php?TatP-1.0 (accessed on 11 November 2019). The longer sequence comprising 63 AA was classified as unlikely to be a Tat signal peptide, but part of the 44 AA sequence was found to score above the cut-off in all but one category. The potential signal peptide cleavage site was predicted to be between positions 36 and 37 (Supplemental Material, Figure S1).
2.2. Heterologous Expression of KaPOx with a Putative Signal Peptide
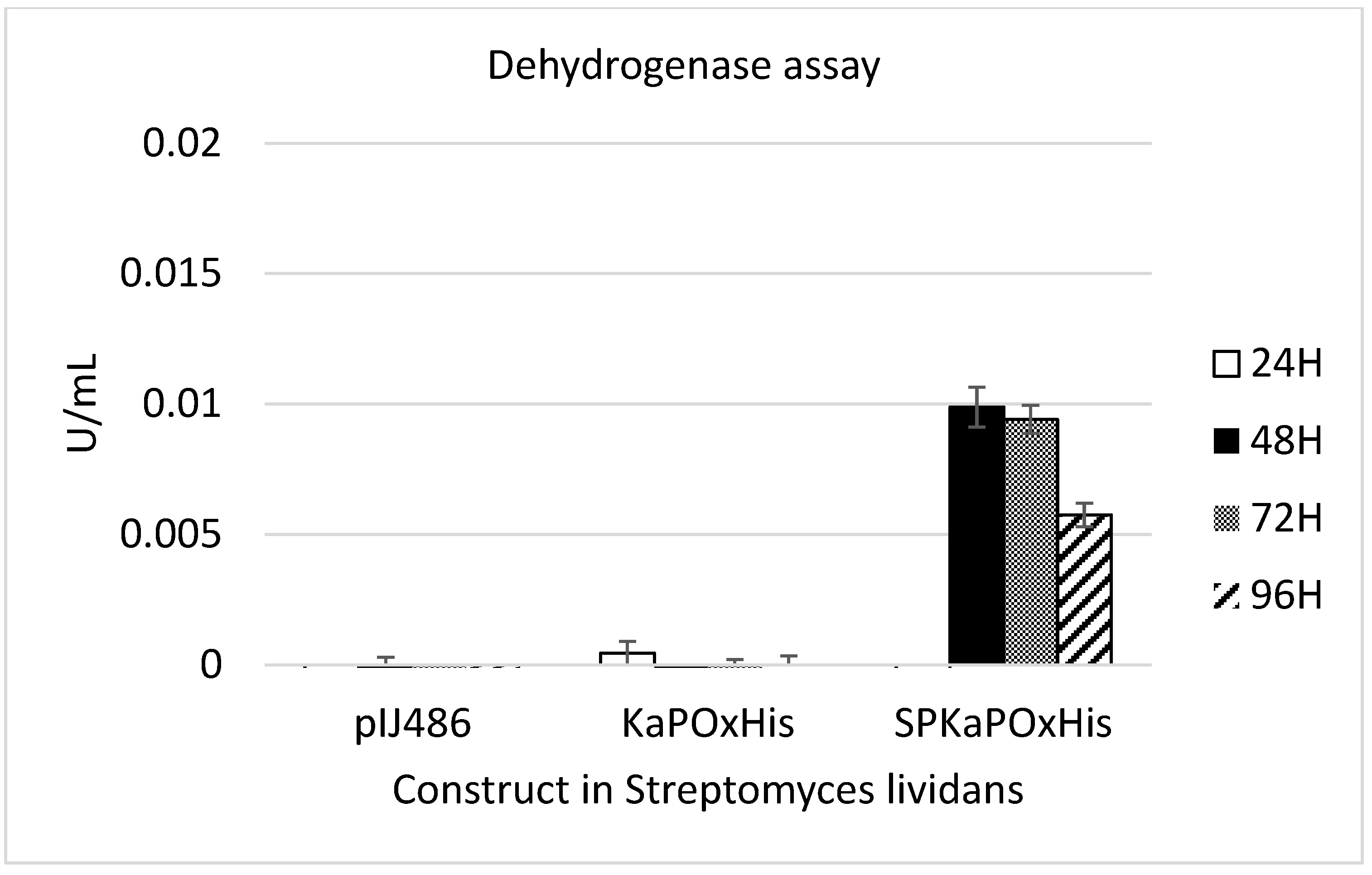
To investigate whether the 44 AA upstream of the originally annotated ATG of KaPOx constitute a functional signal peptide as predicted, constructs with and without this additional sequence (SPKaPOxHis and KaPOxHis, respectively) were prepared and heterologously expressed as described (Figure 2). Expression in B. subtilis did not result in detectable levels of secreted protein with either construct, but the His-tagged protein was detectable in the cell extract by Western blotting (Supplementary Figure S2). Protein purification by affinity chromatography was possible from the cell extract, but not from the supernatant. Yields of purified protein were considerably lower from the extract of cultures expressing the SPKaPOxHis construct (Figure S2). Expression in S. lividans was carried out using the constitutive promoter Pvsi and pIJ486 as plasmid backbone (Figure 2). Secreted KaPOx could be detected in the supernatant of S. lividans carrying the construct with the putative SP by enzymatic assay at a volumetric activity of 0.01 U/mL (dehydrogenase activity; Figure 3) as early as 48 h after start of the cultivation.
2.3. Fusion Protein Approach
Translational fusions of the fluorescent mono-Red Fluorescent Protein (mRFP) C-terminally of KaPOx with a 6× His-Tags added C-terminally of mRFP were constructed (Figure 4).
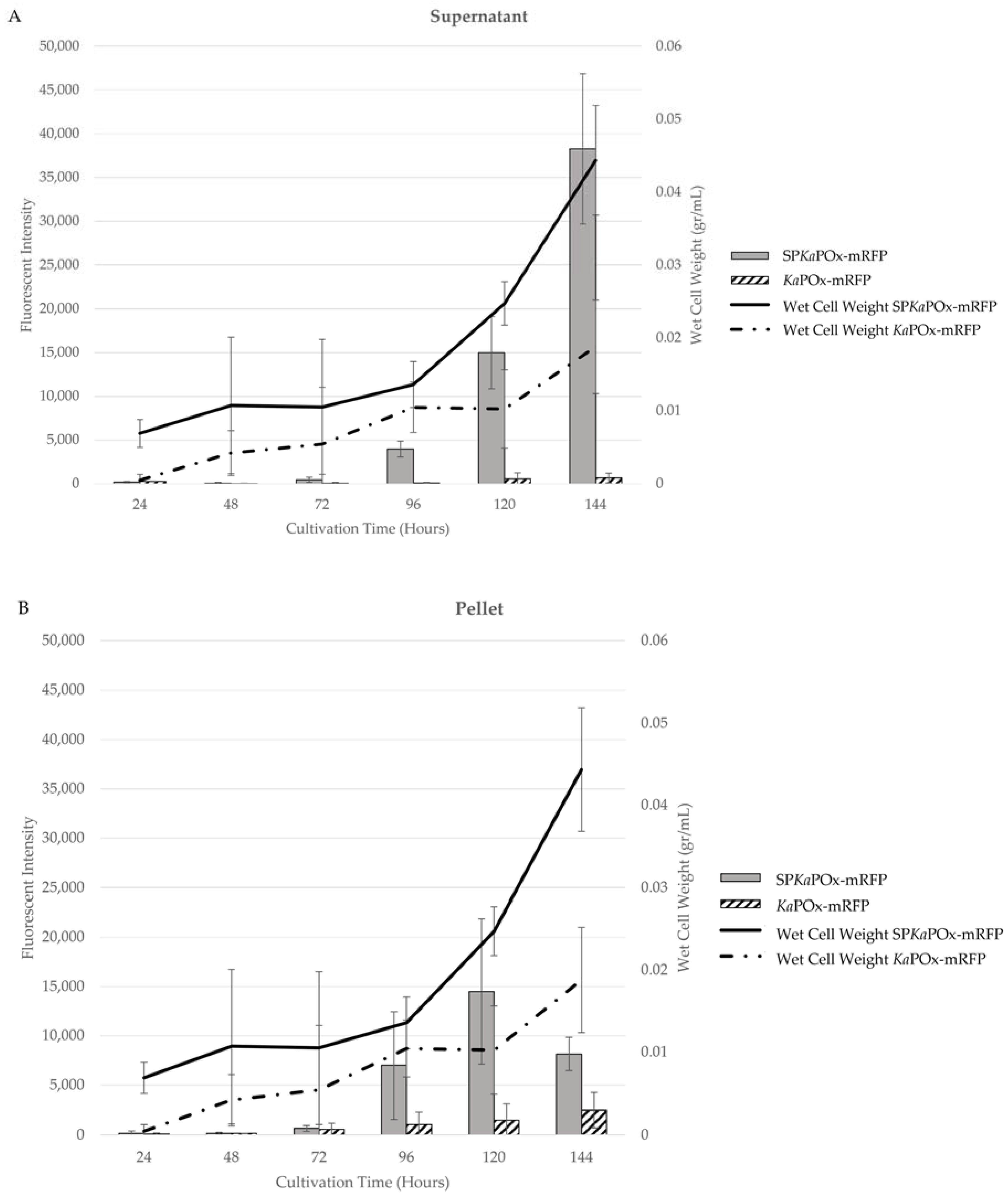
Both fusion constructs (SPKaPOxmRFPHis and KaPOxmRFPHis) were expressed in S. lividans under the control of the Pvsi promoter. In the culture supernatants, SPKaPOxmRFPHis showed a very low level of fluorescence at 72 h that increased constantly in intensity until 144 h. The supernatant from KaPOxmRFPHis showed a much lower fluorescence, detectable only at 120 and 144 h (Figure 5).
Fluorescence was also detected in the cell pellets starting at 72 h in cultures of both constructs. Intensity increased until the end of cultivation at a very low level for cultures harboring KaPOxmRFPHis; for cultures harboring SPKaPOxmRFPHis a stronger increase was measured until 120 h, followed by a decline at 144 h. Wet cell weight (WCW) was monitored over time, and the obtained values were used to normalize the measured fluorescence intensity. A different growth behavior for cells harboring the different constructs was observed: cultures harboring KaPOxmRFPHis showed a constant but slow increase in wet cell weight, with a final value after 144 h, approximately half of that recorded for cultures of SPKaPOxmRFPHis, in which a marked increase after 96 h was observable.
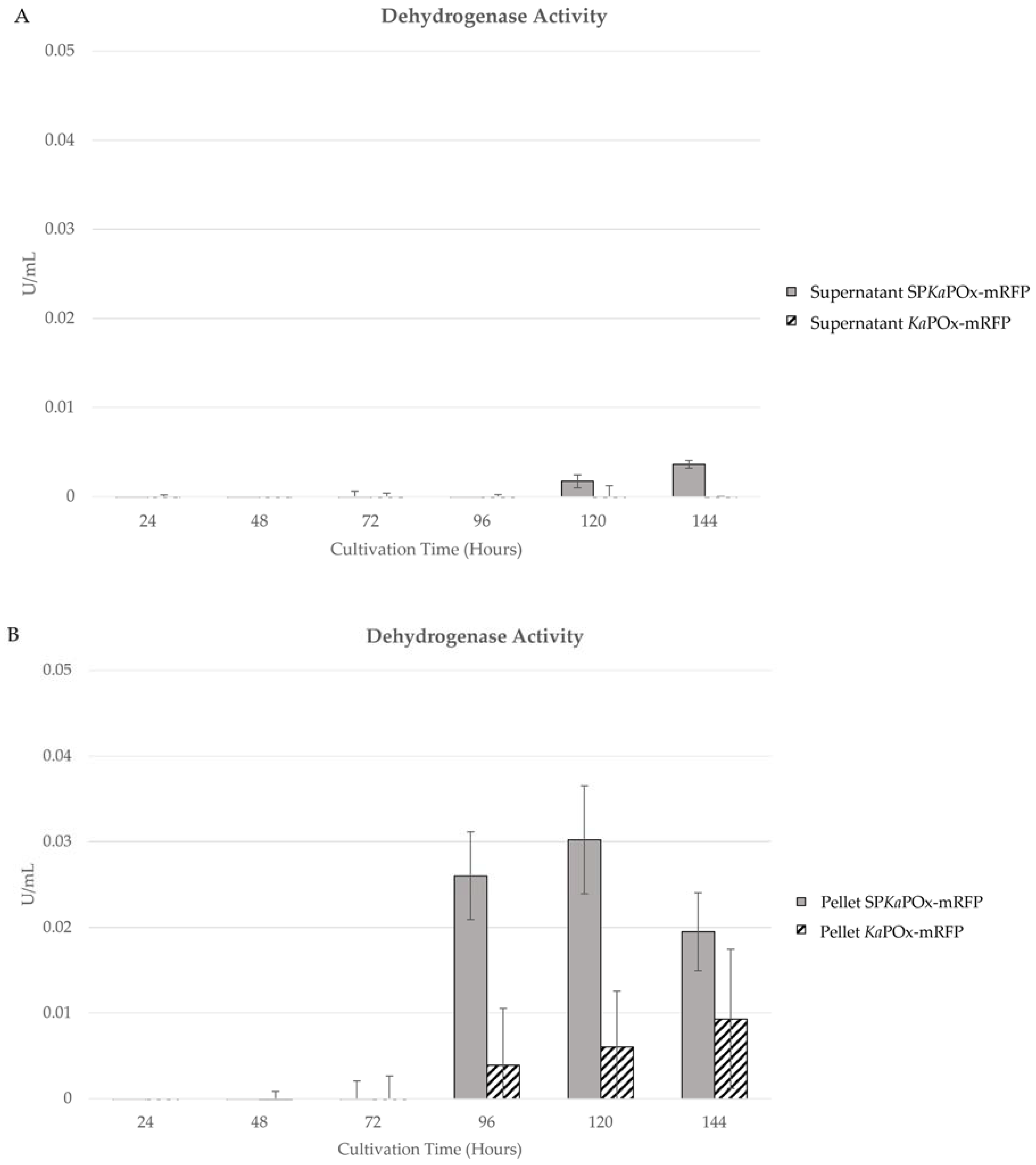
Enzymatic activity assays were also done on the samples from supernatants and pellets of cultures harboring both constructs. Both oxidase as well as dehydrogenase activity could be detected in the pellets starting at 96 h (oxidase activity is not shown, as the values were consistently very low). In the supernatants, no activity was measurable for cultures of KaPOxmRFPHis. Cultures containing SPKaPOxmRFPHis showed low activity at 120 and 144 h (Figure 6A). Enzymatic activities in the cell pellet increased slowly on a low level for KaPOxmRFPHis cultures. In SPKaPOxmRFPHis cultures, activity was higher at 96 and 120 h and declined in the last sample at 144 h. It is notable that the measured activities in the pellet fractions were higher in all corresponding samples than those in the supernatants.
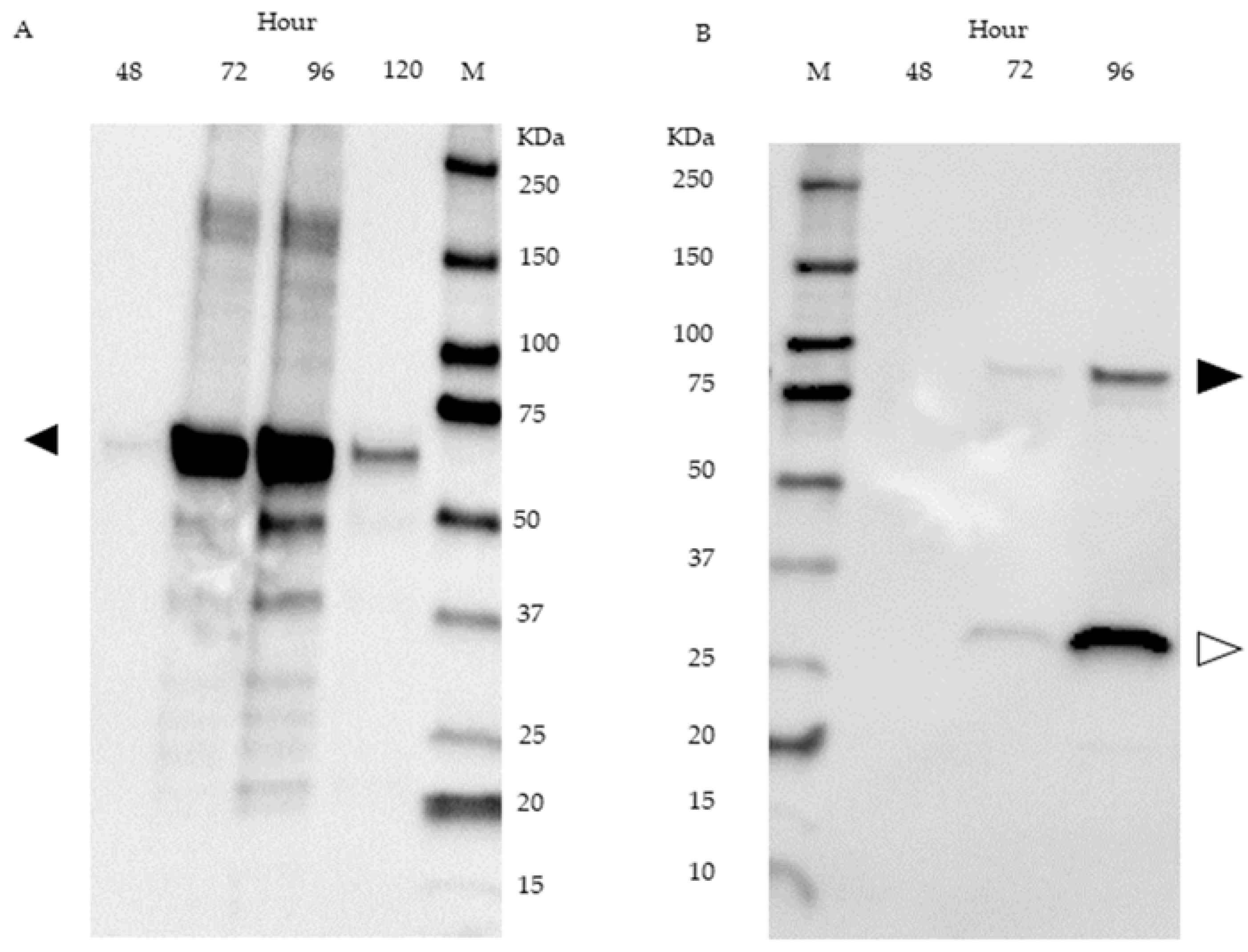
The presence of fusion protein was detected in supernatant samples of SPKaPOxmRFPHis cultures by Western blot using an anti-His-tag antibody. A band corresponding to intact fusion protein is visible in the samples obtained after 72 h and after 96 h of cultivation. In both samples, a notably more intense band corresponding to a molecular weight of 27 kDa is present (Figure 7B). Western blot analysis of SPKaPOxHis derived from the supernatant is shown in Figure 7A.
3. Discussion
Here we expressed pyranose oxidase from K. aureofaciens (KaPOx), a bacterial enzyme from the family Auxiliary Activities 3 (AA3), in the two Gram-positive bacterial expression systems B. subtilis and S. lividans. We used constructs containing KaPOx, as previously expressed intracellularly in E. coli and characterized [6], as well as constructs containing the genomic sequence encoding additional 44 amino acids upstream of and in frame with the previously characterized coding sequence and also starting with an ATG. This upstream sequence was tentatively classified to constitute a Tat signal peptide by computational tools. We could not detect secretory enzyme in cultivations of both constructs expressed in B. subtilis but could purify the tagged protein from the cell extracts of both cultures. When using S. lividans as expression host, secretory enzyme with dehydrogenase activity towards glucose as the electron donor and ferrocenium hexafluorophosphate as the electron acceptor was detected in cultures harboring the construct with the putative signal peptide. The volumetric dehydrogenase activity was low at 0.01 U/mL, and oxidase activity could not be detected. Herzog et al. (2019) showed a 6.6-fold dehydrogenase activity with ferrocenium hexafluorophosphate compared to oxidase activity with molecular oxygen as electron acceptor for recombinant KaPOx produced in E. coli. We conclude that, considering the low dehydrogenase activity of secreted KaPOx produced in S. lividans, oxidase activity is probably below the detection limit.
We subsequently prepared fusion constructs with the gene encoding the fluorescent reporter protein mRFP downstream of the KaPOx-encoding gene (with and without the upstream sequence encoding the putative SP). Fluorescence could be detected in the supernatant after 72 h in cultures expressing the fusion construct with the putative SP, increasing significantly until 144 h and always higher than the intensity in the cell pellet except very early after initial detection (Figure 5A). When constructs without the putative SP were used, a fluorescent signal was detected in the pellet at 72 h, but only another 48 h later in the supernatant, where the intensity remained very low until the last sampling point, and significantly lower than in the cell pellets at all time points (Figure 5B). It is notable that the fluorescence intensity in the cell pellets of SPKaPOxmRFPHis cultures reached a peak at 120 h and decreased for the last sampling point at 144 h. Also notable is the different growth behavior of the cultures expressing the two different constructs: the wet cell weight of SPKaPOxmRFPHis and KaPOxmRFPHis cultures was comparable until 96 h, but increased markedly slower after this in KaPOxmRFPHis cultures to a maximal value of 0.08 g/mL compared to SPKaPOxmRFPHis cultures, which reached 0.140 g/mL. This is concomitant with the appearance of fluorescent fusion protein in significant amounts in the supernatant. These observations suggest that, while (over)expression of both fusion constructs constitutes a metabolic burden for the cells, secretion driven by the signal peptide (and observable in the fluorescent intensity) avoids or alleviates cellular stress through cytoplasmic accumulation of the heterologous fusion protein, resulting in healthier growth and higher biomass formation than in the cells producing fusion protein lacking the putative signal peptide, where the expressed fusion protein accumulates throughout growth.
During six days or 144 h of cultivation, increase of wet cell weight as well as fluorescence intensity relative to WCW increased gradually without fluctuations or abrupt changes. When monitored for longer periods (until 192 h, not shown) an onset of “plateauing” of secretory fluorescence is observable in SPKaPOxmRFPHis cultures, while wet cell weight continues to increase for all cultures and only starts to show a lower increase rate at the last sampling point. This suggests that the cells had not yet reached the stationary phase. Since no sudden increases in extracellular fluorescence with concomitant stagnation or loss of wet cell weight were observed, we conclude that the extracellular fluorescence is a consequence of secretion, not cell death and lysis.
It is notable that the fluorescent signal from the fusion constructs was detectable later than the enzymatic activity in the previous experiments (72 h vs. 48 h). The fusion proteins are larger at 86.5 KDa than the KaPOx mature domain (59.9 KDa). A late secretion for heterologous enzymes in S. lividans was reported by Sianidis et al. (2006) [19] for the 95 KDa xyloglucanase, where secretion peaked at ~120H of cultivation time. It appears plausible that KaPOx is secreted earlier compared to fusion proteins containing the fluorescent reporter.
We also measured enzymatic activity (dehydrogenase) in both cell pellets and supernatants of SPKaPOxmRFPHis and KaPOxmRFPHis cultures and observed a marked discrepancy to the results of the fluorescence measurements. KaPOxmRFPHis cultures followed the same pattern as observed previously, with accumulation of active enzyme in the pellet fraction and essentially no detectable activity in the supernatants. SPKaPOxmRFPHis cultures also showed intracellular accumulation of activity, peaking at 120 h, as did the fluorescence measurement. In the supernatants, however, the activities, while following the same pattern as in the secretory fluorescence (constant increase until 144 h), remained much lower than the measured intracellular activities. Taken by itself, this appears to argue against secretion of active enzymes to meaningful levels. Western blot analysis of supernatant samples revealed intact fusion protein in the supernatant, faintly in samples taken after 72 h and more prominently in samples taken after 96 h. At both time points, a more intense band corresponding to the molecular weight of mRFP (± 27 KDa) is also detected (Figure 7B). We conclude that the lower band represents the product of proteolytic cleavage of secreted fusion protein, namely mRFP, which is detected via the attached His-tag (the rest of the protein, i.e., the KaPOx mature domain, is not detectable, as it does not contain a His-tag). Since mRFP is a small, compact protein composed mostly of β-sheets, it is conceivable that it is released from the fusion protein by extracellular proteases, but stays otherwise intact and fluorescent (thus detectable in the fluorescence measurements), whereas the larger oxidoreductase domain of the fusion protein may be further degraded and rendered inactive. In this case, we can presumably detect all of the secretory fluorescence, but only a fraction of the activity. The appearance of the His-tagged mRFP domain in the supernatant by another pathway, namely cell lysis and subsequent proteolysis, or intracellular proteolysis followed by cell lysis and release, is not plausible at these time points, as this would have been obvious in other parameters. According to the structural model of KaPOx [6], the C-terminus is exposed on the surface of the Rossman domain, facing away from the active site as well as the dimerization interface. An interference of the mRFP domain with dimerization and/or activity appears therefore unlikely, but cannot be entirely ruled out.
Regarding the failure to detect enzymatic activity (either kind) in the experiments with B. subtilis, it has to be noted that the used strain NZ8901 is not optimized for extracellular protease activity [20], which is known to be a major detrimental factor for the expression of heterologous proteins in this organism [21,22]. Since these early experiments were only done with SPKaPOxHis- and KaPOxHis-constructs, which do not allow fluorescent detection (and do not provide a more stable mRFP-domain that can be detected via the His-tag), it is conceivable that secretion driven by the putative signal peptide did, in fact, happen, but remained undetectable due to rapid proteolysis. Additionally, while the gene sequence was adapted to B. subtilis codon usage, a certain incompatibility between the signal peptide and mature domain sequences of SPKaPOx and the B. subtilis secretory machinery has to be considered. K. aureofaciens, which belongs to the phylum Actinobacteria and is closely related to Streptomycetaceae [23], was previously classified as Streptomyces aureofaciens [24]. B. subtilis belongs to the Firmicutes, and differences between the Tat translocase complexes of B. subtilis (comprising two subunits, TatA and TatC) and those of S. lividans and other Streptomyces spp., which comprise three subunits (TatA, TatB and TatC), were reported [25,26]. It is conceivable that the native SP from KaPOx is not properly processed by the B. subtilis Tat system.
In bacteria, Auxiliary Activities Family 3 sequences are generally closely related to fungal POx sequences rather than to sequences from other subfamilies [6,27]. We have shown here that the pyranose oxidase from K. aureofaciens is very likely a secretory enzyme, which supports the discussed biological function as an Auxiliary Activity with biological roles in hydrogen peroxide provision and/or quinone redox cycling, as outlined in the Introduction. This raises the question whether more bacterial POx-like enzymes are secretory enzymes (and whether their annotations in genome data needs to be re-examined and perhaps revised). We performed a BLAST search (tblastn) using KaPOx as the query sequence, selected the 25 sequences with highest similarity and query coverage where upstream sequences were available, plus the characterized enzymes from P. siccitolerans and S. canus, and examined these upstream sequences for putative signal peptide sequences as described. The results are summarized in Table 1: 15 out of 27 sequences extended in frame upstream of the annotated start codon. In four cases, these additional sequences were less than twelve amino acids long. In two cases (both Streptomyces spp.), TatP-1.0 predicted the sequence to be a signal peptide with a score of 5 out of 5. One sequence (from Actinoalloteichus sp., of the family Pseudonocardiaceae) of 100 additional amino acids gave more than one additional sequence, one of which was classified as possibly constituting a signal peptide. The upstream sequence of the gene from P. siccitolerans was predicted with a score of 3 out of 5; two more sequences (from Streptomyces and an Arthrobacter species) resulted in a score of 2 out of 5. The sequences that contain putative upstream signal peptides are distributed across several branches of a phylogenetic tree constructed from extant bacterial POx-like sequences (Supplementary material Figure S4), and an allocation along taxonomic categories is not possible. It appears plausible that bacterial GMC-oxidoreductases, while generally similar to fungal POx sequences, have diversified to a range of biological functions, with some as secretory enzymes with an auxiliary activity in lignocellulose degradation, and others with a cytoplasmic location and a different biological role. Clearly these results are preliminary, and further investigations into the biochemical properties of bacterial POx-like sequences, their subcellular localization and biological function are necessary.
Most fungal POx enzymes have been purified from hyphal extracts [7,9,28,29], although Daniel et al. (1994) [30] proposed an extracellular localization based on microscopic studies. Nishimura et al. (1996) [31] expressed a cDNA from Trametes (Coriolus) versicolor in E. coli and reported that the first 38 amino acids were missing in the translated protein. Hallberg et al. [32] reported that the N-terminal part of Trametes ochracea (multicolor) pyranose oxidase has an unordered conformation and is not resolvable in the crystal structure [32]. These two enzymes show 96% identity, and both their N-terminal parts show similarities to a (bacterial) Tat signal peptide as analyzed by computational tools (TatP-1.0; Supplementary Material Figure S3). Pyranose oxidase has long been discussed as having been acquired by fungi from bacteria via Horizontal Gene Transfer (HGT) [27]. The fact that most fungal POx do not appear to be secretory enzymes (or that the Tat-SP-like sequence at the N-terminus of the T. ochracea and T. versicolor POx is not functional in fungi) does not necessarily contradict this—most lignocellulose-degrading fungi possess other enzymes for these biological functions. It is possible that different bacterial pyranose oxidase sequences were acquired by fungi on separate occasions and have diversified post-HGT for other roles.
4. Materials and Methods
4.1. Strains and Plasmids
Escherichia coli JM109 and MC1061 (MoBiTec GmbH, Göttingen, Germany) were used as an intermediate cloning host. Bacillus subtilis NZ8901 (MoBiTec GmbH) and Streptomyces lividans TK24 were used as an expression host. Vector pUC19 was used for cloning purposes in E. coli, pNZ8901 was used as an expression vector in B. subtilis and pIJ486 was used as a backbone for expression in S. lividans. S. lividans TK24 and pIJ486 were a gift from Dr. Mohamed Belal Hamed (KU Leuven) [33,34].
4.2. Media
Standard Luria Bertani (LB) medium containing Ampicillin (100 µg/mL) or Chloramphenicol (10 µg/mL) was used for E. coli cultivation. Cultivation of B. subtilis was conducted in 2 × YT medium (per liter: 16 g tryptone, 10 g yeast extract, 5 g NaCl) containing chloramphenicol (5 µg/mL) as described in the supplier’s manual. S. lividans was cultivated as described [35] in phage medium (per liter: 0.5 g MgSO4·7H2O, 0.74 g CaCl2·2H2O, 10 g glucose, 5 g tryptone, 5 g yeast extract, 5 g Lab Lemco powder; the pH was adjusted to 7.2 with 5 N NaOH). For S. lividans transformation, R2 medium was used (per liter: 103 g sucrose, 0.25 g K2SO4, 12.12 g MgCl2·6H2O, 0.1 g casamino acids, 1 g yeast extract, 5 g of Lab Lemco powder, 100 mL TES buffer, 2 mL trace element solution, 10 mL 0.5% KH2PO4, and 2% agar. 10 mL filter-sterile 36.8% CaCl2·2H2O and 1 mL filter-sterile 2 mM CuSO4 solution were added after sterilization). The trace element solution contained (per liter) 40 mg ZnCl2, 200 mg FeCl3⋅6H2O, 10 mg CuCl2⋅2H2O, 10 mg MnCl2⋅4H2O, 10 mg Na2B4O7⋅ 10H2O, and 10 mg (NH4)6Mo7O24⋅4H2O) and was filter-sterilized. Thiostrepton was used as a selective marker and added to a final concentration of 50 µg/mL from a stock solution of 50 mg/mL in DMSO. Chemicals and media components were purchased from Sigma Aldrich (St. Louis, MO, USA).
4.3. DNA Manipulation
Genomic extraction was carried out using Monarch® Genomic DNA Purification Kit, and plasmid DNA was isolated using Monarch® Plasmid Miniprep Kit. Polymerase Chain Reaction (PCR) was carried out using Q5® High-Fidelity DNA polymerase with the oligonucleotides listed in Supplementary Table S1. Monarch® PCR & DNA Cleanup Kit and Monarch® DNA Gel Extraction Kit were used to purify PCR products. All extraction and purification kits and all DNA-modifying enzymes and restriction endonuclease were purchased from New England Biolabs (Ipswich, MA, USA).
4.4. Vector Constructions
Constructions based on the shuttle vector pNZ8901 for the B. subtilis expression system were done by amplifying the gene of interest from the genome of K. aureofaciens through standard PCR. Primers pNZ_SPKaPOx_F and KaPOxHis_R were used to amplify spkapox (the KaPOx gene harboring the putative signal peptide sequence and a C-terminal His-tag), while primers pNZ_KaPOx_F and KaPOxHis_R were used to amplify kapox (the KaPOx gene with a C-terminal His-tag). Restriction endonucleases PstI and XbaI were used to digest both PCR products (spkapox and kapox) and pNZ8901 prior to ligation using T4 Ligase. The ligated products were transformed to E. coli MC1061 for propagation and transformed into B. subtilis NZ8901 as circular plasmids isolated from E. coli MC1061.
Vector construction for S. lividans TK24 expression system was conducted in several steps due to the lack of a promoter in the pIJ486 backbone. The constitutive promoter (Pvsi) from the subtilisin inhibitor gene (vsi) was amplified using primers Pvsi_F and Pvsi_R from pIJ486-spsec-mRFP (gift from KU Leuven), digested by HindIII and PstI and ligated to pUC19 digested with the same restriction endonucleases to generate pUC19-Pvsi. Both primer pairs SPKaPOx_F with KaPOxHis_R and KaPOx_F with KaPOxHis_R were used to amplify spkapox and kapox, respectively, from the K. aureofaciens genome. Restriction endonucleases PstI and XbaI were used to digest both PCR products and pUC19-Pvsi prior to ligation to generate pUC19-Pvsi-spkapox and pUC19-Pvsi-kapox. Constructs with the reporter gene mrfp were generated through Gibson Assembly® (New England Biolab). We modified the pIJ486-spsec-mrfp into pIJ486-mrfp by removing the signal peptide sequence, spsec. Primer mRFP_GA_F (carrying an overhang of the Pvsi 3′ end sequence) and mRFP_GA_R (carrying an overhang of the pUC19 sequence) were used to amplify mrfp from pIJ486-spsec-mrfp. Vector pUC19-Pvsi was linearized with PstI, and Gibson Assembly® was used to insert the amplification product to generate pUC19-Pvsi-mrfp. The primer pair (SP)KaPOx_GA_F and KaPOx_GA_R was used to generate the PCR products (sp)kapox with an overhang of the Pvsi sequence on its 5′ end, a linker and an overhang with the mRFP sequence in its 3′ end. The primer pair mRFP_lkk_GA_F and mRFPHis_GA_R was used to generate mrfp with a linker on its 5′ end and a His-tag with a complementary sequence to pUC19 on its 3′ end. The PCR products (sp)kapox-linker and linker-mrfp-his, together with pUC-Pvsi linearized with PstI, were used as a template for the Gibson Assembly® reaction, to generate pUC19-Pvsi-(sp)kapox-mrfp. All intermediate constructs in pUC19-Pvsi were digested with restriction enzymes HindIII and XbaI and ligated into pIJ486 digested with the same restriction enzymes. All constructs used in this study were listed in Table 2.
4.5. Transformation
Chemically competent cells were used to conduct transformation in E. coli [36], and electroporation was used for transformation in B. subtilis following the instructions in the supplier´s manual (MoBiTec GmbH). Transformation of S. lividans TK24 was carried out through protoplast preparation and transformation [35], using thiostrepton (50 µg/mL) as a selection marker.
4.6. Cultivation and Protein Production
B. subtilis harboring recombinant vector pNZ8901 and its derivatives were grown in 50 mL 2xYT medium containing chloramphenicol 5 µg/mL at 37 °C, 200 rpm. Recombinant protein production was induced by adding 1% (v/v) subtilin preparation when OD600 reached 0.8. The subtilin preparation was produced from B. subtilis NZ8963 (MoBiTec GmbH) according to the supplier´s manual. Cultures were harvested at 24 h and 48 h after induction. Then, 50 mL culture was centrifuged and the supernatant separated from the pellet and stored with the addition of protease inhibitor (10 µg/mL phenyl methyl sulfonyl fluoride; PMSF). The pellet was washed three times with phosphate buffer pH 7 and suspended in 5 mL buffer following the last washing step. PMSF was added at 10 µg/mL, and the cells were pre-treated with lysozyme (0.5 mg/mL for one hour at 37 °C) and disrupted by sonication on ice using a Bandelin Sonopuls HD 60 (Bandelin electronic GmbH, Berlin, DE) set at 80 V and 30%-cycle for 3 × 1 min, with two-minute intervals. Recombinant His-tagged protein was purified on 1 mL Ni-NTA columns (Merck, Darmstadt, Germany) following the manufacturers recommendations using Buffer A (50 mM Tris-HCl pH 7.5, 30 mM NaCl, and 30 mM Imidazole) as binding and washing buffer and Buffer B (50 mM Tris-HCl pH 7.5, 30 mM NaCl, and 250 mM Imidazole) as elution buffer.
Cultivation of S. lividans TK24 was conducted in baffled flasks containing 50 mL phage medium with 10 µg/mL thiostrepton at 30 °C, 150 rpm. No induction was required due to the use of the constitutive Pvsi promoter. Samples of 1 mL were obtained every 24 h and centrifuged (4000× g, 10 min, 4 °C). Supernatants were separated and stored at 4 °C with 10 µg/mL PMSF. Wet cell weight of the pellets was determined, and samples were washed three times with 1 mL phosphate buffer pH 7 containing 25 mM NaCl. Treatment with lysozyme and sonication was done as described for B. subtilis (sonication was done for only 1 min). The lysed samples were centrifuged at 13,000 rpm for 5 min at 4 °C, and extracts were stored with 10 µg/mL PMSF.
4.7. SDS-PAGE and Western Blot
Samples were mixed with 2x SDS buffer mix (Sigma), denatured at 95 °C for 3 min and separated on Mini-PROTEAN®TGX Stain-Free™ Precast Gels 4–20% (Bio-Rad, Hercules, CA, USA) at 150 volt for 50 min. Precision Plus Protein™ Western C™ (Bio-Rad) was used as a molecular weight standard. Proteins were transferred to a 0.2 µm Nitrocellulose membrane by dry-blotting in a Trans-Blot® Turbo™ (Bio-Rad) at 1.3 A, and 25 V for 7 min. Proteins were detected using a BSA-free anti-Penta-His-tag mouse monoclonal IgG (Qiagen, Hilden, Germany) as primary antibody and Polyclonal Rabbit-anti-mouse Immunoglobulin/HRP (Agilent, Santa Clara, CA, USA) as secondary antibody according to the manufacturer´s recommendations. Clarity Western ECL (Bio-Rad) was used as substrate. Visualization of both stain-free SDS-PAGE gel and Western blot was done in a ChemiDoc™ XRS+ (BioRad).
4.8. Enzymatic Activity Assay and Fluorescent Signal Analysis
To determine the KaPOx dehydrogenase activity, an enzymatic activity assay was performed using 160 mM glucose as electron donor and ferrocenium hexafluorophosphate 0.16 mM as electron acceptor. For the oxidase activity, atmospheric oxygen was used as electron acceptor. Quantification of oxidase activity was done using 1 mM 2,2′-Azino-bis (3-ethylbenzothiazoline-6-sulfonic acid; ABTS) and Horseradish Peroxidase (Sigma Aldrich; 143 U/mL). Dehydrogenase activity was determined spectrophotometrically at 300 nm, while oxidase activity was determined at 420 nm as described previously [6].
Detection of fluorescence of mRFP was conducted by exciting the samples at 584 nm and analyzing the emission at 603 nm. Both KaPOx enzymatic activities as well as mRFP fluorescent signals were measured in an EnSpire® multimode plate reader (PerkinElmer, Waltham, MA, USA).
Supplementary Materials
The supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms24031975/s1.
Author Contributions
Conceptualization, C.P.; Investigation, L.J.V.; Writing—original draft, L.J.V.; Writing—review & editing, C.P.; Supervision, C.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the AUSTRIAN SCIENCE FUND FWF, grant W1224, and the Austrian Agency for Education and Internationalisation OeAD via an ASEA-UNINET Ernst-Mach-Grant for L. Jessica Virginia. Open Access Funding by the Austrian Science Fund (FWF).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The financial support for L. Jessica Virginia from the Austrian Agency for Education and Internationalisation (OeAD) through an ASEA-UNINET Ernst-Mach-Grant is gratefully acknowledged. L. Jessica Virginia was a member of the Doctoral Program BioToP—Biomolecular Technology of Proteins (supported by the Austrian Science Fund FWF grant W1224). We are grateful to Mohamed Belal Hamed and Anastassios Economou, KU Leuven, for the gift of plasmids and bacterial strains.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cragg, S.M.; Beckham, G.T.; Bruce, N.C.; Bugg, T.D.H.; Distel, D.L.; Dupree, P.; Etxabe, A.G.; Goodell, B.S.; Jellison, J.; McGeehan, J.E.; et al. Lignocellulose degradation mechanisms across the Tree of Life. Curr. Opin. Chem. Biol. 2015, 29, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Polegioni, L.; Tonin, F.; Rosini, E. Lignin-degrading enzymes. FEBS J. 2015, 282, 1190–1213. [Google Scholar] [CrossRef]
- Bugg, T.D.H.; Ahmad, M.; Hardiman, E.M.; Rahmanpour, R. Pathways for degradation of lignin in bacteria and fungi. Nat. Prod. Rep. 2011, 28, 1883. [Google Scholar] [CrossRef]
- Xu, Z.; Lei, P.; Wen, Z.; Jin, M. Recent advances in lignin valorization with bacterial cultures: Microorganisms, metabolic pathways, and bio-products. Biotechnol. Biofuels 2019, 12, 2–19. [Google Scholar] [CrossRef] [Green Version]
- Bissaro, B.; Røhr, Å.K.; Müller, G.; Chylenski, P.; Skaugen, M.; Forsberg, Z.; Horn, S.J.; Vaaje-Kohlstad, G.; Eijsink, V.G.H. Oxidative cleavage of polysaccharides by monocopper enzymes depends on H2O2. Nat. Chem. Biol. 2017, 13, 1123–1128. [Google Scholar] [CrossRef]
- Herzog, P.L.; Sützl, L.; Eisenhut, B.; Maresch, D.; Haltrich, D.; Obinger, C.; Peterbauer, C.K. Versatile Oxidase and Dehydrogenase Activities of Bacterial Pyranose 2-Oxidase Facilitate Redox Cycling with Manganese Peroxidase In Vitro. Appl. Environ. Microbiol. 2019, 85, e00390-19. [Google Scholar] [CrossRef] [Green Version]
- Leitner, C.; Volc, J.; Haltrich, D. Purification and Characterization of Pyranose Oxidase from the White Rot Fungus Trametes multicolor. Appl. Environ. Microbiol. 2001, 67, 3636–3644. [Google Scholar] [CrossRef] [Green Version]
- Pisanelli, I.; Kujawa, M.; Spadiut, O.; Kitt, R.; Halada, P.; Volc, J.; Mozuch, M.D.; Kersten, P.; Haltrich, D.; Peterbauer, C. Pyranose 2-oxidase from Phanerochaete chrysosporium--expression in E. coli and biochemical characterization. J. Biotechnol. 2009, 142, 97–106. [Google Scholar] [CrossRef]
- Ai, M.Q.; Wang, F.F.; Zhang, Y.Z.; Huang, F. Purification of pyranose oxidase from the white rot fungus Irpex lacteus and its cooperation with laccase in lignin degradation. Proc. Biochem. 2014, 49, 2191–2198. [Google Scholar] [CrossRef]
- Wang, F.F.; Huang, F.; Ai, M.Q. Synergetic Depolymerization of Aspen CEL by Pyranose 2-Oxidase and Lignin-degrading Peroxidases. Bioresources 2019, 14, 3481–3494. [Google Scholar] [CrossRef]
- Bugg, T.D.H.; Ahmad, M.; Hardiman, E.M.; Singh, R. The emerging role for bacteria in lignin degradation and bio-product formation. Curr. Opin. Biotechnol. 2011, 22, 394–400. [Google Scholar] [CrossRef]
- Bugg, T.D.H.; James, J.W.; Rashid, G.M.M. Bacterial enzymes for lignin depolymerisation: New biocatalysts for generation of renewable chemicals from biomass. Curr. Opin. Chem. Biol. 2020, 55, 26–33. [Google Scholar] [CrossRef]
- Díaz-García, L.; Bugg, T.D.H.; Jiménez, D.J. Exploring the lignin catabolism potential of soil-derived lignocellulolytic microbial consortia by a gene-centric metagenomic approach. Microb. Ecol. 2020, 80, 885–896. [Google Scholar] [CrossRef]
- Mendes, S.; Banha, C.; Madeira, J.; Santos, D.; Miranda, V.; Manzanera, M.; Ventura, M.R.; van Berkel, W.J.H.; Martins, L.O. Characterization of a bacterial pyranose 2-oxidase from Arthrobacter siccitolerans. J. Mol. Catal. B Enzym. 2016, 133, S34–S43. [Google Scholar] [CrossRef]
- Kostelac, A.; Sützl, L.; Puc, J.; Furlanetto, V.; Divne, C.; Haltrich, D. Biochemical Characterization of Pyranose Oxidase from Streptomyces canus—Towards a Better Understanding of Pyranose Oxidase Homologues in Bacteria. Int. J. Mol. Sci. 2022, 23, 13595. [Google Scholar] [CrossRef]
- Abrera, A.T.; Sützl, L.; Haltrich, D. Pyranose Oxidase: A versatile sugar oxidoreductase for bioelectrochemical applications. Bioelectrochemistry 2020, 132, 107409. [Google Scholar] [CrossRef]
- Whittaker, M.M.; Whittaker, J.W. Streptomyces coelicolor oxidase (SCO2837p): A new free radical metalloenzyme secreted by Streptomyces coelicolor A3(2). Arch. Biochem. Biophys. 2006, 452, 108–118. [Google Scholar] [CrossRef]
- Bendtsen, J.D.; Nielsen, H.; Widdick, D.; Palmer, T.; Brunak, S. Prediction of twin-arginine signal peptides. BMC Bioinform. 2005, 6, 167. [Google Scholar] [CrossRef] [Green Version]
- Sianidis, G.; Pozidis, C.; Becker, F.; Vrancken, K.; Sjoeholm, C.; Karamanou, S.; Takamiya-Wik, M.; Mellaert, L.V.; Schaefer, T.; Anne, J.; et al. Functional large-scale production of a novel Jonesia sp. xyloglucanase by heterologous secretion from Streptomyces lividans. J. Biotechnol. 2006, 121, 498–507. [Google Scholar] [CrossRef]
- Bongers, R.S.; Veening, J.; van Wieringen, M.; Kuipers, O.P.; Kleerebezem, M. Development and Characterization of a Subtilin-Regulated Expression System in Bacillus subtilis: Strict Comtrol of Gene Expression by Addition of Subtilin. Appl. Env. Microbiol. 2005, 71, 8818–8824. [Google Scholar] [CrossRef]
- Harwood, C.; Cranenburgh, R. Bacillus protein secretion: An unfolding story. Trends Microbiol. 2008, 16, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Neef, J.; van Dijl, J.M.; Buist, G. Recombinant protein secretion by Bacillus subtilis and Lactococcus lactis: Pathways, applications and innovation potentials. Essays Biochem. 2021, 65, 187–195. [Google Scholar] [CrossRef]
- Hsiao, N.H.; Kirby, R. Comparative genomics of Streptomyces avermitilis, Streptomyces cattleya, Streptomyces maritimus and Kitasatospora aureofaciens using a Streptomyces coelicolor microarray system. Antonie Leeuwenhoek 2007, 93, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Labeda, D.P.; Dunlap, C.A.; Rong, X.; Huang, Y.; Doroghazi, J.R.; Ju, K.S.; Metcalf, W.W. Phylogenetic relationships in the family Streptomycetaceae using multi-locus sequence analysis. Antonie Leeuwenhoek 2017, 110, 563–583. [Google Scholar] [CrossRef] [PubMed]
- Anne, J.; Vrancken, K.; van Mellaert, L.; van Impe, J.; Bernaerts, K. Protein secretion biotechnology in gram-positive bacteria with special emphasis on Streptomyces lividans. Biochim. Biophys. Acta 2014, 1843, 1750–1761. [Google Scholar] [CrossRef] [PubMed]
- Kolkman, M.A.B.; van der Ploeg, R.; Bertels, M.; van Dijk, M.; van der Laan, J.; van Dijl, J.M.; Ferrari, E. The Twin-Arginine Signal Peptide of Bacillus subtilis YwbN Can Direct either Tat- or Sec-Dependent Secretion of Different Cargo Proteins: Secretion of Active Subtilisin via the B. subtilis Tat Pathway. Appl. Environ. Microbiol. 2008, 74, 7507–7513. [Google Scholar] [CrossRef] [Green Version]
- Sützl, L.; Foley, G.; Gillam, E.M.J.; Bodén, M.; Haltrich, D. The GMC superfamily of oxidoreductases revisited: Analysis and evolution of fungal GMC oxidoreductases. Biotechnol. Biofuels 2019, 12, 118. [Google Scholar] [CrossRef]
- Danneel, H.J.; Rossner, E.; Zeeck, A.; Giffhorn, F. Purification and characterization of a pyranose oxidase from the basidiomycete Peniophora gigantea and chemical analyses of its reaction products. Eur. J. Biochem. 1993, 214, 795–802. [Google Scholar] [CrossRef]
- Machida, Y.; Nakanishi, T. Purification and properties of pyranose oxidase from Coriolus versicolor. Agric. Biol. Chem. 1984, 48, 2463–2470. [Google Scholar] [CrossRef] [Green Version]
- Daniel, G.; Volc, J.; Kubatova, E. Pyranose oxidase, a major source of H2O2 during wood degradation by Phanerochaete chrysosporium, Trametes versicolor, and Oudemansiella mucida. Appl. Environ. Microbiol. 1994, 60, 2524–2532. [Google Scholar] [CrossRef]
- Nishimura, I.; Okada, K.; Koyama, Y. Cloning and expression of pyranose oxidase cDNA from Coriolus versicolor in Escherichia coli. J. Biotechnol. 1996, 52, 11–20. [Google Scholar] [CrossRef]
- Hallberg, B.M.; Leitner, C.; Haltrich, D.; Divne, C. Crystallization and preliminary X-ray diffraction analysis of pyranose 2-oxidase from the white-rot fungus Trametes multicolor. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 197–199. [Google Scholar] [CrossRef]
- Hamed, M.B.; Vrancken, K.; Bilyk, B.; Koepff, J.; Novakova, R.; van Mellaert, L.; Oldiges, M.; Luzhetskyy, A.; Kormanec, J.; Anné, J.; et al. Monitoring Protein Secretion in Streptomyces Using Fluorescent Proteins. Front. Microbiol. 2018, 9, 3019. [Google Scholar] [CrossRef] [Green Version]
- Ward, J.M.; Janssen, G.R.; Kieser, T.; Bibb, M.J.; Buttner, J.M.; Bibb, M.J. Construction and characterisation of a series of multi-copy promoter-probe plasmid vectors for Streptomyces using the aminoglycoside phototransferase gene from Tn5 as indicator. Mol. Gen. Genet. 1986, 203, 468–478. [Google Scholar] [CrossRef]
- Vrancken, K.; Van Mellaert, L.; Anné, J. Cloning and Expression Vectors for a Gram-Positive Host, Streptomyces lividans. Methods Mol. Biol. 2010, 668, 97–107. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Plasmids and Their Usefulness in Molecular Cloning. In Molecular Cloning: A Laboratory Manual, 3rd ed.; Argentine, J., Irwin, N., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001; Volume 1, pp. 116–118. [Google Scholar]
Figure 1.
Results from ORF Finder showing two Methionine (M) in frame in the upstream region (filled triangles), followed by the previously annotated start codon (M in red). The putative cleavage site is shown in blue with an open triangle. The Rossman fold motif is shown in green.
Figure 1.
Results from ORF Finder showing two Methionine (M) in frame in the upstream region (filled triangles), followed by the previously annotated start codon (M in red). The putative cleavage site is shown in blue with an open triangle. The Rossman fold motif is shown in green.

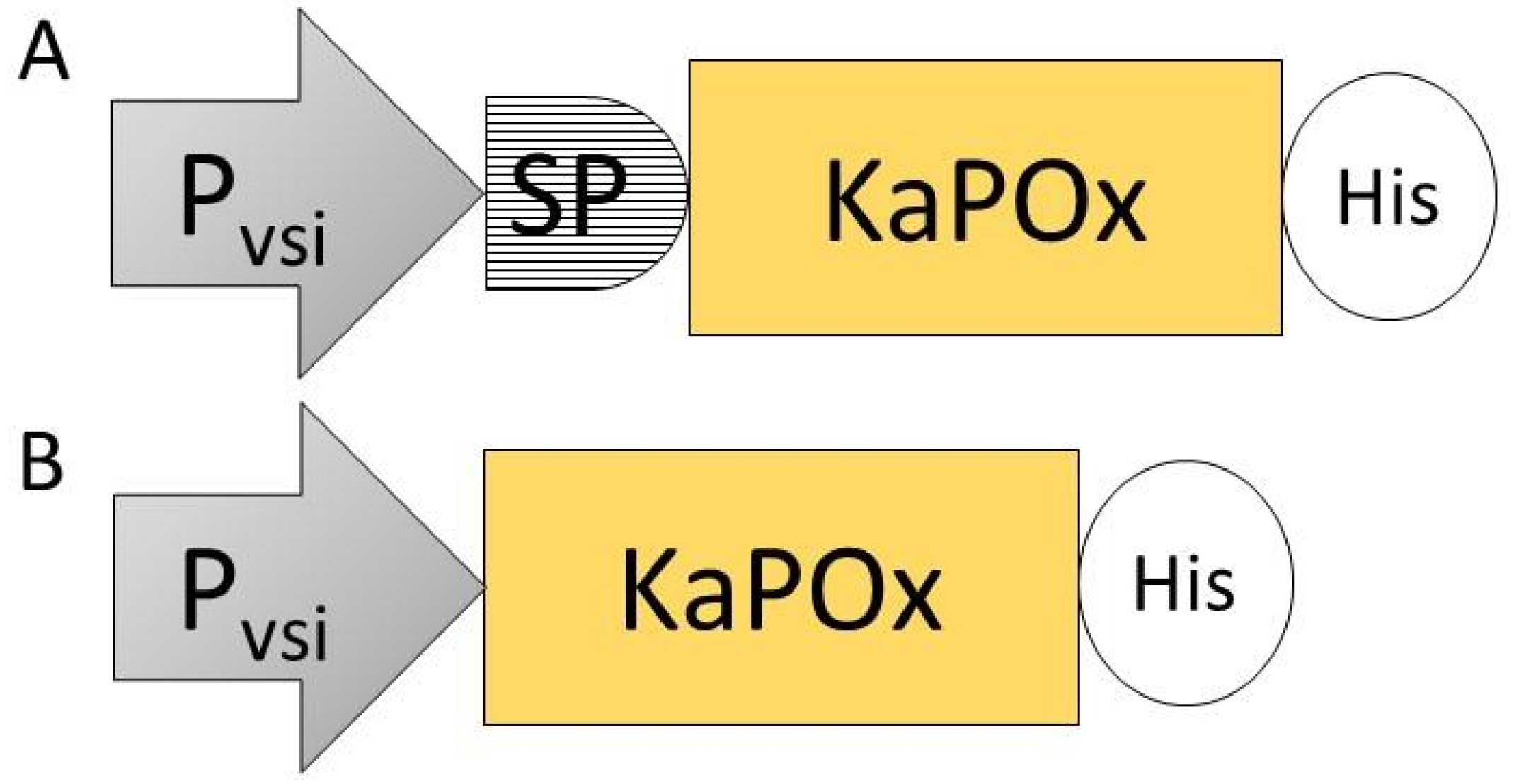
Figure 2.
Expression constructs SPKaPOxHis (A) and KaPOxHis (B), containing the constitutive promoter of the S. venezuelae subtilisin inhibitor gene (Pvsi), the putative signal peptide (SP) identified upstream of the mature domain of pyranose oxidase from K. aureofaciens (KaPOx) and a C-terminal 6× His-tag (His).
Figure 2.
Expression constructs SPKaPOxHis (A) and KaPOxHis (B), containing the constitutive promoter of the S. venezuelae subtilisin inhibitor gene (Pvsi), the putative signal peptide (SP) identified upstream of the mature domain of pyranose oxidase from K. aureofaciens (KaPOx) and a C-terminal 6× His-tag (His).

Figure 3.
Dehydrogenase activity assay of S. lividans supernatant obtained from cultures harboring the empty vector pIJ486, pIJ486-KaPOxHis, and pIJ486-SPKaPOxHis. Samples were taken from the supernatant at 24, 48, 72 and 96 h (H) as indicated.
Figure 3.
Dehydrogenase activity assay of S. lividans supernatant obtained from cultures harboring the empty vector pIJ486, pIJ486-KaPOxHis, and pIJ486-SPKaPOxHis. Samples were taken from the supernatant at 24, 48, 72 and 96 h (H) as indicated.

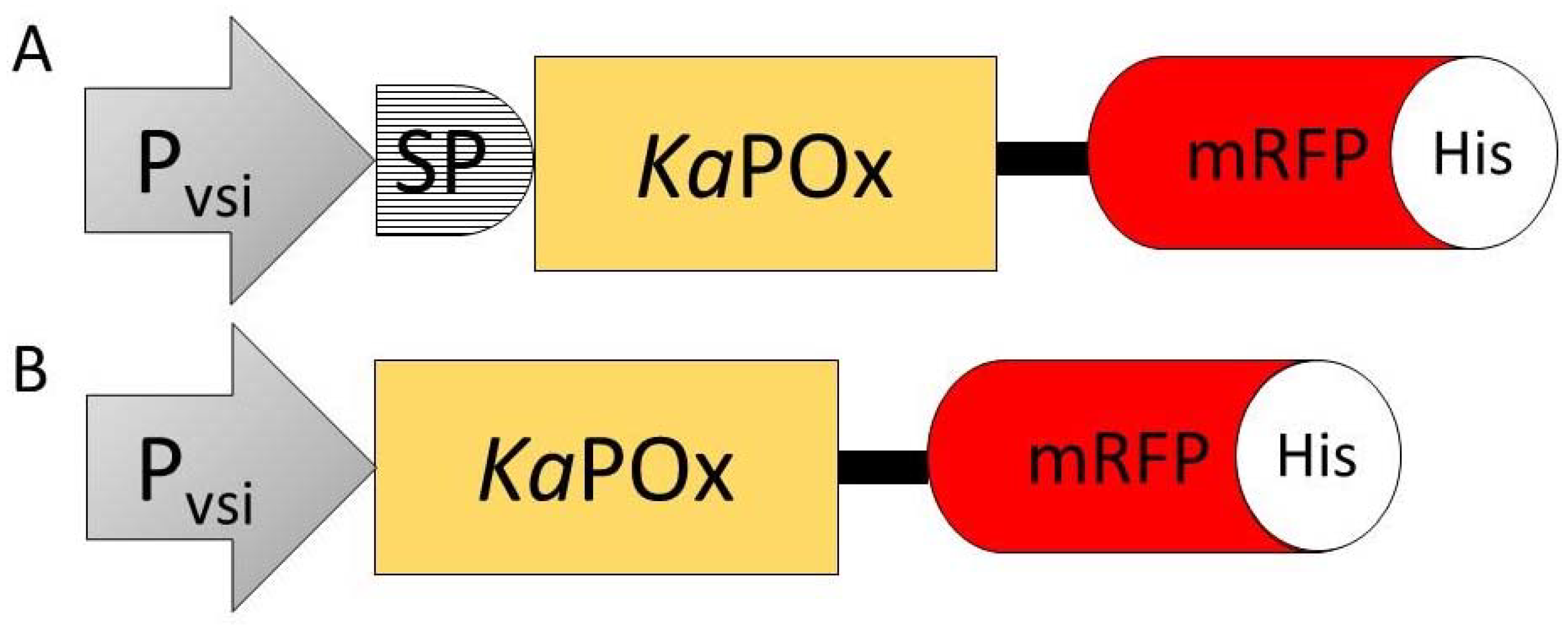
Figure 4.
Constructs SPKaPOxmRFPHis (A) and KaPOxmRFPHis (B) containing the constitutive S. venezuelae Pvsi promoter (Pvsi), the putative signal peptide (SP), the KaPOx mature domain, the mRFP fluorescent protein (mRFP) and a C-terminal 6×His-Tag (His).
Figure 4.
Constructs SPKaPOxmRFPHis (A) and KaPOxmRFPHis (B) containing the constitutive S. venezuelae Pvsi promoter (Pvsi), the putative signal peptide (SP), the KaPOx mature domain, the mRFP fluorescent protein (mRFP) and a C-terminal 6×His-Tag (His).

Figure 5.
Growth curve (in wet cell weight) and mRFP fluorescence intensity from cultures expressing the fusion constructs SPKaPOxmRFPHis and KaPOxmRFPHis in the supernatants (A) and the pellet fractions (B). Fluorescent intensity values are adjusted for the recorded wet cell weight and are the mean results of three cultures.
Figure 5.
Growth curve (in wet cell weight) and mRFP fluorescence intensity from cultures expressing the fusion constructs SPKaPOxmRFPHis and KaPOxmRFPHis in the supernatants (A) and the pellet fractions (B). Fluorescent intensity values are adjusted for the recorded wet cell weight and are the mean results of three cultures.

Figure 6.
Enzymatic activity (dehydrogenase) in samples of the supernatant (A) and the pellet fractions (B) of cultures harboring SPKaPOxmRFPHis and KaPOxmRFPHis. Activity values are normalized for the recorded wet cell weight, as in Figure 5.
Figure 6.
Enzymatic activity (dehydrogenase) in samples of the supernatant (A) and the pellet fractions (B) of cultures harboring SPKaPOxmRFPHis and KaPOxmRFPHis. Activity values are normalized for the recorded wet cell weight, as in Figure 5.

Figure 7.
Western blot of supernatant samples from S. lividans cultures harboring SPKaPOxHis at 48, 72, 96 and 120 h (A) and SPKaPOxmRFPHis at 48, 72 and 96 h (B). A size marker is shown in lane M. The expected molecular weight of intact fusion protein and of the mRFP domain is 86.5 KDa (black arrow) and 26.6 KDa (white arrow), respectively.
Figure 7.
Western blot of supernatant samples from S. lividans cultures harboring SPKaPOxHis at 48, 72, 96 and 120 h (A) and SPKaPOxmRFPHis at 48, 72 and 96 h (B). A size marker is shown in lane M. The expected molecular weight of intact fusion protein and of the mRFP domain is 86.5 KDa (black arrow) and 26.6 KDa (white arrow), respectively.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Prediction of putative Tat signal peptide using TatP-1.0.
| No. | Species | E Value | Per. Ident | Upstream | Length (AA) | TatP-1.0 | Note |
|---|---|---|---|---|---|---|---|
| 1 | Kitasatospora aureofaciens | 0 | 95.24 | v | 44 | Predicted | This study |
| 2 | Mycobacterium lacus | 0 | 65.19 | n.d | |||
| 3 | Actinomyces sp. Lu 9419 | 6.00 × 10−175 | 56.75 | v | 101 | n.d | |
| 4 | Amycolatopsis japonica | 3.00 × 10−168 | 55.29 | n.d | |||
| 5 | Frankia alni ACN14a | 7.00 × 10−152 | 51.06 | n.d | |||
| 6 | Streptomyces sudanensis | 9.00 × 10−140 | 49.01 | n.d | |||
| 7 | Streptomyces davawennsis JCM 4913 | 3.00 × 10−135 | 48.99 | v | 55 | n.d | |
| 8 | Streptomyces alboniger | 8.00 × 10−139 | 48.91 | v | 56 | Predicted | TatP-1.0 score 5 out of 5 |
| 9 | Streptomyces sp. MRC013 | 1.00 × 10−138 | 48.74 | n.d | |||
| 10 | Phytohabitans suffuscus | 4.00 × 10−102 | 48.15 | n.d | n.d | ||
| 11 | Streptomyces cinnabarinus | 2.00 × 10−135 | 48.08 | v | 12 | n.d | |
| 12 | Streptomyces cyaneogriseus subsp. noncyanogenus | 2.00 × 10−93 | 48.04 | v | 52 | n.d | TatP-1.0 score 2 out of 5 |
| 13 | Streptomyces huasconensis | 4.00 × 10−136 | 47.76 | v | 64 | Predicted | TatP-1.0 score 5 out of 5 |
| 14 | Streptomyces tuirus | 6.00 × 10−143 | 46.94 | v | 32 | n.d | |
| 15 | Streptomyces lusitanus | 3.00 × 10−139 | 46.48 | n.d | |||
| 16 | Phytohabitans flavus | 3.00 × 10−125 | 46.23 | n.d | |||
| 17 | Nocardiopsis sp. Mg02 | 4.00 × 10−106 | 38.95 | v | 10 | n.d | |
| 19 | Microbacterium sp. 10M-3C3 | 5.00 × 10−63 | 38.25 | v | 50 | n.d | |
| 20 | Microbacterium atlanticum | 2.00 × 10−60 | 37.59 | v | 41 | n.d | |
| 21 | Microbacterium sp. KUDC0405 | 1.00 × 10−48 | 37.59 | v | 5 | n.d | |
| 22 | Microbacterium testaceum StLB037 | 6.00 × 10−62 | 37.55 | n.d | |||
| 23 | Micromonospora carbonacea | 7.00 × 10−76 | 37.1 | n.d | |||
| 24 | Arthrobacter sp. DNA4 | 6.00 × 10−59 | 37.08 | v | 35 | n.d | TatP-1.0 score 2 out of 5 |
| 25 | Actinoalloteichus sp. AHMU CJ021 | 1.00 × 10−59 | 37.06 | v | 100 | Predicted | More than 1 possibility for TatP-1.0 prediction |
| 26 | Microbacterium sp. XT11 | 7.00 × 10−63 | 37.02 | v | 10 | n.d | |
| 27 | Streptomyces canus | 9.00 × 10−61 | 33.71 | n.d | |||
| 28 | Pseudarthrobacter siccitolerans | 6.00 × 10−64 | 34.89 | v | 60 | Predicted | TatP-1.0 score 3 out of 5 |
Table 2.
Vectors used in this study.
| Name | Recombinant Gene | Host | Origin |
| pUC19 | - | E. coli | NEB |
| pNZ8901 | - | E. coli/B. subtilis | MoBiTec |
| pNZ8901-spkapox | KaPOx with signal peptide (SPKaPOx) | E. coli/B. subtilis | This study |
| pNZ8901-kapox | KaPOx | E. coli/B. subtilis | This study |
| pIJ486 | - | S. lividans TK24 | KU Leuven |
| pIJ486-spsec-mrfp | mRFP with signal peptide | S. lividans TK24 | KU Leuven |
| pUC19-Pvsi | Promoter VSI (Pvsi) | E. coli | This study |
| pUC19-Pvsi-mrfp | Pvsi, mrfp | E. coli | This study |
| pUC19-Pvsi-spkapox | Pvsi, SPKaPOx | E. coli | This study |
| pUC19-Pvsi-kapox | Pvsi, KaPOx | E. coli | This study |
| pUC19-Pvsi-spkapox-mrfp | Pvsi, SPKaPOx, mrfp | E. coli | This study |
| pUC19-Pvsi-kapox-mrfp | Pvsi, KaPOx, mrfp | E. coli | This study |
| pIJ486-mrfp | Pvsi, mrfp | S. lividans TK24 | This study |
| pIJ486-spkapox | Pvsi, SPKaPOx | S. lividans TK24 | This study |
| pIJ486-kapox | Pvsi, KaPOx | S. lividans TK24 | This study |
| pIJ486-spkapox-mrfp | Pvsi, SPKaPOx, mrfp | S. lividans TK24 | This study |
| pIJ486-kapox-mrfp | Pvsi, KaPOx, mrfp | S. lividans TK24 | This study |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Virginia, L.J.; Peterbauer, C. Localization of Pyranose 2-Oxidase from Kitasatospora aureofaciens: A Step Closer to Elucidate a Biological Role. Int. J. Mol. Sci. 2023, 24, 1975. https://doi.org/10.3390/ijms24031975
AMA Style
Virginia LJ, Peterbauer C. Localization of Pyranose 2-Oxidase from Kitasatospora aureofaciens: A Step Closer to Elucidate a Biological Role. International Journal of Molecular Sciences. 2023; 24(3):1975. https://doi.org/10.3390/ijms24031975
Chicago/Turabian StyleVirginia, Ludovika Jessica, and Clemens Peterbauer. 2023. "Localization of Pyranose 2-Oxidase from Kitasatospora aureofaciens: A Step Closer to Elucidate a Biological Role" International Journal of Molecular Sciences 24, no. 3: 1975. https://doi.org/10.3390/ijms24031975
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.