
Inhibitors of the Sialidase NEU3 as Potential Therapeutics for Fibrosis

Department of Biology, Texas A&M University, College Station, TX 77843-3474, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2023, 24(1), 239; https://doi.org/10.3390/ijms24010239
Submission received: 9 November 2022
/
Revised: 9 December 2022
/
Accepted: 13 December 2022
/
Published: 23 December 2022
(This article belongs to the Special Issue Molecular and Cellular Mechanisms of Idiopathic Pulmonary Fibrosis and Interstitial Lung Diseases)
{kind=link}
{kind=link}
Abstract
:Fibrosing diseases are a major medical problem, and are associated with more deaths per year than cancer in the US. Sialidases are enzymes that remove the sugar sialic acid from glycoconjugates. In this review, we describe efforts to inhibit fibrosis by inhibiting sialidases, and describe the following rationale for considering sialidases to be a potential target to inhibit fibrosis. First, sialidases are upregulated in fibrotic lesions in humans and in a mouse model of pulmonary fibrosis. Second, the extracellular sialidase NEU3 appears to be both necessary and sufficient for pulmonary fibrosis in mice. Third, there exist at least three mechanistic ways in which NEU3 potentiates fibrosis, with two of them being positive feedback loops where a profibrotic cytokine upregulates NEU3, and the upregulated NEU3 then upregulates the profibrotic cytokine. Fourth, a variety of NEU3 inhibitors block pulmonary fibrosis in a mouse model. Finally, the high sialidase levels in a fibrotic lesion cause an easily observed desialylation of serum proteins, and in a mouse model, sialidase inhibitors that stop fibrosis reverse the serum protein desialylation. This then indicates that serum protein sialylation is a potential surrogate biomarker for the effect of sialidase inhibitors, which would facilitate clinical trials to test the exciting possibility that sialidase inhibitors could be used as therapeutics for fibrosis.
1. Fibrosis
Fibrosis is a disease that involves an increased amount of scar tissue appearing in an internal organ. In a fibrosing disease, an insult or an injury to an internal organ initiates an uncontrolled wound repair mechanism which leads to a progressive increase in the deposition of scar tissue, eventually leading to organ failure and death [1,2]. There are more than 60 different fibrosing diseases, and 30–45% of deaths in the western world are from diseases where fibrosis was a significant factor [1]. One tissue that can develop fibrosis is the lung [3]. The lungs are directly exposed to external harmful environmental factors present in air, which can cause a recurring injury in the lungs, initiating a repeated wound healing response and eventually causing pulmonary fibrosis [4,5,6,7].
Interstitial lung disease (ILD) is a general term for a group of diseases that includes idiopathic pulmonary fibrosis (IPF), a chronic inflammatory disorder characterized by the accumulation of scar tissue in the lungs [8]. With an incidence rate of 2–30 cases per 100,000 person-years, this translates to a population prevalence of ~3 million people worldwide [8,9]. IPF has a survival rate of only 30% within five years after initial diagnosis, and has an incidence of 1 in 400 in patients older than 65 years [10]. Despite the high prevalence of IPF, there are only two FDA-approved therapeutics which slow down but do not reverse the progression of the disease [11].
2. Sialidases
In eukaryotic cells, some proteins undergo post-translational modification, such as glycosylation adding a chain of sugar molecules onto specific amino acids in the protein sequence [12,13]. In some glycoconjugates, the distal tip or tips (some glycoconjugates are branched) is the sugar sialic acid [14,15,16]. The sialic acid can make a glycoconjugate fully functional and can thus affect physiological and pathological processes [16,17,18,19]. Conversely, loss of the sialic acid can cause the glycoconjugate to lose its functional ability [16,17,20].
The enzymes that remove sialic acid from glycoconjugates are called neuraminidases or sialidases. Sialidases are present in, and sometimes on, viruses, bacteria, protozoa, and mammalian cells [21,22]. Viruses such as influenza require sialidase to release the virus from the sialic acids on the outside of a host cell [23]. The bacterial respiratory pathogen Pseudomonas aeruginosa uses a sialidase to colonize the lungs [24]. The sialidase NanA from Streptococcus pneumoniae causes downregulation of tight junction protein expression in endothelial cells and allows bacterial invasion through the blood–brain barrier, which can then affect the central nervous system [25]. Mammals have four sialidases, named NEU1, NEU2, NEU3, and NEU4. NEU1 is in the lysosome [26,27,28], NEU2 is a soluble, cytosolic enzyme, and NEU4 has 2 isoforms, one in mitochondria, and the other on intracellular membranes [29,30,31]. NEU3 is in endosomes and on the extracellular side of the plasma membrane, and under some conditions, can be released from the membrane to the extracellular environment [32].
Mammalian sialidases play an important role in the catabolism of glycoconjugates, modulation of the sialylation on cell membranes, skeletal muscle architecture, central nervous system function, and immune function [33,34,35]. Immune cells express different sialidases. Human neutrophils contain sialidase enzyme activity and express NEU2 protein [36]. As monocytes differentiate into macrophages, they increase the expression of NEU1 and NEU3 and reduce the expression of NEU4 [37], and T cells have increased NEU1 expression after stimulation [38].
Upregulation of sialidases is associated with inflammation [39,40,41,42,43,44,45]. For instance, NEU1 potentiates airway inflammation in asthma [46], and upregulated levels of NEU1 potentiate inflammation in atherosclerosis [47]. In cardiovascular disease models, both NEU1 and NEU3 are upregulated, and genetic disruption of NEU1 or NEU3 in mice, or treatment of mice with sialidase inhibitors, attenuate atherosclerosis, cardiac hypertrophy, or cardiac fibrosis [48,49,50]. In the LPS-induced air pouch model of inflammation, leukocyte recruitment is reduced in Neu1−/− and Neu3−/− mice, while it is increased in Neu4−/− mice [51]. Neu3−/− mice are also protected from Salmonella enterica Typhimurium-induced intestinal inflammation and colitis [52], and colitis-induced colon cancer [53]. NEU3 is upregulated in, and is important for the survival of, tumor cells in colon [54], renal [55], ovarian [56], and prostate cancers [57].
Sialidase inhibitors have been used to treat inflammation in both preclinical models and in patients. Zanamivir (Relenza) was designed to inhibit influenza sialidases, but at 100 mg/kg, it has shown efficacy in an animal model of arthritis, with reduced joint inflammation and arthritis score, reduced autoantibody production, and increased sialyation of serum IgG [58]. Recently, sialidase inhibitors (oseltamivir and peramivir) have been shown to reduce mortality in patients with severe COVID-19 [59].
In addition, the sialidase NEU1 is upregulated in IPF [60]. Although the specific upregulated sialidase was not identified, 8 of 9 IPF patients had a high sialidase activity in the lung bronchoalveolar lavage (BAL) fluid, while healthy controls showed no detectable sialidase activity [61]. These studies point towards a significant involvement of sialidases in the progression of pulmonary fibrosis.
3. Transforming Growth Factor-β1 (TGF-β1)
TGF-β1 is a homodimer peptide signal that regulates processes such as cell proliferation and growth, apoptosis, and maintaining immune homeostasis [62,63,64]. TGF-β1 upregulation plays a key role in the progression of pulmonary fibrosis [65]. TGF-β1 is synthesized as a precursor polypeptide consisting of latency-associated peptide (LAP) and what will become one of the peptides of the active TGF-β1 product [66]. The precursor polypeptide is processed in the endoplasmic reticulum (ER) within a cell and dimerizes to form a complex of two active TGF-β1 polypeptides enclosed in two LAPs [63]. For further processing, the nascent TGF-β1 is transferred from the ER to the Golgi, where it undergoes glycosylation/sialylation events on three asparagine residues, which are all present on LAP [63,66,67]. This processed complex is secreted from cells alone or in association with the latent TGF-β-binding protein (LTBP) [63,66]. Several mechanisms such as proteolysis, mechanical tension, or changes in pH cause LAP to release active TGF-β1 [66,68]. In addition to the above mechanisms, viral and bacterial sialidases can desialylate LAP causing LAP to change its conformation and releases active TGF-β1 [25,69,70,71,72,73].
The released active TGF-β1 plays an important role in maintaining homeostasis within the lungs [68,74,75]. When homeostasis is disturbed and TGF-β1 is upregulated, it induces epithelial-to-mesenchymal transition (EMT) in the alveolar epithelial cells [76,77], potentiates proliferation and differentiation of fibroblasts to myofibroblasts, and causes deposition of collagen in the surrounding tissue of the lungs [65,78,79,80]. Since these are a few of many hallmarks of pulmonary fibrosis, targeting TGF-β1 can be a potential therapeutic strategy to control pulmonary fibrosis. This strategy has been previously tried, but the strategies that directly block TGF-β1 cause various side effects, often interfering with the vital homeostatic functions of active TGF-β1 [81]. Thus, a better strategy needs to be devised to target the upregulation of TGF-β1 in a fibrotic lesion, rather than global TGF-β1.
4. NEU3 Is Necessary and Sufficient for Pulmonary Fibrosis and Is Elevated in Fibrosis in Other Organs
There is extensive desialylation of glycoconjugates and upregulation of the sialidase NEU3 in the fibrotic lesions of patients with pulmonary fibrosis and mice with bleomycin-induced pulmonary fibrosis [61,82,83,84,85]. For instance, we observed extensive desialylation of glycoconjugates and upregulation of the sialidases NEU1, 2, and 3 in fibrotic lesions in human and male mouse lungs. Most glycoconjugates are outside cells, and of the sialidases, only NEU3 is located on the extracellular side of the plasma membrane. Compared to male and female C57BL/6 mice, male and female Neu3−/− mice showed strongly attenuated bleomycin-induced weight loss, lung damage, inflammation, upregulation of TGF-β1, and fibrosis [83]. In the Neu3−/− mice, bleomycin did not upregulate NEU1 or NEU2, indicating that NEU3 is a key driver of sialidase upregulation [83]. Conversely, aspirations of NEU3 (with levels corresponding to the level of NEU3 in a fibrotic mouse lung) induced fibrosis in male and female mice [86]. These results suggest that NEU3 is necessary and sufficient for pulmonary fibrosis in mice. We found that epithelial cells, endothelial cells, macrophages, and fibroblasts in the fibrotic lungs express NEU3 [82,87], that NEU3, but not other sialidases, is upregulated in BAL fluid from pulmonary fibrosis patients and male and female mice at day 21 after bleomycin ([82,83], and observed elevated NEU3 in human fibrotic heart, kidney, and liver. Together, these results indicate that NEU3 potentiates fibrosis.
5. NEU3 Desialylates and Inactivates the Anti-Fibrotic Serum Protein SAP and SAP Is Desialylated in the Sera of IPF Patients
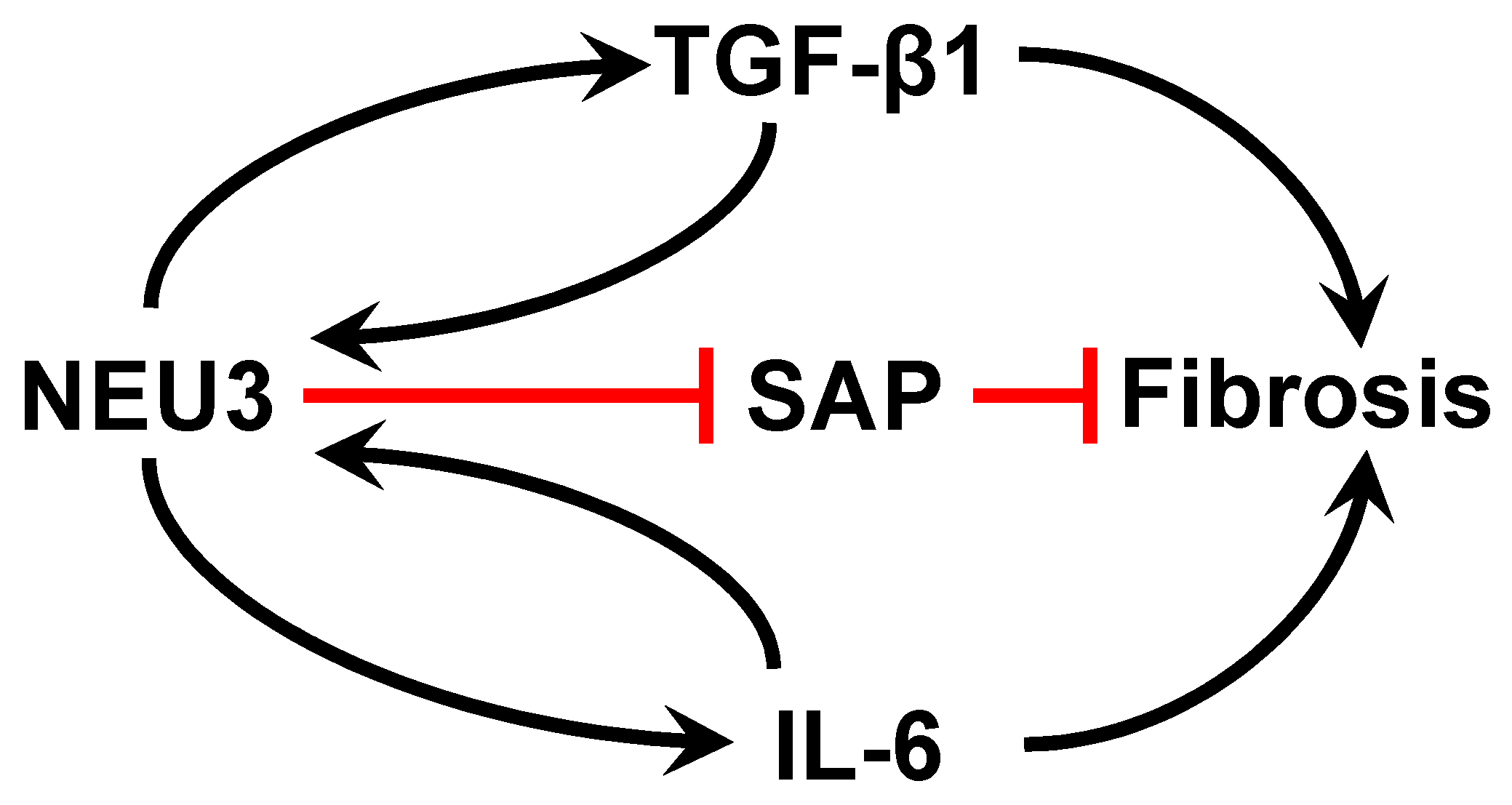
We previously found that a human serum glycoprotein called serum amyloid P (SAP) [88] calms the innate immune system [20,89,90,91,92], and that injections of SAP are therapeutic in mouse and rat models of pulmonary fibrosis [93]. We and others found that SAP injections are therapeutic in a wide variety of other fibrosis models [94,95,96,97,98,99,100,101,102,103,104,105]. Intravenous administration of recombinant SAP (called PRM-151 and zinpentraxin-alfa) was safe in Phase 1 trials [106,107] and works significantly better than the current standard of care in Phase 2 clinical trials for pulmonary fibrosis [108,109,110]. Phase 3 trials for SAP/PRM-151 in pulmonary fibrosis patients are ongoing. SAP is a homopentamer, and each monomer has a branched glycosylation with sialic acid at the distal tips of the glycosylation [88,111]. We found that SAP calms the innate immune system in large part by using its glycosylation to activate the lectin receptor DC-SIGN on innate immune system cells [20], and that desialylation of SAP blocks its ability to calm the innate immune system [20,111]. We found extensive desialylation of SAP in the sera of patients with idiopathic pulmonary fibrosis (IPF), compared to the SAP in the sera of healthy volunteers [111]. In the IPF patients, there was more extensive desialylation of SAP in progressive IPF cases than in stable IPF cases [111]. Compared to SAP from healthy donors, the SAP from IPF patients had a poor biological activity. Enzymatically sialylating the SAP from IPF patient sera made the SAP as active as control SAP, and enzymatically desialylating healthy donor SAP made it less active. Some but not all IPF patients, and none of the control patients, had detectable NEU3 in their sera [111]. Mass spectrometry showed that NEU3 desialylates SAP, suggesting that one way that elevated NEU3 in pulmonary fibrosis potentiates fibrosis is by desialylating the endogenous anti-fibrotic protein SAP (Figure 1).
6. In Addition to Inactivating SAP, NEU3 Activates 2 Positive Feedback Loops to Potentiate Fibrosis
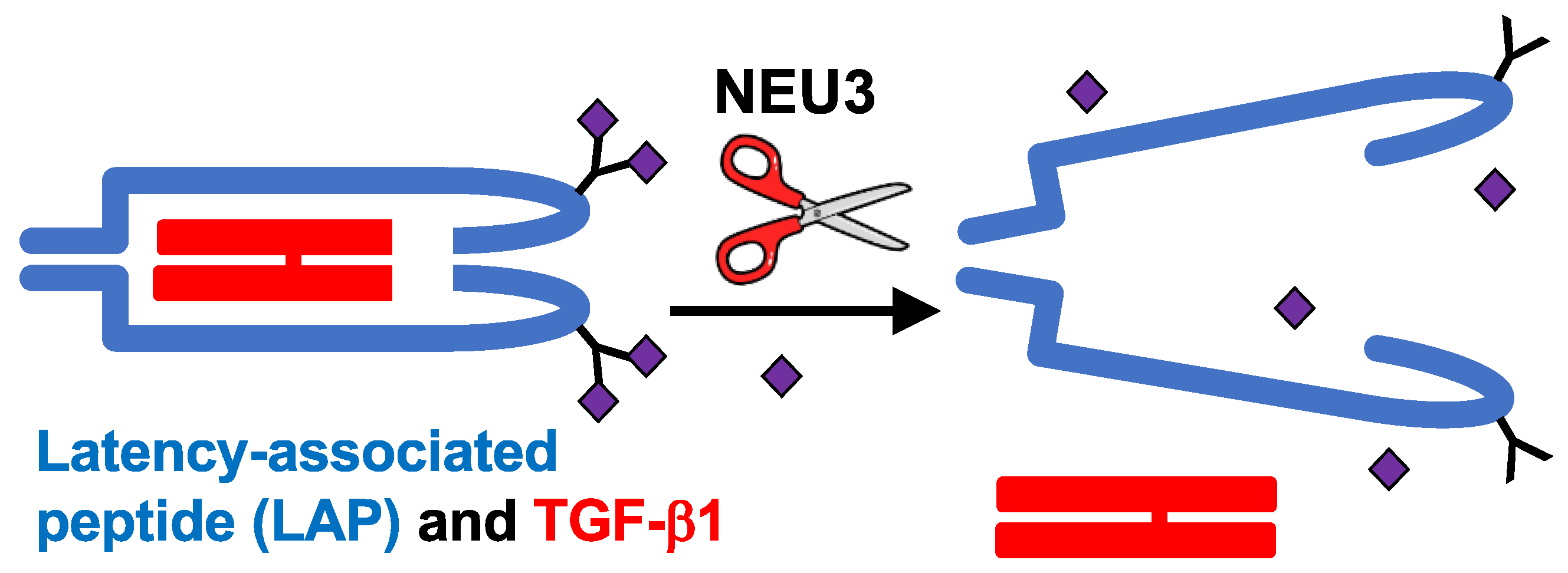
In human peripheral blood mononuclear cells, NEU3 upregulates extracellular accumulation of the profibrotic cytokines IL-6 and IL-1β, and IL-6 upregulates NEU3, suggesting that NEU3 may be part of a positive feedback loop potentiating fibrosis (bottom left of Figure 1 and [83]). Mammalian sialidases cause extracellular accumulation of active TGF-β1 [82]. Recombinant human LAP is sialylated, and we found that recombinant human NEU3 desialylates LAP [84] causing LAP to release active TGF-β1 ([84] and Figure 2). Conversely, TGF-β1 upregulates NEU3 [112]. Thus, in addition to the NEU3 → IL-6 → NEU3 positive feedback loop, a second positive feedback loop involves NEU3 → TGF-β1 → NEU3 (top left of Figure 1). NEU3 also primes human neutrophils [113], suggesting that upregulated NEU3 can contribute to inflammation in addition to fibrosis.
However, NEU3 may also inhibit TGF-β signaling [114,115]. Human cardiac fibroblasts cultured with TGF-β1 for 72 h have reduced NEU3 mRNA levels and reduced sialidase activity (as assessed with 4-MU-NeuAc at pH 3.8), and overexpression of NEU3 reduced TGF-β1-induced collagen production and inhibited TGF-β signaling [114].
7. TGF-β1 Upregulates Translation of NEU3 and Other mRNAs
We observed increased levels of NEU3 protein in fibrotic lesions in human and mouse pulmonary fibrosis [82]. However, RNA-seq and single-cell RNA-seq indicated that there is no significant increase in the levels of NEU3 mRNA in IPF patient lungs or lung cell types [116,117]. This suggested that the increased levels of NEU3 protein in fibrotic lesions could be due to increased translation of NEU3 mRNA. Using sucrose gradients to fractionate RNA-ribosome complexes into monosomes (mRNA with a single ribosome; indicative of a low translation rate per mRNA), and mRNA with multiple ribosomes (polysomes, indicative of high levels of translation) [118], we found that TGF-β1 increases the NEU3 protein in human lung epithelial cells without increasing total levels of NEU3 mRNA, and causes NEU3 mRNA to shift from monosomes to polysomes [112]. This then indicated that TGF-β1 increases levels of the NEU3 protein by increasing NEU3 mRNA translation.
8. Development of Potent NEU3 Inhibitors That Block Fibrosis in a Mouse Model
In the bleomycin model, the general sialidase inhibitors DANA and Oseltamivir given starting at day 10 strongly decreased fibrosis, with collagen and TGF-β1 levels not significantly different from saline controls [82]. These sialidase inhibitors however have ~10 µM IC50’s for NEU3, and are thus low-potency inhibitors. We found that compounds which mimic the transition state of the sialic acid when NEU3 is removing the sialic acid from a glycoconjugate act as potent (some less than 2 nM IC50) NEU3 inhibitors [84]. We tested three of these NEU3 inhibitors in the mouse bleomycin model of pulmonary fibrosis, and all three (with one showing strong efficacy at 0.1 mg/kg) decreased fibrosis and TGF-β1 accumulation in the lung (supporting the model in Figure 1) when administered starting at day 10, with no discernable toxicity [84]. The observation that the upregulated NEU3 in IPF desialylates and inactivates SAP suggests that our new class of NEU3 inhibitors could be useful as stand-alone therapeutics, or therapeutics to bolster the efficacy of SAP. Sialidase inhibitors may also be useful in other diseases. For instance, in mice, injections of the sialidase inhibitor DANA, or the lack of NEU3 (Neu3−/− mice) attenuate diet-induced adipose tissue and liver inflammation, and DANA reduces liver steatosis [104,119].
9. Development of a Surrogate Biomarker for Blocking NEU3 Activity or Upregulation
We also found that several other glycoproteins are desialylated in the sera of IPF patients [111]. In the mouse bleomycin model, there was also a bleomycin-induced increase in serum protein desialylation at 21 days after bleomycin aspiration. NEU3 inhibitors administered starting at day 10 partially reversed the bleomycin-induced increase in serum protein desialylation. This suggests that in clinical trials of a NEU3 inhibitor, increased serum SAP sialylation, or increased general protein sialylation, could be used as an early surrogate marker for NEU3 inhibitor efficacy.
Funding
This work was supported by NIH grant R01 HL132919.
Acknowledgments
We thank Tom Meek, Texas A&M, for the collaboration that identified the new NEU3 inhibitors.
Conflicts of Interest
R.H.G. is a scientific founder of Prosia Therapies, an early-stage company developing NEU3 inhibitors as therapeutics for pulmonary fibrosis. Texas A&M University has published patent applications on the use of sialidase inhibitors to regulate fibrosis. T.R.K., D.P. and R.H.G. are inventors on pending patent applications for the use of sialidase inhibitors as anti-inflammatory, anti-fibrotic, and/or anti-obesity compounds.
References
- Duffield, J.S.; Lupher, M.; Thannickal, V.J.; Wynn, T.A. Host Responses in Tissue Repair and Fibrosis. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 241–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of Idiopathic Pulmonary Fibrosis. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 157–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zisman, D.A.; Keane, M.P.; Belperio, J.A.; Strieter, R.M.; Lynch, J.P., 3rd. Pulmonary fibrosis. Methods Mol. Med. 2005, 117, 3–44. [Google Scholar] [PubMed]
- Hubbard, R.; Lewis, S.; Richards, K.; Johnston, I.; Britton, J. Occupational exposure to metal or wood dust and aetiology of cryptogenic fibrosing alveolitis. Lancet 1996, 347, 284–289. [Google Scholar] [CrossRef]
- Baumgartner, K.B.; Samet, J.M.; Coultas, D.B.; Stidley, C.A.; Hunt, W.C.; Colby, T.V.; Waldron, J.A.; Collaborating Centers. Occupational and environmental risk factors for idiopathic pulmonary fibrosis: A multicenter case-control study. Am. J. Epidemiol. 2000, 152, 307–315. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Zhou, Y.; Gaggar, A.; Duncan, S.R. Fibrosis: Ultimate and proximate causes. J. Clin. Investig. 2014, 124, 4673–4677. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.-F.; Flaherty, K.R.; Lasky, J.A.; et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-based Guidelines for Diagnosis and Management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef]
- Raghu, G.; Rochwerg, B.; Zhang, Y.; Garcia, C.A.; Azuma, A.; Behr, J.; Brozek, J.L.; Collard, H.R.; Cunningham, W.; Homma, S.; et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2015, 192, e3–e19. [Google Scholar] [CrossRef]
- Varki, A.; Cummings, R.D.; Aebi, M.; Packer, N.H.; Seeberger, P.H.; Esko, J.D.; Stanley, P.; Hart, G.; Darvill, A.; Kinoshita, T.; et al. Symbol Nomenclature for Graphical Representations of Glycans. Glycobiology 2015, 25, 1323–1324. [Google Scholar] [CrossRef] [Green Version]
- Spiro, R.G. Glycoproteins. Adv. Protein Chem. 1973, 27, 349–467. [Google Scholar]
- Schwab, I.; Nimmerjahn, F. Intravenous immunoglobulin therapy: How does IgG modulate the immune system? Nat. Rev. Immunol. 2013, 13, 176–189. [Google Scholar] [CrossRef]
- Freire-de-Lima, L.; Oliveira, I.A.; Neves, J.L.; Penha, L.L.; Alisson-Silva, F.; Dias, W.B.; Todeschini, A.R. Sialic acid: A sweet swing between mammalian host and Trypanosoma cruzi. Front. Immunol. 2012, 3, 356. [Google Scholar] [CrossRef] [Green Version]
- Varki, A.; Gagneux, P. Multifarious roles of sialic acids in immunity. Ann. N. Y. Acad. Sci. 2012, 1253, 16–36. [Google Scholar] [CrossRef] [Green Version]
- Varki, N.M.; Varki, A. Diversity in cell surface sialic acid presentations: Implications for biology and disease. Lab. Investig. 2007, 87, 851–857. [Google Scholar] [CrossRef] [Green Version]
- Varki, A. Sialic acids in human health and disease. Trends Mol. Med. 2008, 14, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Varki, A.; Crocker, P.R. I-type Lectins. In Essentials of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Cox, N.; Pilling, D.; Gomer, R.H. DC-SIGN activation mediates the differential effects of SAP and CRP on the innate immune system and inhibits fibrosis in mice. Proc. Natl. Acad. Sci. USA 2015, 112, 8385–8390. [Google Scholar] [CrossRef] [Green Version]
- Pshezhetsky, A.V.; Ashmarina, L.I. Desialylation of surface receptors as a new dimension in cell signaling. Biochem. Biokhimiia 2013, 78, 736–745. [Google Scholar] [CrossRef]
- Miyagi, T.; Takahashi, K.; Hata, K.; Shiozaki, K.; Yamaguchi, K. Sialidase significance for cancer progression. Glycoconj. J. 2012, 29, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Razonable, R.R. Antiviral drugs for viruses other than human immunodeficiency virus. Mayo Clin. Proc. 2011, 86, 1009–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soong, G.; Muir, A.; Gomez, M.I.; Waks, J.; Reddy, B.; Planet, P.; Singh, P.K.; Kaneko, Y.; Wolfgang, M.C.; Hsiao, Y.S.; et al. Bacterial neuraminidase facilitates mucosal infection by participating in biofilm production. J. Clin. Investig. 2006, 116, 2297–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gratz, N.; Loh, L.N.; Mann, B.; Gao, G.; Carter, R.; Rosch, J.; Tuomanen, E.I. Pneumococcal neuraminidase activates TGF-beta signalling. Microbiology 2017, 163, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Bonten, E.; van der Spoel, A.; Fornerod, M.; Grosveld, G.; d’Azzo, A. Characterization of human lysosomal neuraminidase defines the molecular basis of the metabolic storage disorder sialidosis. Genes Dev. 1996, 10, 3156–3169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pshezhetsky, A.V.; Richard, C.; Michaud, L.; Igdoura, S.; Wang, S.; Elsliger, M.A.; Qu, J.; Leclerc, D.; Gravel, R.; Dallaire, L.; et al. Cloning, expression and chromosomal mapping of human lysosomal sialidase and characterization of mutations in sialidosis. Nat. Genet. 1997, 15, 316–320. [Google Scholar] [CrossRef]
- Lillehoj, E.P.; Hyun, S.W.; Feng, C.; Zhang, L.; Liu, A.; Guang, W.; Nguyen, C.; Luzina, I.G.; Atamas, S.P.; Passaniti, A.; et al. NEU1 sialidase expressed in human airway epithelia regulates epidermal growth factor receptor (EGFR) and MUC1 protein signaling. J. Biol. Chem. 2012, 287, 8214–8231. [Google Scholar] [CrossRef] [Green Version]
- D’Avila, F.; Tringali, C.; Papini, N.; Anastasia, L.; Croci, G.; Massaccesi, L.; Monti, E.; Tettamanti, G.; Venerando, B. Identification of lysosomal sialidase NEU1 and plasma membrane sialidase NEU3 in human erythrocytes. J. Cell. Biochem. 2013, 114, 204–211. [Google Scholar] [CrossRef]
- Monti, E.; Erik, B.; D’Azzo, A.; Bresciani, R.; Venerando, B.; Borsani, G.; Schauer, R.; Tettamanti, G. Sialidases in Vertebrates: A Family of Enzymes Tailored for Several Cell Functions. Adv. Carbohydr. Chem. Biochem. 2010, 64, 403–479. [Google Scholar]
- Smutova, V.; Albohy, A.; Pan, X.; Korchagina, E.; Miyagi, T.; Bovin, N.; Cairo, C.W.; Pshezhetsky, A.V. Structural Basis for Substrate Specificity of Mammalian Neuraminidases. PLoS ONE 2014, 9, e106320. [Google Scholar] [CrossRef] [Green Version]
- Zanchetti, G.; Colombi, P.; Manzoni, M.; Anastasia, L.; Caimi, L.; Borsani, G.; Venerando, B.; Tettamanti, G.; Preti, A.; Monti, E.; et al. Sialidase NEU3 is a peripheral membrane protein localized on the cell surface and in endosomal structures. Biochem. J. 2007, 408, 211–219. [Google Scholar] [CrossRef] [Green Version]
- Lewis, A.L.; Lewis, W.G. Host sialoglycans and bacterial sialidases: A mucosal perspective. Cell. Microbiol. 2012, 14, 1174–1182. [Google Scholar] [CrossRef]
- Fanzani, A.; Zanola, A.; Faggi, F.; Papini, N.; Venerando, B.; Tettamanti, G.; Sampaolesi, M.; Monti, E. Implications for the mammalian sialidases in the physiopathology of skeletal muscle. Skelet. Muscle 2012, 2, 23. [Google Scholar] [CrossRef] [Green Version]
- Pshezhetsky, A.V.; Ashmarina, M. Keeping it trim: Roles of neuraminidases in CNS function. Glycoconj. J. 2018, 35, 375–386. [Google Scholar] [CrossRef] [Green Version]
- Hyun, S.W.; Feng, C.; Liu, A.; Lillehoj, E.P.; Trotta, R.; Kingsbury, T.J.; Passaniti, A.; Lugkey, K.N.; Chauhan, S.; Cipollo, J.F.; et al. Altered sialidase expression in human myeloid cells undergoing apoptosis and differentiation. Sci. Rep. 2022, 12, 14173. [Google Scholar] [CrossRef]
- Stamatos, N.M.; Liang, F.; Nan, X.; Landry, K.; Cross, A.S.; Wang, L.X.; Pshezhetsky, A.V. Differential expression of endogenous sialidases of human monocytes during cellular differentiation into macrophages. FEBS J. 2005, 272, 2545–2556. [Google Scholar] [CrossRef]
- Nan, X.; Carubelli, I.; Stamatos, N.M. Sialidase expression in activated human T lymphocytes influences production of IFN-gamma. J. Leukoc. Biol. 2007, 81, 284–296. [Google Scholar] [CrossRef]
- Liang, F.; Seyrantepe, V.; Landry, K.; Ahmad, R.; Ahmad, A.; Stamatos, N.M.; Pshezhetsky, A.V. Monocyte differentiation up-regulates the expression of the lysosomal sialidase, Neu1, and triggers its targeting to the plasma membrane via major histocompatibility complex class II-positive compartments. J. Biol. Chem. 2006, 281, 27526–27538. [Google Scholar] [CrossRef] [Green Version]
- Amith, S.R.; Jayanth, P.; Franchuk, S.; Siddiqui, S.; Seyrantepe, V.; Gee, K.; Basta, S.; Beyaert, R.; Pshezhetsky, A.V.; Szewczuk, M.R. Dependence of pathogen molecule-induced toll-like receptor activation and cell function on Neu1 sialidase. Glycoconj. J. 2009, 26, 1197–1212. [Google Scholar] [CrossRef]
- Amith, S.R.; Jayanth, P.; Franchuk, S.; Finlay, T.; Seyrantepe, V.; Beyaert, R.; Pshezhetsky, A.V.; Szewczuk, M.R. Neu1 desialylation of sialyl alpha-2,3-linked beta-galactosyl residues of TOLL-like receptor 4 is essential for receptor activation and cellular signaling. Cell. Signal. 2010, 22, 314–324. [Google Scholar] [CrossRef]
- Stamatos, N.M.; Carubelli, I.; van de Vlekkert, D.; Bonten, E.J.; Papini, N.; Feng, C.; Venerando, B.; d’Azzo, A.; Cross, A.S.; Wang, L.X.; et al. LPS-induced cytokine production in human dendritic cells is regulated by sialidase activity. J. Leukoc. Biol. 2010, 88, 1227–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Zhang, J.; Bian, H.; Wu, P.; Kuvelkar, R.; Kung, T.T.; Crawley, Y.; Egan, R.W.; Billah, M.M. Induction of lysosomal and plasma membrane-bound sialidases in human T-cells via T-cell receptor. Biochem. J. 2004, 380, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Sakarya, S.; Rifat, S.; Zhou, J.; Bannerman, D.D.; Stamatos, N.M.; Cross, A.S.; Goldblum, S.E. Mobilization of neutrophil sialidase activity desialylates the pulmonary vascular endothelial surface and increases resting neutrophil adhesion to and migration across the endothelium. Glycobiology 2004, 14, 481–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, C.; Zhang, L.; Nguyen, C.; Vogel, S.N.; Goldblum, S.E.; Blackwelder, W.C.; Cross, A.S. Neuraminidase reprograms lung tissue and potentiates lipopolysaccharide-induced acute lung injury in mice. J. Immunol. 2013, 191, 4828–4837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, S.; Maeda, S.; Fukuoka, H.; Wada, T.; Moriya, S.; Mori, A.; Yamaguchi, K.; Senda, S.; Miyagi, T. A crucial role of sialidase Neu1 in hyaluronan receptor function of CD44 in T helper type 2-mediated airway inflammation of murine acute asthmatic model. Clin. Exp. Immunol. 2010, 161, 233–241. [Google Scholar] [CrossRef]
- Sieve, I.; Ricke-Hoch, M.; Kasten, M.; Battmer, K.; Stapel, B.; Falk, C.S.; Leisegang, M.S.; Haverich, A.; Scherr, M.; Hilfiker-Kleiner, D. A positive feedback loop between IL-1beta, LPS and NEU1 may promote atherosclerosis by enhancing a pro-inflammatory state in monocytes and macrophages. Vasc. Pharmacol. 2018, 103–105, 16–28. [Google Scholar] [CrossRef]
- Demina, E.P.; Smutova, V.; Pan, X.; Fougerat, A.; Guo, T.; Zou, C.; Chakraberty, R.; Snarr, B.D.; Shiao, T.C.; Roy, R.; et al. Neuraminidases 1 and 3 Trigger Atherosclerosis by Desialylating Low-Density Lipoproteins and Increasing Their Uptake by Macrophages. J. Am. Heart Assoc. 2021, 10, e018756. [Google Scholar]
- Guo, Z.; Tuo, H.; Tang, N.; Liu, F.Y.; Ma, S.Q.; An, P.; Yang, D.; Wang, M.Y.; Fan, D.; Yang, Z.; et al. Neuraminidase 1 deficiency attenuates cardiac dysfunction, oxidative stress, fibrosis, inflammatory via AMPK-SIRT3 pathway in diabetic cardiomyopathy mice. Int. J. Biol. Sci. 2022, 18, 826–840. [Google Scholar] [CrossRef]
- Chen, Q.Q.; Ma, G.; Liu, J.F.; Cai, Y.Y.; Zhang, J.Y.; Wei, T.T.; Pan, A.; Jiang, S.; Xiao, Y.; Xiao, P.; et al. Neuraminidase 1 is a driver of experimental cardiac hypertrophy. Eur. Heart J. 2021, 42, 3770–3782. [Google Scholar] [CrossRef]
- Howlader, M.A.; Demina, E.P.; Samarani, S.; Guo, T.; Caillon, A.; Ahmad, A.; Pshezhetsky, A.V.; Cairo, C.W. The Janus-like role of neuraminidase isoenzymes in inflammation. FASEB J. 2022, 36, e22285. [Google Scholar] [CrossRef]
- Yang, W.H.; Westman, J.S.; Heithoff, D.M.; Sperandio, M.; Cho, J.W.; Mahan, M.J.; Marth, J.D. Neu3 neuraminidase induction triggers intestinal inflammation and colitis in a model of recurrent human food-poisoning. Proc. Natl. Acad. Sci. USA 2021, 118, e2100937118. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Shiozaki, K.; Moriya, S.; Koseki, K.; Wada, T.; Tateno, H.; Sato, I.; Asano, M.; Iwakura, Y.; Miyagi, T. Reduced Susceptibility to Colitis-Associated Colon Carcinogenesis in Mice Lacking Plasma Membrane-Associated Sialidase. PLoS ONE 2012, 7, e41132. [Google Scholar] [CrossRef] [Green Version]
- Kakugawa, Y.; Wada, T.; Yamaguchi, K.; Yamanami, H.; Ouchi, K.; Sato, I.; Miyagi, T. Up-regulation of plasma membrane-associated ganglioside sialidase (Neu3) in human colon cancer and its involvement in apoptosis suppression. Proc. Natl. Acad. Sci. USA 2002, 99, 10718–10723. [Google Scholar] [CrossRef]
- Ueno, S.; Saito, S.; Wada, T.; Yamaguchi, K.; Satoh, M.; Arai, Y.; Miyagi, T. Plasma Membrane-associated Sialidase Is Up-regulated in Renal Cell Carcinoma and Promotes Interleukin-6-induced Apoptosis Suppression and Cell Motility. J. Biol. Chem. 2006, 281, 7756–7764. [Google Scholar] [CrossRef] [Green Version]
- Nomura, H.; Tamada, Y.; Miyagi, T.; Suzuki, A.; Taira, M.; Suzuki, N.; Susumu, N.; Irimura, T.; Aoki, D. Expression of NEU3 (plasma membrane-associated sialidase) in clear cell adenocarcinoma of the ovary: Its relationship with T factor of pTNM classification. Oncol. Res. 2006, 16, 289–297. [Google Scholar] [CrossRef]
- Kawamura, S.; Sato, I.; Wada, T.; Yamaguchi, K.; Li, Y.; Li, D.; Zhao, X.; Ueno, S.; Aoki, H.; Tochigi, T.; et al. Plasma membrane-associated sialidase (NEU3) regulates progression of prostate cancer to androgen-independent growth through modulation of androgen receptor signaling. Cell Death Differ. 2011, 19, 170. [Google Scholar] [CrossRef]
- Sehnert, B.; Mietz, J.; Rzepka, R.; Buchholz, S.; Maul-Pavicic, A.; Schaffer, S.; Nimmerjahn, F.; Voll, R.E. Neuraminidase Inhibitor Zanamivir Ameliorates Collagen-Induced Arthritis. Int. J. Mol. Sci. 2021, 22, 1428. [Google Scholar] [CrossRef]
- Wu, J.; Zhao, M.; Wei, H.; Li, C.; Hu, D.; Zheng, L.; Wang, D.W. Neuraminidase inhibitor treatment is associated with decreased mortality in COVID-19 patients: A retrospective analysis. Eur. Heart J.-Cardiovasc. Pharmacother. 2022, 8, 392–401. [Google Scholar] [CrossRef]
- Luzina, I.G.; Lockatell, V.; Hyun, S.W.; Kopach, P.; Kang, P.H.; Noor, Z.; Liu, A.; Lillehoj, E.P.; Lee, C.; Miranda-Ribera, A.; et al. Elevated expression of NEU1 sialidase in idiopathic pulmonary fibrosis provokes pulmonary collagen deposition, lymphocytosis, and fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L940–L954. [Google Scholar] [CrossRef] [Green Version]
- Lambré, C.R.; Pilatte, Y.; Le Maho, S.; Greffard, A.; De Crémoux, H.; Bignon, J. Sialidase activity and antibodies to sialidase-treated autologous erythrocytes in bronchoalveolar lavages from patients with idiopathic pulmonary fibrosis or sarcoidosis. Clin. Exp. Immunol. 1988, 73, 230–235. [Google Scholar]
- Roberts, A.B.; Sporn, M.B. Physiological actions and clinical applications of transforming growth factor-beta (TGF-beta). Growth Factors 1993, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-beta and TGF-beta-Related Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. [Google Scholar] [CrossRef] [PubMed]
- Wahl, S.M.; Wen, J.; Moutsopoulos, N. TGF-β: A mobile purveyor of immune privilege. Immunol. Rev. 2006, 213, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheppard, D. Transforming growth factor beta: A central modulator of pulmonary and airway inflammation and fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Hinck, A.P.; Mueller, T.D.; Springer, T.A. Structural Biology and Evolution of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2016, 8, a022103. [Google Scholar] [CrossRef] [Green Version]
- Moremen, K.W.; Tiemeyer, M.; Nairn, A.V. Vertebrate protein glycosylation: Diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 2012, 13, 448–462. [Google Scholar] [CrossRef] [Green Version]
- Travis, M.A.; Sheppard, D. TGF-β Activation and Function in Immunity. Annu. Rev. Immunol. 2014, 32, 51–82. [Google Scholar] [CrossRef] [Green Version]
- Schultz-Cherry, S.; Hinshaw, V.S. Influenza virus neuraminidase activates latent transforming growth factor beta. J. Virol. 1996, 70, 8624–8629. [Google Scholar] [CrossRef] [Green Version]
- Carlson, C.M.; Turpin, E.A.; Moser, L.A.; O’Brien, K.B.; Cline, T.D.; Jones, J.C.; Tumpey, T.M.; Katz, J.M.; Kelley, L.A.; Gauldie, J.; et al. Transforming growth factor-beta: Activation by neuraminidase and role in highly pathogenic H5N1 influenza pathogenesis. PLoS Pathog. 2010, 6, e1001136. [Google Scholar] [CrossRef] [Green Version]
- Grillet, F.; Behr, J.; Calame, P.; Aubry, S.; Delabrousse, E. Acute pulmonary embolism associated with COVID-19 pneumonia detected by pulmonary CT angiography. Radiology 2020, 296, E186–E188. [Google Scholar] [CrossRef] [Green Version]
- Sha, X.; Brunner, A.M.; Purchio, A.F.; Gentry, L.E. Transforming growth factor beta 1: Importance of glycosylation and acidic proteases for processing and secretion. Mol. Endocrinol. 1989, 3, 1090–1098. [Google Scholar] [CrossRef] [Green Version]
- Miyazono, K.; Heldin, C.H. Role for carbohydrate structures in TGF-beta 1 latency. Nature 1989, 338, 158–160. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Kökény, G.; Calvier, L.; Hansmann, G. PPARγ and TGFβ-Major Regulators of Metabolism, Inflammation, and Fibrosis in the Lungs and Kidneys. Int. J Mol. Sci. 2021, 22, 10431. [Google Scholar] [CrossRef]
- Salton, F.; Volpe, M.C.; Confalonieri, M. Epithelial–Mesenchymal Transition in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Medicina 2019, 55, 83. [Google Scholar] [CrossRef] [Green Version]
- Kasai, H.; Allen, J.T.; Mason, R.M.; Kamimura, T.; Zhang, Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res. 2005, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Yue, X.; Shan, B.; Lasky, J.A. TGF-β: Titan of Lung Fibrogenesis. Curr. Enzym. Inhib. 2010, 6, 67–77. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Eickelberg, O. The Impact of TGF-β on Lung Fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef]
- Moore, B.B.; Lawson, W.E.; Oury, T.D.; Sisson, T.H.; Raghavendran, K.; Hogaboam, C.M. Animal Models of Fibrotic Lung Disease. Am. J. Respir. Cell Mol. Biol. 2013, 49, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Datta, A.; Scotton, C.J.; Chambers, R.C. Novel therapeutic approaches for pulmonary fibrosis. Br. J. Pharm. 2011, 163, 141–172. [Google Scholar] [CrossRef] [Green Version]
- Karhadkar, T.R.; Pilling, D.; Cox, N.; Gomer, R.H. Sialidase inhibitors attenuate pulmonary fibrosis in a mouse model. Sci. Rep. 2017, 7, 15069. [Google Scholar] [CrossRef] [Green Version]
- Karhadkar, T.R.; Chen, W.; Gomer, R.H. Attenuated pulmonary fibrosis in sialidase-3 knockout (Neu3(-/-)) mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L165–L179. [Google Scholar] [CrossRef] [PubMed]
- Karhadkar, T.R.; Meek, T.D.; Gomer, R.H. Inhibiting sialidase-induced TGF-β1 activation attenuates pulmonary fibrosis in mice. J. Pharm. Exp. 2021, 376, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Lillehoj, E.P.; Hyun, S.W.; Feng, C.; Zhang, L.; Liu, A.; Guang, W.; Nguyen, C.; Sun, W.; Luzina, I.G.; Webb, T.J.; et al. Human airway epithelia express catalytically active NEU3 sialidase. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L876–L886. [Google Scholar] [CrossRef] [PubMed]
- Pilling, D.; Sahlberg, K.; Karhadkar, T.R.; Chen, W.; Gomer, R.H. The sialidase NEU3 promotes pulmonary fibrosis in mice. Respir. Res. 2022, 23, 215. [Google Scholar] [CrossRef]
- Pilling, D.; Sahlberg, K.; Chen, W.; Gomer, R.H. Changes in lung sialidases in male and female mice after bleomycin aspiration. Exp. Lung Res. 2022, 48, 291–304. [Google Scholar] [CrossRef]
- Pepys, M.B.; Rademacher, T.W.; Amatayakul-Chantler, S.; Williams, P.; Noble, G.E.; Hutchinson, W.L.; Hawkins, P.N.; Nelson, S.R.; Gallimore, J.R.; Herbert, J. Human serum amyloid P component is an invariant constituent of amyloid deposits and has a uniquely homogeneous glycostructure. Proc. Natl. Acad. Sci. USA 1994, 91, 5602–5606. [Google Scholar] [CrossRef] [Green Version]
- Pilling, D.; Buckley, C.D.; Salmon, M.; Gomer, R.H. Inhibition of fibrocyte differentiation by serum amyloid P. J. Immunol. 2003, 17, 5537–5546. [Google Scholar] [CrossRef] [Green Version]
- Maharjan, A.S.; Roife, D.; Brazill, D.; Gomer, R.H. Serum amyloid P inhibits granulocyte adhesion. Fibrogenesis Tissue Repair 2013, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Crawford, J.R.; Pilling, D.; Gomer, R.H. FcγRI mediates serum amyloid P inhibition of fibrocyte differentiation. J. Leukoc. Biol. 2012, 92, 699–711. [Google Scholar] [CrossRef] [Green Version]
- Pilling, D.; Gomer, R.H. Persistent Lung Inflammation and Fibrosis in Serum Amyloid P Component (Apcs −/−) Knockout Mice. PLoS ONE 2014, 9, e93730. [Google Scholar] [CrossRef] [Green Version]
- Pilling, D.; Roife, D.; Wang, M.; Ronkainen, S.D.; Crawford, J.R.; Travis, E.L.; Gomer, R.H. Reduction of bleomycin-induced pulmonary fibrosis by serum amyloid P. J. Immunol. 2007, 179, 4035–4044. [Google Scholar] [CrossRef] [Green Version]
- Cox, N.; Pilling, D.; Gomer, R.H. Serum amyloid P: A systemic regulator of the innate immune response. J. Leukoc. Biol. 2014, 96, 739–743. [Google Scholar] [CrossRef]
- Verstovsek, S.; Manshouri, T.; Pilling, D.; Bueso-Ramos, C.E.; Newberry, K.J.; Prijic, S.; Knez, L.; Bozinovic, K.; Harris, D.M.; Spaeth, E.L.; et al. Role of neoplastic monocyte-derived fibrocytes in primary myelofibrosis. J. Exp. Med. 2016, 213, 1723–1740. [Google Scholar] [CrossRef]
- Pilling, D.; Gomer, R.H. The Development of Serum Amyloid P as a Possible Therapeutic. Front. Immunol. 2018, 9, 2328. [Google Scholar] [CrossRef] [Green Version]
- Moreira, A.P.; Cavassani, K.A.; Hullinger, R.; Rosada, R.S.; Fong, D.J.; Murray, L.; Hesson, D.P.; Hogaboam, C.M. Serum amyloid P attenuates M2 macrophage activation and protects against fungal spore-induced allergic airway disease. J. Allergy Clin. Immunol. 2010, 126, 712–721.e7. [Google Scholar] [CrossRef]
- Murray, L.; Kramer, M.; Hesson, D.; Watkins, B.; Fey, E.; Argentieri, R.; Shaheen, F.; Knight, D.; Sonis, S. Serum amyloid P ameliorates radiation-induced oral mucositis and fibrosis. Fibrogenesis Tissue Repair 2010, 3, 11. [Google Scholar] [CrossRef] [Green Version]
- Haudek, S.B.; Xia, Y.; Huebener, P.; Lee, J.M.; Carlson, S.; Crawford, J.R.; Pilling, D.; Gomer, R.H.; Trial, J.; Frangogiannis, N.G.; et al. Bone Marrow-derived Fibroblast Precursors Mediate Ischemic Cardiomyopathy in Mice. Proc. Natl. Acad. Sci. USA 2006, 103, 18284–18289. [Google Scholar] [CrossRef] [Green Version]
- Murray, L.A.; Chen, Q.; Kramer, M.S.; Hesson, D.P.; Argentieri, R.L.; Peng, X.; Gulati, M.; Homer, R.J.; Russell, T.; van Rooijen, N.; et al. TGF-beta driven lung fibrosis is macrophage dependent and blocked by Serum amyloid P. Int. J. Biochem. Cell Biol. 2011, 43, 154–162. [Google Scholar] [CrossRef]
- Ji, Z.; Ke, Z.J.; Geng, J.G. SAP suppresses the development of experimental autoimmune encephalomyelitis in C57BL/6 mice. Immunol. Cell Biol. 2012, 90, 388–395. [Google Scholar] [CrossRef] [Green Version]
- Santhiago, M.R.; Singh, V.; Barbosa, F.L.; Agrawal, V.; Wilson, S.E. Monocyte development inhibitor PRM-151 decreases corneal myofibroblast generation in rabbits. Exp. Eye Res. 2011, 93, 810–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castano, A.P.; Lin, S.L.; Surowy, T.; Nowlin, B.T.; Turlapati, S.A.; Patel, T.; Singh, A.; Li, S.; Lupher, M.L., Jr.; Duffield, J.S. Serum amyloid P inhibits fibrosis through Fc gamma R-dependent monocyte-macrophage regulation in vivo. Sci. Transl. Med. 2009, 1, 5ra13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilling, D.; Cox, N.; Thomson, M.A.; Karhadkar, T.R.; Gomer, R.H. Serum Amyloid P and a Dendritic Cell Specific Intercellular Adhesion Molecule-3 Grabbing Nonintegrin Ligand Inhibit High-Fat Diet Induced Adipose Tissue and Liver Inflammation and Steatosis in Mice. Am. J. Pathol. 2019, 189, 2400–2413. [Google Scholar] [CrossRef] [PubMed]
- Cong, M.; Zhao, W.; Liu, T.; Wang, P.; Fan, X.; Zhai, Q.; Bao, X.; Zhang, D.; You, H.; Kisseleva, T.; et al. Protective effect of human serum amyloid P on CCl4-induced acute liver injury in mice. Int. J. Mol. Med. 2017, 40, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Dillingh, M.R.; van den Blink, B.; Moerland, M.; van Dongen, M.G.J.; Levi, M.; Kleinjan, A.; Wijsenbeek, M.S.; Lupher, M.L., Jr.; Harper, D.M.; Getsy, J.A.; et al. Recombinant human serum amyloid P in healthy volunteers and patients with pulmonary fibrosis. Pulm. Pharmacol. Ther. 2013, 26, 672–676. [Google Scholar] [CrossRef]
- van den Blink, B.; Dillingh, M.R.; Ginns, L.C.; Morrison, L.D.; Moerland, M.; Wijsenbeek, M.; Trehu, E.G.; Bartholmai, B.J.; Burggraaf, J. Recombinant human pentraxin-2 therapy in patients with idiopathic pulmonary fibrosis: Safety, pharmacokinetics and exploratory efficacy. Eur. Respir. J. 2016, 47, 889–897. [Google Scholar] [CrossRef] [Green Version]
- Raghu, G.; van den Blink, B.; Hamblin, M.J.; Brown, A.W.; Golden, J.A.; Ho, L.A.; Wijsenbeek, M.S.; Vasakova, M.; Pesci, A.; Antin-Ozerkis, D.E.; et al. Effect of Recombinant Human Pentraxin 2 vs Placebo on Change in Forced Vital Capacity in Patients With Idiopathic Pulmonary Fibrosis: A Randomized Clinical Trial. JAMA 2018, 319, 2299–2307. [Google Scholar] [CrossRef] [Green Version]
- Raghu, G.; van den Blink, B.; Hamblin, M.J.; Brown, A.W.; Golden, J.A.; Ho, L.A.; Wijsenbeek, M.S.; Vasakova, M.; Pesci, A.; Antin-Ozerkis, D.E.; et al. Long-term treatment with recombinant human pentraxin 2 protein in patients with idiopathic pulmonary fibrosis: An open-label extension study. Lancet Respir. Med. 2019, 7, 657–664. [Google Scholar] [CrossRef]
- Bose, P.; Masarova, L.; Verstovsek, S. Novel Concepts of Treatment for Patients with Myelofibrosis and Related Neoplasms. Cancers 2020, 12, 2891. [Google Scholar] [CrossRef]
- Chen, W.; Karhadkar, T.R.; Ryu, C.; Herzog, E.L.; Gomer, R.H. Reduced Sialylation and Bioactivity of the Antifibrotic Protein Serum Amyloid P in the Sera of Patients with Idiopathic Pulmonary Fibrosis. ImmunoHorizons 2020, 4, 352. [Google Scholar] [CrossRef]
- Chen, W.; Lamb, T.M.; Gomer, R.H. TGF-β1 increases sialidase 3 expression in human lung epithelial cells by decreasing its degradation and upregulating its translation. Exp. Lung Res. 2020, 46, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Kirolos, S.A.; Pilling, D.; Gomer, R.H. The extracellular sialidase NEU3 primes neutrophils. J. Leukoc. Biol. 2022, 112, 1399–1411. [Google Scholar] [CrossRef] [PubMed]
- Ghiroldi, A.; Piccoli, M.; Creo, P.; Cirillo, F.; Rota, P.; D’Imperio, S.; Ciconte, G.; Monasky, M.M.; Micaglio, E.; Garatti, A.; et al. Role of sialidase Neu3 and ganglioside GM3 in cardiac fibroblasts activation. Biochem. J. 2020, 477, 3401–3415. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, T.; Yamamoto, K. Sialidase NEU3 and its pathological significance. Glycoconj. J 2022, 39, 677–683. [Google Scholar] [CrossRef]
- Nance, T.; Smith, K.S.; Anaya, V.; Richardson, R.; Ho, L.; Pala, M.; Mostafavi, S.; Battle, A.; Feghali-Bostwick, C.; Rosen, G.; et al. Transcriptome Analysis Reveals Differential Splicing Events in IPF Lung Tissue. PLoS ONE 2014, 9, e92111. [Google Scholar] [CrossRef] [Green Version]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.-I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef]
- Panda, A.C.; Martindale, J.L.; Gorospe, M. Polysome Fractionation to Analyze mRNA Distribution Profiles. Bio-Protocol 2017, 7, e2126. [Google Scholar] [CrossRef] [Green Version]
- Pilling, D.; Karhadkar, T.R.; Gomer, R.H. High-fat diet-induced adipose tissue and liver inflammation and steatosis in mice are reduced by inhibiting sialidases. Am. J. Pathol. 2021, 191, 131–143. [Google Scholar] [CrossRef]
Figure 1.
Summary of pathways used by NEU3 to potentiate fibrosis.

Figure 2.
Summary of the NEU3 → TGF-β1 pathway in Figure 1. NEU3 removes sialic acids (purple diamonds) from the LAP protein (blue) that sequesters TGF-β1 (red) in the extracellular environment, causing the LAP to release active TGF-β1.
Figure 2.
Summary of the NEU3 → TGF-β1 pathway in Figure 1. NEU3 removes sialic acids (purple diamonds) from the LAP protein (blue) that sequesters TGF-β1 (red) in the extracellular environment, causing the LAP to release active TGF-β1.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Karhadkar, T.R.; Chen, W.; Pilling, D.; Gomer, R.H. Inhibitors of the Sialidase NEU3 as Potential Therapeutics for Fibrosis. Int. J. Mol. Sci. 2023, 24, 239. https://doi.org/10.3390/ijms24010239
AMA Style
Karhadkar TR, Chen W, Pilling D, Gomer RH. Inhibitors of the Sialidase NEU3 as Potential Therapeutics for Fibrosis. International Journal of Molecular Sciences. 2023; 24(1):239. https://doi.org/10.3390/ijms24010239
Chicago/Turabian StyleKarhadkar, Tejas R., Wensheng Chen, Darrell Pilling, and Richard H. Gomer. 2023. "Inhibitors of the Sialidase NEU3 as Potential Therapeutics for Fibrosis" International Journal of Molecular Sciences 24, no. 1: 239. https://doi.org/10.3390/ijms24010239
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.