
1,2-Hydrogenation and Transhydrogenation Catalyzed by 3-Ketosteroid Δ1-Dehydrogenase from Sterolibacterium denitrificans—Kinetics, Isotope Labelling and QM:MM Modelling Studies
 , , and
, , and 
Abstract
:
1. Introduction
2. Results and Discussion
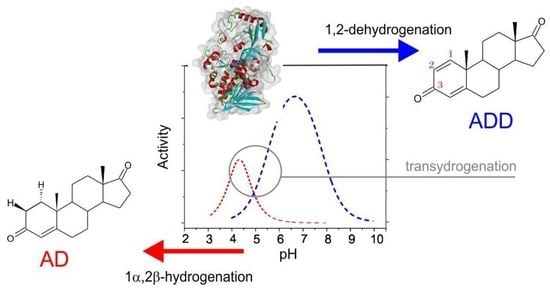
2.1. 1,2-Hydrogenation
2.1.1. pH Dependency
2.1.2. Synthesis of 1α,2β-Deuterated-AD
2.2. Transhydrogenation
3. Material and Methods
3.1. Materials
3.2. Protein Expression and Purification
3.3. Spectrophotometric Assays
3.4. 1,2-Hydrogenation
3.5. Transhydrogenation
3.6. HPLC and LC-MS Detection
3.7. Product Purification and NMR Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rohman, A.; Dijkstra, B.W. The Role and Mechanism of Microbial 3-Ketosteroid Δ1-Dehydrogenases in Steroid Breakdown. J. Steroid Biochem. Mol. Biol. 2019, 191, 105366. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, B.W.; van Oosterwijk, N.; Rohman, A. Structure and Catalytic Mechanism of 3-Ketosteroid Dehydrogenases. Procedia Chem. 2016, 18, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wu, D.; Yang, T.; Xu, M.; Rao, Z. Over-Expression of Mycobacterium Neoaurum 3-Ketosteroid-Δ1-Dehydrogenase in Corynebacterium Crenatum for Efficient Bioconversion of 4-Androstene-3,17-Dione to Androst-1,4-Diene-3,17-Dione. Electron. J. Biotechnol. 2016, 24, 84–90. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Liu, X.; Wang, Y.; Han, Y.; Sun, J.; Shi, J.; Zhang, B. Identification, Function, and Application of 3-Ketosteroid Δ1-Dehydrogenase Isozymes in Mycobacterium Neoaurum DSM 1381 for the Production of Steroidic Synthons. Microb. Cell Fact. 2018, 17, 77. [Google Scholar] [CrossRef] [Green Version]
- Yao, K.; Xu, L.Q.; Wang, F.Q.; Wei, D.Z. Characterization and Engineering of 3-Ketosteroid-△1-Dehydrogenase and 3-Ketosteroid-9α-Hydroxylase in Mycobacterium Neoaurum ATCC 25795 to Produce 9α-Hydroxy-4-Androstene-3,17-Dione through the Catabolism of Sterols. Metab. Eng. 2014, 24, 181–191. [Google Scholar] [CrossRef]
- He, K.; Sun, H.; Song, H. Engineering Phytosterol Transport System in Mycobacterium Sp. Strain MS136 Enhances Production of 9α-Hydroxy-4-Androstene-3,17-Dione. Biotechnol. Lett. 2018, 40, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Brzezinska, M.; Szulc, I.; Brzostek, A.; Klink, M.; Kielbik, M.; Sulowska, Z.; Pawelczyk, J.; Dziadek, J. The Role of 3-Ketosteroid 1(2)-Dehydrogenase in the Pathogenicity of Mycobacterium Tuberculosis. BMC Microbiol. 2013, 13, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohman, A.; Van Oosterwijk, N.; Thunnissen, A.M.W.H.; Dijkstra, B.W. Crystal Structure and Site-Directed Mutagenesis of 3-Ketosteroid Δ1-Dehydrogenase from Rhodococcus Erythropolis SQ1 Explain Its Catalytic Mechanism. J. Biol. Chem. 2013, 288, 35559–35568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wójcik, P.; Glanowski, M.; Mrugała, B.; Procner, M.; Zastawny, O. Structure, Mutagenesis and QM: MM Modelling of 3-Ketosteroid Δ1—Dehydrogenase from Sterolibacterium Denitrificans—The Role of New Membrane-Associated Domain and Proton-Relay System in Catalysis. ChemRxiv 2022. [Google Scholar] [CrossRef]
- Itagaki, E.; Matushita, H.; Hatta, T. Steroid Transhydrogenase Activity of 3-Ketosteroid-δ-Dehydrogenase from Nocardia Corallina. J. Biochem. 1990, 108, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Wojtkiewicz, A.M.; Wójcik, P.; Procner, M.; Flejszar, M.; Oszajca, M.; Hochołowski, M.; Tataruch, M.; Mrugała, B.; Janeczko, T.; Szaleniec, M. The Efficient Δ1-Dehydrogenation of a Wide Spectrum of 3-Ketosteroids in a Broad PH Range by 3-Ketosteroid Dehydrogenase from Sterolibacterium Denitrificans. J. Steroid Biochem. Mol. Biol. 2020, 202, 105731. [Google Scholar] [CrossRef]
- Meemken, F.; Baiker, A. Recent Progress in Heterogeneous Asymmetric Hydrogenation of C=O and C=C Bonds on Supported Noble Metal Catalysts. Chem. Rev. 2017, 117, 11522–11569. [Google Scholar] [CrossRef]
- Hagiwara, H. Chemo- and Enantioselective Catalytic Hydrogenation of α,β-Unsaturated Ketones and Aldehydes as a Tool to Introduce Chiral Centers at α- or β-Positions of Ketones. Nat. Prod. Commun. 2018, 13, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Glanowski, M.; Wójcik, P.; Procner, M.; Borowski, T.; Lupa, D.; Mielczarek, P.; Oszajca, M.; Swiderek, K.; Moliner, V.; Bojarski, A.J.; et al. Enzymatic Δ1-Dehydrogenation of 3-Ketosteroids-Reconciliation of Kinetic Isotope Effects with the Reaction Mechanism. ACS Catal. 2021, 11, 8211–8225. [Google Scholar] [CrossRef]
- Itagaki, E.; Wakabayashi, T.; Hatta, T. Purification and Characterization of 3-Ketosteroid-Delta 1-Dehydrogenase from Nocardia Corallina. Biochim. Biophys. Acta Rev. Cancer 1990, 1038, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Ravasio, N.; Antenori, M.; Gargano, M.; Mastrorilli, P. CuSiO2: An Improved Catalyst for the Chemoselective Hydrogenation of α,β-Unsaturated Ketones. Tetrahedron Lett. 1996, 37, 3529–3532. [Google Scholar] [CrossRef]
- Hayano, M.; Ringold, H.J.; Stefanovic, V.; Gut, M.; Dorfman, R.I. The Stereochemical Course of Enzymatic Steroid 1,2-Dehydrogenation. Biochem. Biophys. Res. Commun. 1961, 4, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Sofińska, K.; Wojtkiewicz, A.M.; Wójcik, P.; Zastawny, O.; Guzik, M.; Winiarska, A.; Waligórski, P.; Cieśla, M.; Barbasz, J.; Szaleniec, M. Investigation of Quaternary Structure of Aggregating 3-Ketosteroid Dehydrogenase from Sterolibacterium Denitrificans: In the Pursuit of Consensus of Various Biophysical Techniques. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1027–1039. [Google Scholar] [CrossRef] [PubMed]
- Welte, C.; Deppenmeier, U. Proton Translocation in Methanogens, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2011; Volume 494, ISBN 9780123851123. [Google Scholar]
- Tipton, K.F.; Dixon, H.B.F. Effects of pH on Enzymes. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1979; Volume 63, pp. 183–234. ISBN 0076-6879. [Google Scholar]
- Verheyden, K.; Noppe, H.; Zorn, H.; Van Immerseel, F.; Bussche, J.V.; Wille, K.; Bekaert, K.; Janssen, C.R.; De Brabander, H.F.; Vanhaecke, L. Endogenous Boldenone-Formation in Cattle: Alternative Invertebrate Organisms to Elucidate the Enzymatic Pathway and the Potential Role of Edible Fungi on Cattle’s Feed. J. Steroid Biochem. Mol. Biol. 2010, 119, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Hamada, H.; Naka, S.; Kurban, H. Stereoselective Reduction in the Biotransformation of Androstane Derivatives by Cell Suspension Cultures of Marchantia Polymorpha. Chem. Lett. 1993, 22, 2111–2112. [Google Scholar] [CrossRef]
- Groh, H.; Komel, R.; Deppmeyer, V.; Schade, W.; Hörhold, C. Steroid Transforming Enzymes from Microorganisms: The Reverse Reaction of the Steroid-1-Dehydrogenase from Nocardia. J. Steroid Biochem. 1980, 13, 1413–1415. [Google Scholar] [CrossRef]
- Qian, Z.; Huang, X.; Wang, Q. Stabilizing Benzyl Viologen Radical Cation by Cucurbit[7]Uril Rotaxanation. Dye. Pigment. 2017, 145, 365–370. [Google Scholar] [CrossRef]
- Kiddle, G.R.; Harris, R.; Homans, S.W. Heteronuclear Overhauser Effects in Carbohydrates. J. Biomol. NMR 1998, 11, 289–294. [Google Scholar] [CrossRef]
- Covington, A.K.; Paabo, M.; Robinson, R.A.; Bates, R.G. Use of the Glass Electrode in Deuterium Oxide and the Relation between the Standardized PD (PaD) Scale and the Operational PH in Heavy Water. Anal. Chem. 1968, 40, 700–706. [Google Scholar] [CrossRef]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of p K a Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; SØndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical p K a Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Dupradeau, F.Y.; Cézard, C.; Lelong, R.; Stanislawiak, E.; Pêcher, J.; Delepine, J.C.; Cieplak, P. R.E.DD.B.: A Database for RESP and ESP Atomic Charges, and Force Field Libraries. Nucleic Acids Res. 2008, 36, 360–367. [Google Scholar] [CrossRef] [Green Version]
- Field, M.J.; Albe, M.; Bret, C.; Proust-De Martin, F.; Thomas, A. The Dynamo Library for Molecular Simulations Using Hybrid Quantum Mechanical and Molecular Mechanical Potentials. J. Comput. Chem. 2000, 21, 1088–1100. [Google Scholar] [CrossRef]
- Krzemińska, A.; Paneth, P.; Moliner, V.; Świderek, K. Binding Isotope Effects as a Tool for Distinguishing Hydrophobic and Hydrophilic Binding Sites of HIV-1 RT. J. Phys. Chem. B 2015, 119, 917–927. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical Sampling Distributions in Monte Carlo Free-Energy Estimation: Umbrella Sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. THE Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules. I. The Method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Baker, J.; Kessi, A.; Delley, B. The Generation and Use of Delocalized Internal Coordinates in Geometry Optimization. J. Chem. Phys. 1996, 105, 192–212. [Google Scholar] [CrossRef]
- Martí, S.; Moliner, V.; Tuñón, I. Improving the QM/MM Description of Chemical Processes: A Dual Level Strategy To Explore the Potential Energy Surface in Very Large Systems. J. Chem. Theory Comput. 2005, 1, 1008–1016. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ∆(E + ZPE) (kcal/mol) | ||||
|---|---|---|---|---|
| 1α,2β-d2-AD | AD | |||
| 2β Attack | 2α Attack | 2β Attack | 2α Attack | |
| E:S | 0 | 0 | 0 | 0 |
| TS1 | 10.85 | 14.19 | 9.94 | 14.05 |
| E:I | 3.13 | 3.13 | 2.92 | 2.92 |
| TS2 | 13.39 | 13.50 | 12.59 | 12.59 |
| E:P | −9.63 | −9.46 | −9.45 | −9.45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wojtkiewicz, A.M.; Glanowski, M.; Waligórski, P.; Janeczko, T.; Szaleniec, M. 1,2-Hydrogenation and Transhydrogenation Catalyzed by 3-Ketosteroid Δ1-Dehydrogenase from Sterolibacterium denitrificans—Kinetics, Isotope Labelling and QM:MM Modelling Studies. Int. J. Mol. Sci. 2022, 23, 14660. https://doi.org/10.3390/ijms232314660
Wojtkiewicz AM, Glanowski M, Waligórski P, Janeczko T, Szaleniec M. 1,2-Hydrogenation and Transhydrogenation Catalyzed by 3-Ketosteroid Δ1-Dehydrogenase from Sterolibacterium denitrificans—Kinetics, Isotope Labelling and QM:MM Modelling Studies. International Journal of Molecular Sciences. 2022; 23(23):14660. https://doi.org/10.3390/ijms232314660
Chicago/Turabian StyleWojtkiewicz, Agnieszka M., Michał Glanowski, Piotr Waligórski, Tomasz Janeczko, and Maciej Szaleniec. 2022. "1,2-Hydrogenation and Transhydrogenation Catalyzed by 3-Ketosteroid Δ1-Dehydrogenase from Sterolibacterium denitrificans—Kinetics, Isotope Labelling and QM:MM Modelling Studies" International Journal of Molecular Sciences 23, no. 23: 14660. https://doi.org/10.3390/ijms232314660