
Detection of Measurable Residual Disease Biomarkers in Extracellular Vesicles from Liquid Biopsies of Multiple Myeloma Patients—A Proof of Concept

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results and Discussion
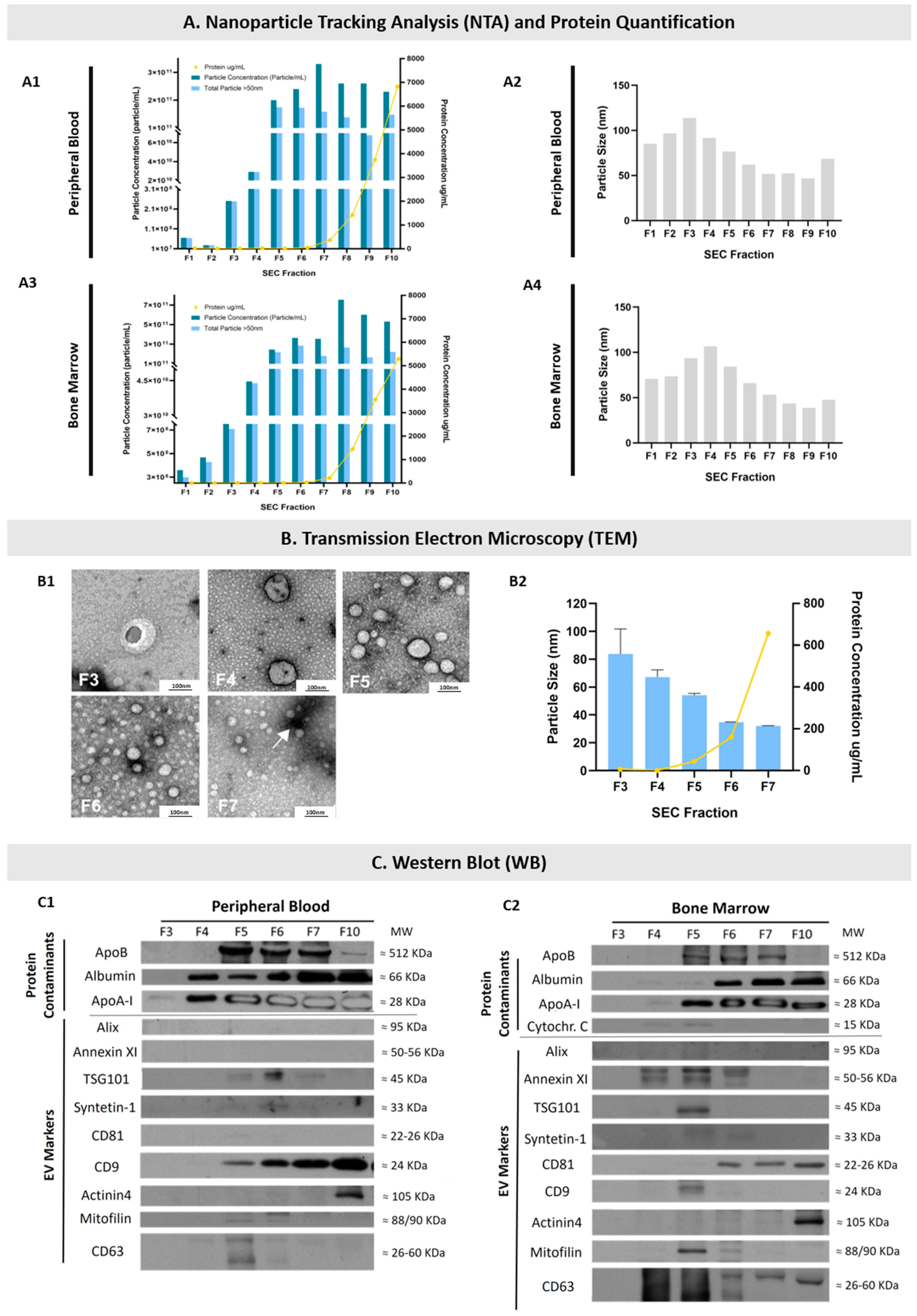
2.1. EVs Were Isolated from the PB and BM of MM Patients
2.2. EVs Were Isolated from Diagnosis and Remission PB and BM Samples of MM Patients
2.2.1. EVs Isolated from Diagnostic PB and BM Samples Present MM Markers in Their Cargo
2.2.2. MM Markers Can Be Found in EVs from Both BM and PB Samples
2.2.3. EVs from Diagnosis and Remission PB and BM Patient Samples Present Different Levels of MM Markers
2.3. EVs from Healthy Controls Do Not Present Most of the MM Markers
3. Material and Methods
3.1. Patients and Samples
3.2. Minimal Residual Disease (MRD) Determination by Flow Cytometry
3.3. Platelet-Poor Plasma (PPP) Isolation
3.4. Extracellular Vesicles (EVs) Isolation from Platelet-Poor Plasma (PPP)
3.4.1. Size Exclusion Chromatography (SEC)
3.4.2. Ultrafiltration (UF)
3.5. Protein Quantification
3.6. EVs Characterization
3.6.1. Nanoparticle Tracking Analysis (NTA)
3.6.2. Transmission Electron Microscopy (TEM)
3.6.3. Western Blot (WB)
3.7. Multiple Myeloma (MM) Marker Analysis by Western Blot (WB)
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AS-PCR | allele-specific oligonucleotide polymerase chain reaction |
| ASCT | autologous stem cell transplant |
| BM | bone marrow |
| CAR-T cells | chimeric antigen receptor-T cells |
| CHUSJ | Centro Hospitalar Universitário São João |
| cfDNA | cell-free DNA |
| CTCs | circulating tumor cells |
| ctDNA | circulating tumor DNA |
| CR | complete response |
| D100 | day 100 after ASCT |
| ECL | enhanced chemiluminescence |
| EMD | extramedullary disease |
| EVs | Extracellular Vesicles |
| FDG-PET/CT | 18-fluoro-2-deoxyglucos positron emission tomography/computed tomography |
| FISH | Fluorescence in situ hybridization |
| FPM | Fuji Medical Film Processor |
| Ig | immunoglobulin |
| IMWG | International Myeloma Working Group |
| ISS | International Staging System |
| K | immunoglobulin kappa (κ) chain |
| L | immunoglobulin lambda (λ) chain |
| LOD | limit of detection |
| MFC | multiparameter flow cytometry |
| MM | Multiple Myeloma |
| MRD | measurable residual disease |
| MSCs | BM mesenchymal stem cells |
| MW | molecular weight |
| NGF | next-generation flow |
| NGS | next-generation sequencing |
| NTA | Nanoparticle Tracking Analysis |
| OS | overall survival |
| PB | peripheral blood |
| PBS | phosphate-buffered saline |
| PC | plasma cells |
| PFS | progression free-survival |
| PPP | Platelet-Poor Plasma |
| PRP | Platelet-Rich Plasma |
| R-ISS | revised-ISS |
| RT | room temperature |
| sCR | stringent complete response |
| SEC | Size Exclusion Chromatography |
| TBS-T | Tris-buffered saline solution with 0.1% Tween-20 |
| TEM | Transmission Electron Microscopy |
| UF | ultrafiltration |
| UF | ultrafiltrated pool from SEC fractions 3 to 6 |
| VEGF | vascular endothelial growth factor |
| VGPR | very good partial response |
| WB | Western Blot |
References
- Thorsteinsdottir, S.; Dickman, P.W.; Landgren, O.; Blimark, C.; Hultcrantz, M.; Turesson, I.; Björkholm, M.; Kristinsson, S.Y. Dramatically improved survival in multiple myeloma patients in the recent decade: Results from a Swedish population-based study. Haematologica 2018, 103, e412–e415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Otero, P.; Paiva, B.; San-Miguel, J.F. Roadmap to cure multiple myeloma. Cancer Treat. Rev. 2021, 100, 102284. [Google Scholar] [CrossRef] [PubMed]
- Luskin, M.R.; Murakami, M.A.; Manalis, S.R.; Weinstock, D.M. Targeting minimal residual disease: A path to cure? Nat. Rev. Cancer 2018, 18, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Pérez, C.; Sola, M.; Teruel-Montoya, R.; García-Malo, M.D.; Ortuño, F.J.; Vicente, V.; de Arriba, F.; Jerez, A. Minimal Residual Disease in Multiple Myeloma: Something Old, Something New. Cancers 2021, 13, 4332. [Google Scholar] [CrossRef]
- Oliva, S.; Gambella, M.; Gilestro, M.; Muccio, V.E.; Gay, F.; Drandi, D.; Ferrero, S.; Passera, R.; Pautasso, C.; Bernardini, A.; et al. Minimal residual disease after transplantation or lenalidomide-based consolidation in myeloma patients: A prospective analysis. Oncotarget 2017, 8, 5924–5935. [Google Scholar] [CrossRef] [Green Version]
- Mina, R.; Belotti, A.; Petrucci, M.T.; Zambello, R.; Capra, A.; Di Lullo, G.; Ronconi, S.; Pescosta, N.; Grasso, M.; Monaco, F.; et al. Bortezomib-dexamethasone as maintenance therapy or early retreatment at biochemical relapse versus observation in relapsed/refractory multiple myeloma patients: A randomized phase II study. Blood Cancer J. 2020, 10, 58. [Google Scholar] [CrossRef]
- Suzuki, K.; Nishiwaki, K.; Yano, S. Treatment Strategy for Multiple Myeloma to Improve Immunological Environment and Maintain MRD Negativity. Cancers 2021, 13, 4867. [Google Scholar] [CrossRef]
- Anderson, K.C.; Auclair, D.; Kelloff, G.J.; Sigman, C.C.; Avet-Loiseau, H.; Farrell, A.T.; Gormley, N.J.; Kumar, S.K.; Landgren, O.; Munshi, N.C.; et al. The Role of Minimal Residual Disease Testing in Myeloma Treatment Selection and Drug Development: Current Value and Future Applications. Clin. Cancer Res. 2017, 23, 3980–3993. [Google Scholar] [CrossRef] [Green Version]
- Landgren, O.; Devlin, S.; Boulad, M.; Mailankody, S. Role of MRD status in relation to clinical outcomes in newly diagnosed multiple myeloma patients: A meta-analysis. Bone Marrow Transplant. 2016, 51, 1565–1568. [Google Scholar] [CrossRef] [Green Version]
- Munshi, N.C.; Avet-Loiseau, H.; Anderson, K.C.; Neri, P.; Paiva, B.; Samur, M.; Dimopoulos, M.; Kulakova, M.; Lam, A.; Hashim, M.; et al. A large meta-analysis establishes the role of MRD negativity in long-term survival outcomes in patients with multiple myeloma. Blood Adv. 2020, 4, 5988–5999. [Google Scholar] [CrossRef]
- Munshi, N.C.; Avet-Loiseau, H.; Rawstron, A.C.; Owen, R.G.; Child, J.A.; Thakurta, A.; Sherrington, P.; Samur, M.K.; Georgieva, A.; Anderson, K.C.; et al. Association of Minimal Residual Disease With Superior Survival Outcomes in Patients With Multiple Myeloma: A Meta-analysis. JAMA Oncol. 2017, 3, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Oliva, S.; D’Agostino, M.; Boccadoro, M.; Larocca, A. Clinical Applications and Future Directions of Minimal Residual Disease Testing in Multiple Myeloma. Front. Oncol. 2020, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Paiva, B.; Puig, N.; Cedena, M.T.; Rosiñol, L.; Cordón, L.; Vidriales, M.B.; Burgos, L.; Flores-Montero, J.; Sanoja-Flores, L.; Lopez-Anglada, L.; et al. Measurable Residual Disease by Next-Generation Flow Cytometry in Multiple Myeloma. J. Clin. Oncol. 2020, 38, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.; Lonial, S.; Bladé, J.; Mateos, M.V.; et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016, 17, e328–e346. [Google Scholar] [CrossRef]
- Lahuerta, J.J.; Paiva, B.; Vidriales, M.B.; Cordón, L.; Cedena, M.T.; Puig, N.; Martinez-Lopez, J.; Rosiñol, L.; Gutierrez, N.C.; Martín-Ramos, M.L.; et al. Depth of Response in Multiple Myeloma: A Pooled Analysis of Three PETHEMA/GEM Clinical Trials. J. Clin. Oncol. 2017, 35, 2900–2910. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, R.; Muchtar, E.; Kumar, S.K.; Jevremovic, D.; Buadi, F.K.; Dingli, D.; Dispenzieri, A.; Hayman, S.R.; Hogan, W.J.; Kapoor, P.; et al. Impact of Post-Transplant Response and Minimal Residual Disease on Survival in Myeloma with High-Risk Cytogenetics. Biol. Blood Marrow Transplant. 2017, 23, 598–605. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Li, F.; Zhou, X.; Mei, J.; Song, P.; An, Z.; Zhao, Q.; Guo, X.; Wang, X.; Zhai, Y. Achieving minimal residual disease-negative by multiparameter flow cytometry may ameliorate a poor prognosis in MM patients with high-risk cytogenetics: A retrospective single-center analysis. Ann. Hematol. 2019, 98, 1185–1195. [Google Scholar] [CrossRef]
- Mateos, M.V.; Dimopoulos, M.A.; Cavo, M.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lucio, P.; Nagy, Z.; Kaplan, P.; et al. Daratumumab plus Bortezomib, Melphalan, and Prednisone for Untreated Myeloma. N. Engl. J. Med. 2018, 378, 518–528. [Google Scholar] [CrossRef]
- Perrot, A.; Lauwers-Cances, V.; Corre, J.; Robillard, N.; Hulin, C.; Chretien, M.L.; Dejoie, T.; Maheo, S.; Stoppa, A.M.; Pegourie, B.; et al. Minimal residual disease negativity using deep sequencing is a major prognostic factor in multiple myeloma. Blood 2018, 132, 2456–2464. [Google Scholar] [CrossRef] [Green Version]
- Avet-Loiseau, H.; San-Miguel, J.; Casneuf, T.; Iida, S.; Lonial, S.; Usmani, S.Z.; Spencer, A.; Moreau, P.; Plesner, T.; Weisel, K.; et al. Evaluation of Sustained Minimal Residual Disease Negativity With Daratumumab-Combination Regimens in Relapsed and/or Refractory Multiple Myeloma: Analysis of POLLUX and CASTOR. J. Clin. Oncol. 2021, 39, 1139–1149. [Google Scholar] [CrossRef]
- Burgos, L.; Puig, N.; Cedena, M.T.; Mateos, M.V.; Lahuerta, J.J.; Paiva, B.; San-Miguel, J.F. Measurable residual disease in multiple myeloma: Ready for clinical practice? J. Hematol. Oncol. 2020, 13, 82. [Google Scholar] [CrossRef]
- Costa, L.J.; Derman, B.A.; Bal, S.; Sidana, S.; Chhabra, S.; Silbermann, R.; Ye, J.C.; Cook, G.; Cornell, R.F.; Holstein, S.A.; et al. International harmonization in performing and reporting minimal residual disease assessment in multiple myeloma trials. Leukemia 2021, 35, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Mina, R.; Oliva, S.; Boccadoro, M. Minimal Residual Disease in Multiple Myeloma: State of the Art and Future Perspectives. J. Clin. Med. 2020, 9, 2142. [Google Scholar] [CrossRef] [PubMed]
- Flores-Montero, J.; Sanoja-Flores, L.; Paiva, B.; Puig, N.; García-Sánchez, O.; Böttcher, S.; van der Velden, V.H.J.; Pérez-Morán, J.J.; Vidriales, M.B.; García-Sanz, R.; et al. Next Generation Flow for highly sensitive and standardized detection of minimal residual disease in multiple myeloma. Leukemia 2017, 31, 2094–2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanoja-Flores, L.; Flores-Montero, J.; Puig, N.; Contreras-Sanfeliciano, T.; Pontes, R.; Corral-Mateos, A.; García-Sánchez, O.; Díez-Campelo, M.; Pessoa de Magalhães, R.J.; García-Martín, L.; et al. Blood monitoring of circulating tumor plasma cells by next generation flow in multiple myeloma after therapy. Blood 2019, 134, 2218–2222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riva, G.; Nasillo, V.; Ottomano, A.M.; Bergonzini, G.; Paolini, A.; Forghieri, F.; Lusenti, B.; Barozzi, P.; Lagreca, I.; Fiorcari, S.; et al. Multiparametric Flow Cytometry for MRD Monitoring in Hematologic Malignancies: Clinical Applications and New Challenges. Cancers 2021, 13, 4582. [Google Scholar] [CrossRef]
- Oliva, S.; Genuardi, E.; Belotti, A.; Frascione, P.M.M.; Galli, M.; Capra, A.; Offidani, M.; Vozella, F.; Zambello, R.; Auclair, D.; et al. Minimal Residual Disease Evaluation By Multiparameter Flow Cytometry and Next Generation Sequencing in the Forte Trial for Newly Diagnosed Multiple Myeloma Patients. Blood 2019, 134, 4322. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Bene, M.C.; Wuilleme, S.; Corre, J.; Attal, M.; Arnulf, B.; Garderet, L.; Macro, M.; Stoppa, A.-M.; Delforge, M.; et al. Concordance of Post-consolidation Minimal Residual Disease Rates by Multiparametric Flow Cytometry and Next-generation Sequencing in CASSIOPEIA. Clin. Lymphoma Myeloma Leuk. 2019, 19, e3–e4. [Google Scholar] [CrossRef]
- Lu, Y.Y.; Chen, J.H.; Lin, W.Y.; Liang, J.A.; Wang, H.Y.; Tsai, S.C.; Kao, C.H. FDG PET or PET/CT for detecting intramedullary and extramedullary lesions in multiple Myeloma: A systematic review and meta-analysis. Clin. Nucl. Med. 2012, 37, 833–837. [Google Scholar] [CrossRef]
- Moreau, P.; Attal, M.; Caillot, D.; Macro, M.; Karlin, L.; Garderet, L.; Facon, T.; Benboubker, L.; Escoffre-Barbe, M.; Stoppa, A.M.; et al. Prospective Evaluation of Magnetic Resonance Imaging and [18F]Fluorodeoxyglucose Positron Emission Tomography-Computed Tomography at Diagnosis and Before Maintenance Therapy in Symptomatic Patients With Multiple Myeloma Included in the IFM/DFCI 2009 Trial: Results of the IMAJEM Study. J. Clin. Oncol. 2017, 35, 2911–2918. [Google Scholar] [CrossRef]
- Rasche, L.; Chavan, S.S.; Stephens, O.W.; Patel, P.H.; Tytarenko, R.; Ashby, C.; Bauer, M.; Stein, C.; Deshpande, S.; Wardell, C.; et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat. Commun. 2017, 8, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neurooncol. 2013, 113, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowal, J.; Tkach, M.; Théry, C. Biogenesis and secretion of exosomes. Curr Opin Cell Biol 2014, 29, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yáñez-Mó, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; El Andaloussi, S.; Wood, M.J. Exosomes and microvesicles: Extracellular vesicles for genetic information transfer and gene therapy. Hum. Mol. Genet. 2012, 21, R125–R134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xavier, C.P.R.; Castro, I.; Caires, H.R.; Ferreira, D.; Cavadas, B.; Pereira, L.; Santos, L.L.; Oliveira, M.J.; Vasconcelos, M.H. Chitinase 3-like-1 and fibronectin in the cargo of extracellular vesicles shed by human macrophages influence pancreatic cancer cellular response to gemcitabine. Cancer Lett. 2021, 501, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Rodrigues, V.; Di Luca, A.; Mleczko, J.; Meleady, P.; Henry, M.; Pesic, M.; Cabrera, D.; van Liempd, S.; Lima, R.T.; O’Connor, R.; et al. Identification of the metabolic alterations associated with the multidrug resistant phenotype in cancer and their intercellular transfer mediated by extracellular vesicles. Sci. Rep. 2017, 7, 44541. [Google Scholar] [CrossRef] [Green Version]
- Sousa, D.; Lima, R.T.; Vasconcelos, M.H. Intercellular Transfer of Cancer Drug Resistance Traits by Extracellular Vesicles. Trends Mol. Med. 2015, 21, 595–608. [Google Scholar] [CrossRef]
- Lopes-Rodrigues, V.; Di Luca, A.; Sousa, D.; Seca, H.; Meleady, P.; Henry, M.; Lima, R.T.; O’Connor, R.; Vasconcelos, M.H. Multidrug resistant tumour cells shed more microvesicle-like EVs and less exosomes than their drug-sensitive counterpart cells. Biochim. Biophys. Acta 2016, 1860, 618–627. [Google Scholar] [CrossRef]
- D’Souza-Schorey, C.; Clancy, J.W. Tumor-derived microvesicles: Shedding light on novel microenvironment modulators and prospective cancer biomarkers. Genes Dev. 2012, 26, 1287–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasconcelos, M.H.; Caires, H.R.; Ābols, A.; Xavier, C.P.R.; Linē, A. Extracellular vesicles as a novel source of biomarkers in liquid biopsies for monitoring cancer progression and drug resistance. Drug Resist. Updat 2019, 47, 100647. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Tumor-Derived Exosomes and Their Role in Cancer Progression. Adv. Clin. Chem. 2016, 74, 103–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saltarella, I.; Lamanuzzi, A.; Apollonio, B.; Desantis, V.; Bartoli, G.; Vacca, A.; Frassanito, M.A. Role of Extracellular Vesicle-Based Cell-to-Cell Communication in Multiple Myeloma Progression. Cells 2021, 10, 3185. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.; Liao, Y.; Hosseinifard, H.; Imani, S.; Wen, Q.L. Diagnostic Role of Extracellular Vesicles in Cancer: A Comprehensive Systematic Review and Meta-Analysis. Front. Cell Dev. Biol. 2021, 9, 705791. [Google Scholar] [CrossRef]
- Caivano, A.; Laurenzana, I.; De Luca, L.; La Rocca, F.; Simeon, V.; Trino, S.; D’Auria, F.; Traficante, A.; Maietti, M.; Izzo, T.; et al. High serum levels of extracellular vesicles expressing malignancy-related markers are released in patients with various types of hematological neoplastic disorders. Tumour Biol. 2015, 36, 9739–9752. [Google Scholar] [CrossRef]
- Azmi, A.S.; Bao, B.; Sarkar, F.H. Exosomes in cancer development, metastasis, and drug resistance: A comprehensive review. Cancer Metastasis Rev. 2013, 32, 623–642. [Google Scholar] [CrossRef] [Green Version]
- Jena, B.C.; Mandal, M. The emerging roles of exosomes in anti-cancer drug resistance and tumor progression: An insight towards tumor-microenvironment interaction. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188488. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Y.; Gong, H.; Luo, S.; Cui, Y. The Role of Exosomes and Their Applications in Cancer. Int. J. Mol. Sci. 2021, 22, 2204. [Google Scholar] [CrossRef]
- Xavier, C.P.R.; Caires, H.R.; Barbosa, M.A.G.; Bergantim, R.; Guimarães, J.E.; Vasconcelos, M.H. The Role of Extracellular Vesicles in the Hallmarks of Cancer and Drug Resistance. Cells 2020, 9, 1141. [Google Scholar] [CrossRef]
- Khalife, J.; Sanchez, J.F.; Pichiorri, F. The Emerging Role of Extracellular Vesicle-Associated RNAs in the Multiple Myeloma Microenvironment. Front. Oncol. 2021, 11, 689538. [Google Scholar] [CrossRef] [PubMed]
- Umezu, T.; Imanishi, S.; Yoshizawa, S.; Kawana, C.; Ohyashiki, J.H.; Ohyashiki, K. Induction of multiple myeloma bone marrow stromal cell apoptosis by inhibiting extracellular vesicle miR-10a secretion. Blood Adv. 2019, 3, 3228–3240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hendrix, A.; Hernot, S.; Lemaire, M.; De Bruyne, E.; Van Valckenborgh, E.; Lahoutte, T.; De Wever, O.; Vanderkerken, K.; Menu, E. Bone marrow stromal cell–derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood 2014, 124, 555–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhu, X.J.; Zeng, C.; Wu, P.H.; Wang, H.X.; Chen, Z.C.; Li, Q.B. Microvesicles secreted from human multiple myeloma cells promote angiogenesis. Acta Pharmacol. Sin. 2014, 35, 230–238. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Kosaka, N.; Takahiro, O.; Hattori, Y. Understanding the Role of Extracellular Vesicles in Lenalidomide-Resistance Multiple Myeloma. Blood 2018, 132, 1887. [Google Scholar] [CrossRef]
- Fontana, F.; Carollo, E.; Melling, G.E.; Carter, D.R.F. Extracellular Vesicles: Emerging Modulators of Cancer Drug Resistance. Cancers 2021, 13, 749. [Google Scholar] [CrossRef]
- Laurenzana, I.; Trino, S.; Lamorte, D.; Girasole, M.; Dinarelli, S.; De Stradis, A.; Grieco, V.; Maietti, M.; Traficante, A.; Statuto, T.; et al. Analysis of Amount, Size, Protein Phenotype and Molecular Content of Circulating Extracellular Vesicles Identifies New Biomarkers in Multiple Myeloma. Int. J. Nanomed. 2021, 16, 3141–3160. [Google Scholar] [CrossRef]
- Colombo, M.; Giannandrea, D.; Lesma, E.; Basile, A.; Chiaramonte, R. Extracellular Vesicles Enhance Multiple Myeloma Metastatic Dissemination. Int. J. Mol. Sci. 2019, 20, 3236. [Google Scholar] [CrossRef] [Green Version]
- Morandi, F.; Marimpietri, D.; Horenstein, A.L.; Bolzoni, M.; Toscani, D.; Costa, F.; Castella, B.; Faini, A.C.; Massaia, M.; Pistoia, V.; et al. Microvesicles released from multiple myeloma cells are equipped with ectoenzymes belonging to canonical and non-canonical adenosinergic pathways and produce adenosine from ATP and NAD+. Oncoimmunology 2018, 7, e1458809. [Google Scholar] [CrossRef] [Green Version]
- Kvorning, S.L.; Nielsen, M.C.; Andersen, N.F.; Hokland, M.; Andersen, M.N.; Møller, H.J. Circulating extracellular vesicle-associated CD163 and CD206 in multiple myeloma. Eur. J. Haematol. 2020, 104, 409–419. [Google Scholar] [CrossRef]
- Gurunathan, S.; Kang, M.H.; Jeyaraj, M.; Qasim, M.; Kim, J.H. Review of the Isolation, Characterization, Biological Function, and Multifarious Therapeutic Approaches of Exosomes. Cells 2019, 8, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boing, A.N.; van der Pol, E.; Grootemaat, A.E.; Coumans, F.A.; Sturk, A.; Nieuwland, R. Single-step isolation of extracellular vesicles by size-exclusion chromatography. J. Extracell. Vesicles 2014, 3, 23430. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Gao, X.; Xing, X.; Huang, H.; Tang, Q.; Ma, S.; Xu, X.; Liang, C.; Li, M.; Liao, L.; et al. An Isolation System to Collect High Quality and Purity Extracellular Vesicles from Serum. Int. J. Nanomed. 2021, 16, 6681–6692. [Google Scholar] [CrossRef] [PubMed]
- Stranska, R.; Gysbrechts, L.; Wouters, J.; Vermeersch, P.; Bloch, K.; Dierickx, D.; Andrei, G.; Snoeck, R. Comparison of membrane affinity-based method with size-exclusion chromatography for isolation of exosome-like vesicles from human plasma. J. Transl. Med. 2018, 16, 1. [Google Scholar] [CrossRef]
- Gaspar, L.S.; Santana, M.M.; Henriques, C.; Pinto, M.M.; Ribeiro-Rodrigues, T.M.; Girão, H.; Nobre, R.J.; Pereira de Almeida, L. Simple and Fast SEC-Based Protocol to Isolate Human Plasma-Derived Extracellular Vesicles for Transcriptional Research. Mol. Ther. Methods Clin. Dev. 2020, 18, 723–737. [Google Scholar] [CrossRef]
- Benedikter, B.J.; Bouwman, F.G.; Vajen, T.; Heinzmann, A.C.A.; Grauls, G.; Mariman, E.C.; Wouters, E.F.M.; Savelkoul, P.H.; Lopez-Iglesias, C.; Koenen, R.R.; et al. Ultrafiltration combined with size exclusion chromatography efficiently isolates extracellular vesicles from cell culture media for compositional and functional studies. Sci Rep. 2017, 10, 15297. [Google Scholar] [CrossRef] [Green Version]
- Yuana, Y.; Koning, R.I.; Kuil, M.E.; Rensen, P.C.; Koster, A.J.; Bertina, R.M.; Osanto, S. Cryo-electron microscopy of extracellular vesicles in fresh plasma. J. Extracell. Vesicles 2013, 2, 21494. [Google Scholar] [CrossRef]
- Nordin, J.Z.; Lee, Y.; Vader, P.; Mäger, I.; Johansson, H.J.; Heusermann, W.; Wiklander, O.P.; Hällbrink, M.; Seow, Y.; Bultema, J.J.; et al. Ultrafiltration with size-exclusion liquid chromatography for high yield isolation of extracellular vesicles preserving intact biophysical and functional properties. Nanomedicine 2015, 11, 879–883. [Google Scholar] [CrossRef] [Green Version]
- van Eijndhoven, M.A.; Zijlstra, J.M.; Groenewegen, N.J.; Drees, E.E.; van Niele, S.; Baglio, S.R.; Koppers-Lalic, D.; van der Voorn, H.; Libregts, S.F.; Wauben, M.H.; et al. Plasma vesicle miRNAs for therapy response monitoring in Hodgkin lymphoma patients. JCI Insight 2016, 1, e89631. [Google Scholar] [CrossRef]
- Tao, S.C.; Guo, S.C. Role of extracellular vesicles in tumour microenvironment. Cell Commun. Signal. 2020, 18, 163. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalla, P.V.; Santos, J.; Milthorpe, B.K.; Padula, M.P. Selectively-Packaged Proteins in Breast Cancer Extracellular Vesicles Involved in Metastasis. Int. J. Mol. Sci. 2020, 21, 4990. [Google Scholar] [CrossRef] [PubMed]
- Burgos, L.; Paiva, B. EuroFlow-Based Next-Generation Flow Cytometry for Detection of Circulating Tumor Cells and Minimal Residual Disease in Multiple Myeloma. Methods Mol. Biol. 2018, 1792, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Jelinek, T.; Sevcikova, T.; Zihala, D.; Popkova, T.; Kapustova, V.; Broskevicova, L.; Capkova, L.; Rihova, L.; Bezdekova, R.; Sevcikova, S.; et al. Limited efficacy of daratumumab in multiple myeloma with extramedullary disease. Leukemia 2021, 36, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Ise, M.; Matsubayashi, K.; Tsujimura, H.; Kumagai, K. Loss of CD38 Expression in Relapsed Refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2016, 16, e59–e64. [Google Scholar] [CrossRef]
- Costa, F.; Dalla Palma, B.; Giuliani, N. CD38 Expression by Myeloma Cells and Its Role in the Context of Bone Marrow Microenvironment: Modulation by Therapeutic Agents. Cells 2019, 8, 1632. [Google Scholar] [CrossRef] [Green Version]
- Akhmetzyanova, I.; McCarron, M.J.; Parekh, S.; Chesi, M.; Bergsagel, P.L.; Fooksman, D.R. Dynamic CD138 surface expression regulates switch between myeloma growth and dissemination. Leukemia 2020, 34, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Gonsalves, W.I.; Timm, M.M.; Rajkumar, S.V.; Morice, W.G.; Dispenzieri, A.; Buadi, F.K.; Lacy, M.Q.; Dingli, D.; Leung, N.; Kapoor, P.; et al. The prognostic significance of CD45 expression by clonal bone marrow plasma cells in patients with newly diagnosed multiple myeloma. Leuk. Res. 2016, 44, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Arendt, B.K.; Walters, D.K.; Wu, X.; Tschumper, R.C.; Huddleston, P.M.; Henderson, K.J.; Dispenzieri, A.; Jelinek, D.F. Increased expression of extracellular matrix metalloproteinase inducer (CD147) in multiple myeloma: Role in regulation of myeloma cell proliferation. Leukemia 2012, 26, 2286–2296. [Google Scholar] [CrossRef] [Green Version]
- Arendt, B.K.; Walters, D.K.; Wu, X.; Tschumper, R.C.; Jelinek, D.F. Multiple myeloma dell-derived microvesicles are enriched in CD147 expression and enhance tumor cell proliferation. Oncotarget 2014, 5, 5686–5699. [Google Scholar] [CrossRef]
- Bjorklund, C.C.; Baladandayuthapani, V.; Lin, H.Y.; Jones, R.J.; Kuiatse, I.; Wang, H.; Yang, J.; Shah, J.J.; Thomas, S.K.; Wang, M.; et al. Evidence of a role for CD44 and cell adhesion in mediating resistance to lenalidomide in multiple myeloma: Therapeutic implications. Leukemia 2014, 28, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harshman, S.W.; Canella, A.; Ciarlariello, P.D.; Agarwal, K.; Branson, O.E.; Rocci, A.; Cordero, H.; Phelps, M.A.; Hade, E.M.; Dubovsky, J.A.; et al. Proteomic characterization of circulating extracellular vesicles identifies novel serum myeloma associated markers. J. Proteom. 2016, 136, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldschmidt, J.M.; Vijaykumar, T.; Knoechel, B.; Lohr, J.G. Tracking myeloma tumor DNA in peripheral blood. Best Pract. Res. Clin. Haematol. 2020, 33, 101146. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Yokoyama, K.; Imoto, S.; Tojo, A. Role of Circulating Tumor DNA in Hematological Malignancy. Cancers 2021, 13, 2078. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, B.; Caetano, J.; Barahona, F.; Lopes, R.; Carneiro, E.; Costa-Silva, B.; João, C. Liquid biopsies for multiple myeloma in a time of precision medicine. J. Mol. Med. 2020, 98, 513–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mithraprabhu, S.; Chen, M.; Savvidou, I.; Reale, A.; Spencer, A. Liquid biopsy: An evolving paradigm for the biological characterisation of plasma cell disorders. Leukemia 2021, 35, 2771–2783. [Google Scholar] [CrossRef] [PubMed]
- Mazzotti, C.; Buisson, L.; Maheo, S.; Perrot, A.; Chretien, M.L.; Leleu, X.; Hulin, C.; Manier, S.; Hébraud, B.; Roussel, M.; et al. Myeloma MRD by deep sequencing from circulating tumor DNA does not correlate with results obtained in the bone marrow. Blood Adv. 2018, 2, 2811–2813. [Google Scholar] [CrossRef] [Green Version]
- Diamandis, E.P.; Fiala, C. Can circulating tumor DNA be used for direct and early stage cancer detection? F1000Research 2017, 6, 2129. [Google Scholar] [CrossRef] [Green Version]
- Manier, S.; Park, J.; Capelletti, M.; Bustoros, M.; Freeman, S.S.; Ha, G.; Rhoades, J.; Liu, C.J.; Huynh, D.; Reed, S.C.; et al. Whole-exome sequencing of cell-free DNA and circulating tumor cells in multiple myeloma. Nat. Commun. 2018, 9, 1691. [Google Scholar] [CrossRef] [Green Version]
- Waldschmidt, J.M.; Anand, P.; Knoechel, B.; Lohr, J.G. Comprehensive characterization of circulating and bone marrow-derived multiple myeloma cells at minimal residual disease. Semin. Hematol. 2018, 55, 33–37. [Google Scholar] [CrossRef]
- Mishima, Y.; Paiva, B.; Shi, J.; Park, J.; Manier, S.; Takagi, S.; Massoud, M.; Perilla-Glen, A.; Aljawai, Y.; Huynh, D.; et al. The Mutational Landscape of Circulating Tumor Cells in Multiple Myeloma. Cell Rep. 2017, 19, 218–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, L.; Laurenzana, I.; Trino, S.; Lamorte, D.; Caivano, A.; Musto, P. An update on extracellular vesicles in multiple myeloma: A focus on their role in cell-to-cell cross-talk and as potential liquid biopsy biomarkers. Expert Rev. Mol. Diagn. 2019, 19, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Mithraprabhu, S.; Ramachandran, M.; Choi, K.; Khong, T.; Spencer, A. Utility of Circulating Cell-Free RNA Analysis for the Characterization of Global Transcriptome Profiles of Multiple Myeloma Patients. Cancers 2019, 11, 887. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Hurley, J.; Roberts, D.; Chakrabortty, S.K.; Enderle, D.; Noerholm, M.; Breakefield, X.O.; Skog, J.K. Exosome-based liquid biopsies in cancer: Opportunities and challenges. Ann. Oncol. 2021, 32, 466–477. [Google Scholar] [CrossRef]
- Lone, S.N.; Nisar, S.; Masoodi, T.; Singh, M.; Rizwan, A.; Hashem, S.; El-Rifai, W.; Bedognetti, D.; Batra, S.K.; Haris, M.; et al. Liquid biopsy: A step closer to transform diagnosis, prognosis and future of cancer treatments. Mol. Cancer 2022, 21, 79. [Google Scholar] [CrossRef]
- Krishnan, S.R.; Luk, F.; Brown, R.D.; Suen, H.; Kwan, Y.; Bebawy, M. Isolation of Human CD138(+) Microparticles from the Plasma of Patients with Multiple Myeloma. Neoplasia 2016, 18, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.Y.; Tian, M.Y.; Deng, L.; Wang, Y.S.; Xing, R.; Liu, H.; Fu, R. The potential diagnostic power of CD138+ microparticles from the plasma analysis for multiple myeloma clinical monitoring. Hematol. Oncol. 2019, 37, 401–408. [Google Scholar] [CrossRef]
- Rajeev Krishnan, S.; De Rubis, G.; Suen, H.; Joshua, D.; Lam Kwan, Y.; Bebawy, M. A liquid biopsy to detect multidrug resistance and disease burden in multiple myeloma. Blood Cancer J. 2020, 10, 37. [Google Scholar] [CrossRef] [Green Version]
- Koumpis, E.; Tassi, I.; Malea, T.; Papathanasiou, K.; Papakonstantinou, I.; Serpanou, A.; Tsolas, E.; Kapsali, E.; Vassilakopoulos, T.P.; Papoudou-Bai, A.; et al. CD56 expression in multiple myeloma: Correlation with poor prognostic markers but no effect on outcome. Pathol. Res. Pract. 2021, 225, 153567. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, H.; Tao, Q.; Zhang, C.; Yang, D.; Qin, H.; Xiong, S.; Tao, L.; Wu, F.; Zhang, J.; et al. Absence of both CD56 and CD117 expression on malignant plasma cells is related with a poor prognosis in patients with newly diagnosed multiple myeloma. Leuk. Res. 2016, 40, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.-V.; Martínez, B.P.; González-Calle, V. High-risk multiple myeloma: How to treat at diagnosis and relapse? Hematology 2021, 2021, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhou, X.; Zhu, J.W.; Ye, J.N.; Guo, H.F.; Sun, C. Association of CD117 and HLA-DR expression with shorter overall survival and/or progression-free survival in patients with multiple myeloma treated with bortezomib and thalidomide combination treatment without transplantation. Oncol. Lett. 2018, 16, 5655–5666. [Google Scholar] [CrossRef] [PubMed]
- Di Noto, G.; Paolini, L.; Zendrini, A.; Radeghieri, A.; Caimi, L.; Ricotta, D. C-src enriched serum microvesicles are generated in malignant plasma cell dyscrasia. PLoS ONE 2013, 8, e70811. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, A.; Adhikarla, V.; Poku, E.K.; Palmer, J.; Chaudhry, A.; Biglang-Awa, V.E.; Bowles, N.; Nathwani, N.; Rosenzweig, M.; Sahebi, F.; et al. Identifying CD38+ cells in patients with multiple myeloma: First-in-human imaging using copper-64-labeled daratumumab. Blood Adv. 2020, 4, 5194–5202. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Moreau, P.; Terpos, E.; Mateos, M.V.; Zweegman, S.; Cook, G.; Delforge, M.; Hájek, R.; Schjesvold, F.; Cavo, M.; et al. Multiple myeloma: EHA-ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2021, 32, 309–322. [Google Scholar] [CrossRef]
- Stetler-Stevenson, M.; Paiva, B.; Stoolman, L.; Lin, P.; Jorgensen, J.L.; Orfao, A.; Van Dongen, J.; Rawstron, A.C. Consensus guidelines for myeloma minimal residual disease sample staining and data acquisition. Cytometry Part B Clin. Cytom. 2016, 90, 26–30. [Google Scholar] [CrossRef]
- Arroz, M.; Came, N.; Lin, P.; Chen, W.; Yuan, C.; Lagoo, A.; Monreal, M.; de Tute, R.; Vergilio, J.A.; Rawstron, A.C.; et al. Consensus guidelines on plasma cell myeloma minimal residual disease analysis and reporting. Cytometry Part B Clin. Cytom. 2016, 90, 31–39. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | MM Isotype | ISS | FISH | EMD | % of PCs at Diagnosis | MM Markers by WB at EVs PB/BM at Diagnosis | IMWG Response Criteria at D100 | MRD at D100 by Flow Cytometry | % of PCs at D100 | MM Markers by WB at EVs PB/BM at D100 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PB | BM | CD38 | CD138 | K | L | BM | BM | PB | BM | CD38 | CD138 | K | L | |||||
| 01 | IgG/K | 2 | no abnormality | Yes | ≤0.05 | ≤10 | wd/wd | nd | ++/++ | +/++ | sCR | negative | 0 | ≤0.05 | wd/− | nd | +/+ | ++/+ |
| 02 | IgG/K | 3 | amp1q21 | No | 0 | ≥40 and ≤50 | ++/++ | nd | ++/++ | ++/++ | sCR | negative | 0 | ≥0.05 and ≤0.1 | +/+ | nd | +/+ | +/+ |
| 03 | IgG/K | 3 | t(4;14) | Yes | ≤0.05 | ≤10 | −/− | nd | ++/++ | +/++ | sCR | positive | ≤0.01 | ≥0.1 and ≤0.5 | wd/wd | nd | +/+ | +/wd |
| 04 | IgA/L | 3 | t(4;14), del17p | No | ≤0.05 | ≥10 and ≤20 | ++/++ | +/++ | ++/++ | ++/++ | VGPR | positive | ≤0.01 | ≥0.1 and ≤0.5 | ++/++ | ++/++ | +/+ | +/+ |
| 05 | IgG/L | 2 | no abnormality | No | ≤0.002 | ≥10 and ≤20 | ++/++ | nd | +/wd | ++/++ | sCR | positive | 0 | ≤0.05 | ++/+ | nd | ++/++ | ++/++ |
| 06 | L | 3 | no abnormality | No | ≤0.002 | ≥20 and ≤30 | +/+ | nd | ++/++ | +/+ | sCR | negative | 0 | ≤0.05 | ++/++ | nd | ++/++ | +/+ |
| 07 | K | 3 | del17p | Yes | 0 | ≤10 | nd | ++/+ | +/++ | +/++ | CR | negative | 0 | ≤0.05 | nd | ++/+ | +/+ | ++/++ |
| 08 | IgG/L | 3 | amp1q21 | No | ≤0.05 | ≥40 and ≤50 | +/++ | +/++ | wd/+ | +/+ | sCR | negative | ≤0.01 | ≤0.05 | wd/wd | −/− | wd/wd | ++/++ |
| 09 | IgG/K | 2 | no abnormality | No | 0 | ≥30 and ≤40 | +/wd | nd | ++/++ | +/+ | sCR | negative | 0 | ≤0.05 | +/wd | nd | ++/++ | +/+ |
| 10 | K | 3 | t(4;14) | Yes | 0 | ≥10 and ≤20 | wd/− | nd | ++/++ | wd/+ | CR | positive | 0 | ≤0.05 | wd/wd | nd | +/+ | ++/++ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bergantim, R.; Peixoto da Silva, S.; Polónia, B.; Barbosa, M.A.G.; Albergaria, A.; Lima, J.; Caires, H.R.; Guimarães, J.E.; Vasconcelos, M.H. Detection of Measurable Residual Disease Biomarkers in Extracellular Vesicles from Liquid Biopsies of Multiple Myeloma Patients—A Proof of Concept. Int. J. Mol. Sci. 2022, 23, 13686. https://doi.org/10.3390/ijms232213686
Bergantim R, Peixoto da Silva S, Polónia B, Barbosa MAG, Albergaria A, Lima J, Caires HR, Guimarães JE, Vasconcelos MH. Detection of Measurable Residual Disease Biomarkers in Extracellular Vesicles from Liquid Biopsies of Multiple Myeloma Patients—A Proof of Concept. International Journal of Molecular Sciences. 2022; 23(22):13686. https://doi.org/10.3390/ijms232213686
Chicago/Turabian StyleBergantim, Rui, Sara Peixoto da Silva, Bárbara Polónia, Mélanie A. G. Barbosa, André Albergaria, Jorge Lima, Hugo R. Caires, José E. Guimarães, and M. Helena Vasconcelos. 2022. "Detection of Measurable Residual Disease Biomarkers in Extracellular Vesicles from Liquid Biopsies of Multiple Myeloma Patients—A Proof of Concept" International Journal of Molecular Sciences 23, no. 22: 13686. https://doi.org/10.3390/ijms232213686