
Mitochondrial Effects of Common Cardiovascular Medications: The Good, the Bad and the Mixed

 and
and
Abstract
:1. Introduction
2. Mitochondrial Effects of the Main Classes of Drugs Used in Cardiovascular Diseases
2.1. Sympathomimetics
Isoprotenerol
2.2. Antiarrhytmics
2.2.1. Class I (Na-Channel Blockers)
- Quinidine
- Lidocaine
- Phenytoin
- Propafenone
2.2.2. Class II (β-Blockers)
- Carvedilol
- Nebivolol
- Metoprolol
- Atenolol
- Propanolol
- Timolol
- Esmolol
2.2.3. Class III (K-Channel Blockers)
- Amiodarone
- Dronedarone
- Ibutilide
- Sotalol
- Dofetilide
2.2.4. Class IV (Ca-Channel Blockers)
- Verapamil
- Diltiazem
2.2.5. Others
- Adenosine
- Digitalis
2.3. Renin-Angiotensin-Aldosterone System (RAAS) Blockers
2.3.1. Angiotensin-Converting Enzyme Inhibitors (ACEI)
- Ramipril
- Zofenopril
- Perindopril
- Captopril
- Trandolapril
- Lisinopril
- Enalapril
2.3.2. Angiotensin Receptor Blockers (ARBs)
- Valsartan
- Losartan
- Candesartan
- Irbesartan
- Telmisartan
- Olmesartan
- Azilsartan
2.3.3. Angiotensin Receptor Neprilysin Inhibitor (ARNi): Sacubitril/Valsartan
2.4. Calcium Channel Blockers-Dihydropyridines
Amlodipine
2.5. Antithrombotic Agents
2.5.1. Acetyl-Salicylic Acid
2.5.2. Clopidogrel
2.5.3. Ticagrelor
2.5.4. Prasugrel and Ticlopidine
2.6. Oral Anticoagulants
2.6.1. Coumarin Derivatives
2.6.2. Direct Oral Anticoagulants
2.7. Diuretics
2.7.1. Loop Diuretics
2.7.2. Antagonists of Aldosterone
2.7.3. Epithelial Sodium Channel Blockers
2.8. Statins
2.9. Direct Vasodilators
2.9.1. Organic Nitrates
2.9.2. Molsidomine
2.9.3. Hydralazine
2.9.4. Sodium Nitroprusside
2.9.5. Minoxidil
2.10. Biguanides
2.11. Sodium-Glucose Cotransporter 2 (SGLT2) Inhibitors
2.11.1. Empagliflozin
2.11.2. Dapagliflozin
2.11.3. Canagliflozin
2.12. Glucagon-like Peptide-1 Receptor Agonists (GLP-1 RAs)
2.12.1. Liraglutide
2.12.2. Exenatide
2.12.3. Dulaglutide
2.12.4. Semaglutide
2.12.5. Lixisenatide
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACEI | Angiotensin-converting enzyme inhibitors |
| ADP | Adenosine diphosphate |
| AMPK | AMP-activated protein kinase |
| ARBs | Angiotensin receptor blockers |
| ARNi | Angiotensin receptor neprilysin inhibitor |
| ATP | Adenosine triphosphate |
| Bcl-2 | B-cell lymphoma 2 |
| Bax | Bcl-2-associated X protein |
| Cyt. c | Cytochrome c |
| CI | Complex I |
| CII | Complex II |
| ETS | Electron transport system |
| eNOS-PGC-1α | Endothelial nitric oxide synthase 3-Peroxisome proliferator-activated receptor-gamma coactivator-1alpha |
| GLP-1 RAs | Glucagon-like peptide-1 receptor agonists |
| LEAK | Non-phosphorylating respiration |
| LDL | Low-density lipoprotein |
| mtDNA | Mitochondrial deoxyribonucleic acid |
| mPTP | Mitochondrial permeability transition pore |
| NADH | Nicotinamide adenine dinucleotide reduced form |
| OXPHOS | Oxidative phosphorylation |
| RAAS | Renin-angiotensin-aldosterone system |
| ROS | Reactive oxygen species |
| SGLT2 | Sodium-glucose cotransporter 2 inhibitors |
| TNFα | Tumor necrosis factor α |
References
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., 2nd; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mottis, A.; Herzig, S.; Auwerx, J. Mitocellular communication: Shaping health and disease. Science 2019, 366, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Vegas, A.; Sanchez-Aguilera, P.; Krycer, J.R.; Morales, P.E.; Monsalves-Alvarez, M.; Cifuentes, M.; Rothermel, B.A.; Lavandero, S. Is Mitochondrial Dysfunction a Common Root of Noncommunicable Chronic Diseases? Endocr. Rev. 2020, 41, bnaa005. [Google Scholar] [CrossRef]
- Siasos, G.; Tsigkou, V.; Kosmopoulos, M.; Theodosiadis, D.; Simantiris, S.; Tagkou, N.M.; Tsimpiktsioglou, A.; Stampouloglou, P.K.; Oikonomou, E.; Mourouzis, K.; et al. Mitochondria and cardiovascular diseases-from pathophysiology to treatment. Ann. Transl. Med. 2018, 6, 256. [Google Scholar] [CrossRef]
- Mason, F.E.; Pronto, J.R.D.; Alhussini, K.; Maack, C.; Voigt, N. Cellular and mitochondrial mechanisms of atrial fibrillation. Basic Res. Cardiol. 2020, 115, 72. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Dietl, A.; Maack, C. Targeting Mitochondrial Calcium Handling and Reactive Oxygen Species in Heart Failure. Curr. Heart Fail. Rep. 2017, 14, 338–349. [Google Scholar] [CrossRef]
- Pool, L.; Wijdeveld, L.; de Groot, N.M.S.; Brundel, B. The Role of Mitochondrial Dysfunction in Atrial Fibrillation: Translation to Druggable Target and Biomarker Discovery. Int. J. Mol. Sci. 2021, 22, 8463. [Google Scholar] [CrossRef]
- Marzetti, E.; Csiszar, A.; Dutta, D.; Balagopal, G.; Calvani, R.; Leeuwenburgh, C. Role of mitochondrial dysfunction and altered autophagy in cardiovascular aging and disease: From mechanisms to therapeutics. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H459–H476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varga, Z.V.; Ferdinandy, P.; Liaudet, L.; Pacher, P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1453–H1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoker, M.L.; Newport, E.; Hulit, J.C.; West, A.P.; Morten, K.J. Impact of pharmacological agents on mitochondrial function: A growing opportunity? Biochem. Soc. Trans. 2019, 47, 1757–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abolbashari, M.; Macaulay, T.E.; Whayne, T.F.; Mukherjee, D.; Saha, S. Polypharmacy in Cardiovascular Medicine: Problems and Promises! Cardiovasc. Hematol. Agents Med. Chem. 2017, 15, 31–39. [Google Scholar] [CrossRef]
- Will, Y.; Shields, J.E.; Wallace, K.B. Drug-Induced Mitochondrial Toxicity in the Geriatric Population: Challenges and Future Directions. Biology 2019, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- Dykens, J.A.; Will, Y. The significance of mitochondrial toxicity testing in drug development. Drug Discov. Today 2007, 12, 777–785. [Google Scholar] [CrossRef]
- Niyazov, D.M.; Kahler, S.G.; Frye, R.E. Primary Mitochondrial Disease and Secondary Mitochondrial Dysfunction: Importance of Distinction for Diagnosis and Treatment. Mol. Syndromol. 2016, 7, 122–137. [Google Scholar] [CrossRef] [Green Version]
- Vuda, M.; Kamath, A. Drug induced mitochondrial dysfunction: Mechanisms and adverse clinical consequences. Mitochondrion 2016, 31, 63–74. [Google Scholar] [CrossRef]
- Will, Y.; Dykens, J. Mitochondrial toxicity assessment in industry—A decade of technology development and insight. Expert Opin. Drug. Metab. Toxicol. 2014, 10, 1061–1067. [Google Scholar] [CrossRef]
- Hargreaves, I.P.; Al Shahrani, M.; Wainwright, L.; Heales, S.J. Drug-Induced Mitochondrial Toxicity. Drug Saf. 2016, 39, 661–674. [Google Scholar] [CrossRef]
- Dykens, J.A.; Marroquin, L.D.; Will, Y. Strategies to reduce late-stage drug attrition due to mitochondrial toxicity. Expert Rev. Mol. Diagn. 2007, 7, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Truong, D.; Shangari, N.; O’Brien, P.J. Drug-induced mitochondrial toxicity. Expert Opin. Drug. Metab. Toxicol. 2005, 1, 655–669. [Google Scholar] [CrossRef] [PubMed]
- Penman, S.L.; Carter, A.S.; Chadwick, A.E. Investigating the importance of individual mitochondrial genotype in susceptibility to drug-induced toxicity. Biochem. Soc. Trans. 2020, 48, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Sieprath, T.; Corne, T.D.; Willems, P.H.; Koopman, W.J.; De Vos, W.H. Integrated High-Content Quantification of Intracellular ROS Levels and Mitochondrial Morphofunction. Adv. Anat. Embryol. Cell Biol. 2016, 219, 149–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forini, F.; Canale, P.; Nicolini, G.; Iervasi, G. Mitochondria-Targeted Drug Delivery in Cardiovascular Disease: A Long Road to Nano-Cardio Medicine. Pharmaceutics 2020, 12, 1122. [Google Scholar] [CrossRef]
- Weissman, D.; Maack, C. Redox signaling in heart failure and therapeutic implications. Free Radic. Biol. Med. 2021, 171, 345–364. [Google Scholar] [CrossRef]
- Maack, C.; Eschenhagen, T.; Hamdani, N.; Heinzel, F.R.; Lyon, A.R.; Manstein, D.J.; Metzger, J.; Papp, Z.; Tocchetti, C.G.; Yilmaz, M.B.; et al. Treatments targeting inotropy. Eur. Heart J. 2019, 40, 3626–3644. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.; Costa, D.; Lima, J.L.; Fernandes, E. Antioxidant activity of beta-blockers: An effect mediated by scavenging reactive oxygen and nitrogen species? Bioorganic Med. Chem. 2006, 14, 4568–4577. [Google Scholar] [CrossRef]
- Djanani, A.; Kaneider, N.C.; Meierhofer, C.; Sturn, D.; Dunzendorfer, S.; Allmeier, H.; Wiedermann, C.J. Inhibition of neutrophil migration and oxygen free radical release by metipranolol and timolol. Pharmacology 2003, 68, 198–203. [Google Scholar] [CrossRef]
- Miyamoto, N.; Izumi, H.; Miyamoto, R.; Kubota, T.; Tawara, A.; Sasaguri, Y.; Kohno, K. Nipradilol and timolol induce Foxo3a and peroxiredoxin 2 expression and protect trabecular meshwork cells from oxidative stress. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2777–2784. [Google Scholar] [CrossRef]
- Sozmen, N.N.; Tuncay, E.; Bilginoglu, A.; Turan, B. Profound cardioprotection with timolol in a female rat model of aging-related altered left ventricular function. Can. J. Physiol. Pharmacol. 2011, 89, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.L.; Huang, X.; Yang, Y.; Zhao, Y.J.; Wei, C.X.; Zhao, M. Ibutilide protects against cardiomyocytes injury via inhibiting endoplasmic reticulum and mitochondrial stress pathways. Heart Vessel. 2017, 32, 208–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bețiu, A.M.; Chamkha, I.; Gustafsson, E.; Meijer, E.; Avram, V.F.; Åsander Frostner, E.; Ehinger, J.K.; Petrescu, L.; Muntean, D.M.; Elmér, E. Cell-Permeable Succinate Rescues Mitochondrial Respiration in Cellular Models of Amiodarone Toxicity. Int. J. Mol. Sci. 2021, 22, 11786. [Google Scholar] [CrossRef] [PubMed]
- Hamed, K.H.; Hu, C.; Dai, D.Z.; Yu, F.; Dai, Y. CPU228, a derivative of dofetilide, relieves cardiac dysfunction by normalizing FKBP12.6, NADPH oxidase and protein kinase C epsilon in the myocardium. J. Pharm. Pharmacol. 2010, 62, 77–83. [Google Scholar] [CrossRef]
- Jangholi, E.; Sharifi, Z.N.; Hoseinian, M.; Zarrindast, M.R.; Rahimi, H.R.; Mowla, A.; Aryan, H.; Javidi, M.A.; Parsa, Y.; Ghaffarpasand, F.; et al. Verapamil Inhibits Mitochondria-Induced Reactive Oxygen Species and Dependent Apoptosis Pathways in Cerebral Transient Global Ischemia/Reperfusion. Oxidative Med. Cell. Longev. 2020, 2020, 5872645. [Google Scholar] [CrossRef]
- Kedziora-Kornatowska, K.; Szram, S.; Kornatowski, T.; Szadujkis-Szadurski, L.; Kedziora, J.; Bartosz, G. The effect of verapamil on the antioxidant defence system in diabetic kidney. Clin. Chim. Acta Int. J. Clin. Chem. 2002, 322, 105–112. [Google Scholar] [CrossRef]
- Kröner, A.; Seitelberger, R.; Schirnhofer, J.; Bernecker, O.; Mallinger, R.; Hallström, S.; Ploner, M.; Podesser, B.K. Diltiazem during reperfusion preserves high energy phosphates by protection of mitochondrial integrity. Eur. J. Cardio-Thorac. Surg. Off. J. Eur. Assoc. Cardio-Thorac. Surg. 2002, 21, 224–231. [Google Scholar] [CrossRef] [Green Version]
- Kavanaugh, K.M.; Aisen, A.M.; Fechner, K.P.; Wroblewski, L.; Chenevert, T.L.; Buda, A.J. Effects of diltiazem on phosphate metabolism in ischemic and reperfused myocardium using phosphorus31 nuclear magnetic resonance spectroscopy in vivo. Am. Heart J. 1989, 118, 1210–1219. [Google Scholar] [CrossRef] [Green Version]
- Zografos, P.; Watts, J.A. Shifts in calcium in ischemic and reperfused rat hearts: A cytochemical and morphometric study of the effects of diltiazem. Am. J. Cardiovasc. Pathol. 1990, 3, 155–165. [Google Scholar]
- Koller, P.T.; Bergmann, S.R. Reduction of lipid peroxidation in reperfused isolated rabbit hearts by diltiazem. Circ. Res. 1989, 65, 838–846. [Google Scholar] [CrossRef] [Green Version]
- Crenesse, D.; Tornieri, K.; Laurens, M.; Heurteaux, C.; Cursio, R.; Gugenheim, J.; Schmid-Alliana, A. Diltiazem reduces apoptosis in rat hepatocytes subjected to warm hypoxia-reoxygenation. Pharmacology 2002, 65, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.; Cargnoni, A.; Curello, S.; Ceconi, C.; Boraso, A.; Visioli, O. Protection of the ischemic myocardium by the converting-enzyme inhibitor zofenopril: Insight into its mechanism of action. J. Cardiovasc. Pharmacol. 1992, 20, 694–704. [Google Scholar] [PubMed]
- Zhu, Z.; Li, H.; Chen, W.; Cui, Y.; Huang, A.; Qi, X. Perindopril Improves Cardiac Function by Enhancing the Expression of SIRT3 and PGC-1α in a Rat Model of Isoproterenol-Induced Cardiomyopathy. Front. Pharmacol. 2020, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Mamou, Z.; Chahine, M.; Rhondali, O.; Dehina, L.; Chevalier, P.; Descotes, J.; Bui-Xuan, B.; Romestaing, C.; Timour, Q. Effects of amlodipine and perindoprilate on the structure and function of mitochondria in ventricular cardiomyocytes during ischemia-reperfusion in the pig. Fundam. Clin. Pharmacol. 2015, 29, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, A.; Shetty, S.; Shirole, T.; Jagtap, A.G. Potential of protease inhibitor in 3-nitropropionic acid induced Huntington’s disease like symptoms: Mitochondrial dysfunction and neurodegeneration. Neurotoxicology 2014, 45, 139–148. [Google Scholar] [CrossRef]
- Toga, W.; Tanonaka, K.; Takeo, S. Changes in Hsp60 level of the failing heart following acute myocardial infarction and the effect of long-term treatment with trandolapril. Biol. Pharm. Bull. 2007, 30, 105–110. [Google Scholar] [CrossRef] [Green Version]
- Sanbe, A.; Tanonaka, K.; Kobayasi, R.; Takeo, S. Effects of long-term therapy with ACE inhibitors, captopril, enalapril and trandolapril, on myocardial energy metabolism in rats with heart failure following myocardial infarction. J. Mol. Cell. Cardiol. 1995, 27, 2209–2222. [Google Scholar] [CrossRef]
- De Cavanagh, E.M.; Inserra, F.; Ferder, L.; Fraga, C.G. Enalapril and captopril enhance glutathione-dependent antioxidant defenses in mouse tissues. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 278, R572–R577. [Google Scholar] [CrossRef]
- Piotrkowski, B.; Fraga, C.G.; de Cavanagh, E.M. Mitochondrial function and nitric oxide metabolism are modified by enalapril treatment in rat kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1494–R1501. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Sirago, G.; Pesce, V.; Lezza, A.M.S.; Calvani, R.; Bossola, M.; Villani, E.R.; Landi, F.; Leeuwenburgh, C.; Bernabei, R.; et al. Administration of Enalapril Started Late in Life Attenuates Hypertrophy and Oxidative Stress Burden, Increases Mitochondrial Mass, and Modulates Mitochondrial Quality Control Signaling in the Rat Heart. Biomolecules 2018, 8, 177. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, S.A.; Paramesha, B.; Meghwani, H.; Kumar Reddy, M.P.; Arava, S.K.; Banerjee, S.K. Allyl Methyl Sulfide Preserved Pressure Overload-Induced Heart Failure Via Modulation of Mitochondrial Function. Biomed. Pharmacother. Biomed. Pharmacother. 2021, 138, 111316. [Google Scholar] [CrossRef] [PubMed]
- Hiona, A.; Lee, A.S.; Nagendran, J.; Xie, X.; Connolly, A.J.; Robbins, R.C.; Wu, J.C. Pretreatment with angiotensin-converting enzyme inhibitor improves doxorubicin-induced cardiomyopathy via preservation of mitochondrial function. J. Thorac. Cardiovasc. Surg. 2011, 142, 396–403.e393. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, Z.L.; Crane, J.A.; Jordan, K.L.; Pawar, A.S.; Textor, S.C.; Lerman, A.; Lerman, L.O. Valsartan regulates myocardial autophagy and mitochondrial turnover in experimental hypertension. Hypertension 2014, 64, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Qiang, G.; Zhang, L.; Yang, X.; Xuan, Q.; Shi, L.; Zhang, H.; Chen, B.; Li, X.; Zu, M.; Zhou, D.; et al. Effect of valsartan on the pathological progression of hepatic fibrosis in rats with type 2 diabetes. Eur. J. Pharmacol. 2012, 685, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Clark, S.E.; Thyfault, J.P.; Uptergrove, G.M.; Li, W.; Whaley-Connell, A.T.; Ferrario, C.M.; Sowers, J.R.; Ibdah, J.A. Oxidative stress-mediated mitochondrial dysfunction contributes to angiotensin II-induced nonalcoholic fatty liver disease in transgenic Ren2 rats. Am. J. Pathol. 2009, 174, 1329–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cavanagh, E.M.; Toblli, J.E.; Ferder, L.; Piotrkowski, B.; Stella, I.; Inserra, F. Renal mitochondrial dysfunction in spontaneously hypertensive rats is attenuated by losartan but not by amlodipine. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R1616–R1625. [Google Scholar] [CrossRef] [Green Version]
- De Cavanagh, E.M.; Inserra, F.; Ferder, L. Angiotensin II blockade: A strategy to slow ageing by protecting mitochondria? Cardiovasc. Res. 2011, 89, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.M.; Wang, C.H.; Chang, Z.Y.; Huang, T.H.; Lee, T.Y. Losartan Attenuates Insulin Resistance and Regulates Browning Phenomenon of White Adipose Tissue in ob/ob Mice. Curr. Issues Mol. Biol. 2021, 43, 1828–1843. [Google Scholar] [CrossRef]
- Uchikado, Y.; Ikeda, Y.; Sasaki, Y.; Iwabayashi, M.; Akasaki, Y.; Ohishi, M. Association of Lectin-Like Oxidized Low-Density Lipoprotein Receptor-1 With Angiotensin II Type 1 Receptor Impacts Mitochondrial Quality Control, Offering Promise for the Treatment of Vascular Senescence. Front. Cardiovasc. Med. 2021, 8, 788655. [Google Scholar] [CrossRef]
- Wang, Z.; Niu, Q.; Peng, X.; Li, M.; Liu, K.; Liu, Y.; Liu, J.; Jin, F.; Li, X.; Wei, Y. Candesartan cilexetil attenuated cardiac remodeling by improving expression and function of mitofusin 2 in SHR. Int. J. Cardiol. 2016, 214, 348–357. [Google Scholar] [CrossRef]
- Gaur, V.; Kumar, A. Neuroprotective potentials of candesartan, atorvastatin and their combination against stroke induced motor dysfunction. Inflammopharmacology 2011, 19, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Pai, P.Y.; Lin, Y.Y.; Yu, S.H.; Lin, C.Y.; Liou, Y.F.; Wu, X.B.; Wong, J.K.S.; Huang, C.Y.; Lee, S.D. Angiotensin II receptor blocker irbesartan attenuates sleep apnea-induced cardiac apoptosis and enhances cardiac survival and Sirtuin 1 upregulation. Sleep Breath. Schlaf Atm. 2022, 26, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Ding, J.; Lai, Q.; Wang, X.; Li, A.; Liu, S. Irbesartan Ameliorates Lipid Deposition by Enhancing Autophagy via PKC/AMPK/ULK1 Axis in Free Fatty Acid Induced Hepatocytes. Front. Physiol. 2019, 10, 681. [Google Scholar] [CrossRef] [PubMed]
- Ray, B.; Ramesh, G.; Verma, S.R.; Ramamurthy, S.; Tuladhar, S.; Mahalakshmi, A.M.; Essa, M.M.; Chidambaram, S.B. Effects of Telmisartan, an AT1 receptor antagonist, on mitochondria-specific genes expression in a mouse MPTP model of Parkinsonism. Front. Biosci. 2021, 26, 262–271. [Google Scholar] [CrossRef]
- Zhan, X.; Chen, W.; Chen, J.; Lei, C.; Wei, L. Telmisartan Mitigates High-Glucose-Induced Injury in Renal Glomerular Endothelial Cells (rGECs) and Albuminuria in Diabetes Mice. Chem. Res. Toxicol. 2021, 34, 2079–2086. [Google Scholar] [CrossRef]
- Wang, B.; Xiong, S.; Lin, S.; Xia, W.; Li, Q.; Zhao, Z.; Wei, X.; Lu, Z.; Wei, X.; Gao, P.; et al. Enhanced Mitochondrial Transient Receptor Potential Channel, Canonical Type 3-Mediated Calcium Handling in the Vasculature From Hypertensive Rats. J. Am. Heart Assoc. 2017, 6, e005812. [Google Scholar] [CrossRef]
- Takeuchi, K.; Yamamoto, K.; Ohishi, M.; Takeshita, H.; Hongyo, K.; Kawai, T.; Takeda, M.; Kamide, K.; Kurtz, T.W.; Rakugi, H. Telmisartan modulates mitochondrial function in vascular smooth muscle cells. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2013, 36, 433–439. [Google Scholar] [CrossRef] [Green Version]
- Thorwald, M.; Rodriguez, R.; Lee, A.; Martinez, B.; Peti-Peterdi, J.; Nakano, D.; Nishiyama, A.; Ortiz, R.M. Angiotensin receptor blockade improves cardiac mitochondrial activity in response to an acute glucose load in obese insulin resistant rats. Redox Biol. 2018, 14, 371–378. [Google Scholar] [CrossRef]
- Takada, S.; Kinugawa, S.; Hirabayashi, K.; Suga, T.; Yokota, T.; Takahashi, M.; Fukushima, A.; Homma, T.; Ono, T.; Sobirin, M.A.; et al. Angiotensin II receptor blocker improves the lowered exercise capacity and impaired mitochondrial function of the skeletal muscle in type 2 diabetic mice. J. Appl. Physiol. 2013, 114, 844–857. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Mao, P.; Wang, J.; Wang, T.; Xie, C.H. Azilsartan, an angiotensin II type 1 receptor blocker, attenuates tert-butyl hydroperoxide-induced endothelial cell injury through inhibition of mitochondrial dysfunction and anti-inflammatory activity. Neurochem. Int. 2016, 94, 48–56. [Google Scholar] [CrossRef]
- Gupta, V.; Dhull, D.K.; Joshi, J.; Kaur, S.; Kumar, A. Neuroprotective potential of azilsartan against cerebral ischemic injury: Possible involvement of mitochondrial mechanisms. Neurochem. Int. 2020, 132, 104604. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Braza, J.; Mende, U.; Choudhary, G.; Zhang, P. Cardioprotective effects of early intervention with sacubitril/valsartan on pressure overloaded rat hearts. Sci. Rep. 2021, 11, 16542. [Google Scholar] [CrossRef]
- Sabbah, H.N.; Zhang, K.; Gupta, R.C.; Xu, J.; Singh-Gupta, V. Effects of Angiotensin-Neprilysin Inhibition in Canines with Experimentally Induced Cardiorenal Syndrome. J. Card. Fail. 2020, 26, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Mutaf, I.; Habif, S.; Turgan, N.; Parildar, Z.; Ozmen, D.; Bayindir, O.; Uysal, A. Amlodipine and glutathione cycle in hypercholesterolaemia. Acta Cardiol. 2004, 59, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Pronobesh, C.; Dagagi, A.V.; Pallab, C.; Kumar, W.A. Protective role of the calcium channel blocker amlodipine against mitochondrial injury in ischemia and reperfusion injury of rat liver. Acta Pharm. 2008, 58, 421–428. [Google Scholar] [CrossRef] [Green Version]
- Mason, R.P.; Trumbore, M.W.; Mason, P.E. [Membrane biophysical interaction of amlodipine and antioxidant properties]. Drugs 2000, 59, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.A.; Chattopadhyay, P.; Abid, M.; Pawdey, A.; Kishore, K.; Wahi, A.K. Protective effects of amlodipine on mitochondrial injury in ischemic reperfused rat heart. J. Environ. Biol. 2012, 33, 591–595. [Google Scholar] [PubMed]
- Park, H.H.; Han, M.H.; Choi, H.; Lee, Y.J.; Kim, J.M.; Cheong, J.H.; Ryu, J.I.; Lee, K.Y.; Koh, S.H. Mitochondria damaged by Oxygen Glucose Deprivation can be Restored through Activation of the PI3K/Akt Pathway and Inhibition of Calcium Influx by Amlodipine Camsylate. Sci. Rep. 2019, 9, 15717. [Google Scholar] [CrossRef] [Green Version]
- Olgar, Y.; Tuncay, E.; Billur, D.; Durak, A.; Ozdemir, S.; Turan, B. Ticagrelor reverses the mitochondrial dysfunction through preventing accumulated autophagosomes-dependent apoptosis and ER stress in insulin-resistant H9c2 myocytes. Mol. Cell. Biochem. 2020, 469, 97–107. [Google Scholar] [CrossRef]
- Olgar, Y.; Durak, A.; Degirmenci, S.; Tuncay, E.; Billur, D.; Ozdemir, S.; Turan, B. Ticagrelor alleviates high-carbohydrate intake induced altered electrical activity of ventricular cardiomyocytes by regulating sarcoplasmic reticulum-mitochondria miscommunication. Mol. Cell. Biochem. 2021, 476, 3827–3844. [Google Scholar] [CrossRef]
- Torramade-Moix, S.; Palomo, M.; Vera, M.; Jerez, D.; Moreno-Castaño, A.B.; Zafar, M.U.; Rovira, J.; Diekmann, F.; Garcia-Pagan, J.C.; Escolar, G.; et al. Apixaban Downregulates Endothelial Inflammatory and Prothrombotic Phenotype in an In Vitro Model of Endothelial Dysfunction in Uremia. Cardiovasc. Drugs Ther. 2021, 35, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Bukowska, A.; Schild, L.; Bornfleth, P.; Peter, D.; Wiese-Rischke, C.; Gardemann, A.; Isermann, B.; Walles, T.; Goette, A. Activated clotting factor X mediates mitochondrial alterations and inflammatory responses via protease-activated receptor signaling in alveolar epithelial cells. Eur. J. Pharmacol. 2020, 869, 172875. [Google Scholar] [CrossRef] [PubMed]
- Kintner, D.B.; Luo, J.; Gerdts, J.; Ballard, A.J.; Shull, G.E.; Sun, D. Role of Na+-K+-Cl- cotransport and Na+/Ca2+ exchange in mitochondrial dysfunction in astrocytes following in vitro ischemia. Am. J. Physiol. Cell Physiol. 2007, 292, C1113–C1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Kintner, D.B.; Begum, G.; Algharabli, J.; Cengiz, P.; Shull, G.E.; Liu, X.J.; Sun, D. Endoplasmic reticulum Ca2+ signaling and mitochondrial Cyt c release in astrocytes following oxygen and glucose deprivation. J. Neurochem. 2010, 114, 1436–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.Y.; Suh, K.S.; Jung, W.W.; Chin, S.O. Spironolactone Attenuates Methylglyoxal-induced Cellular Dysfunction in MC3T3-E1 Osteoblastic Cells. J. Korean Med. Sci. 2021, 36, e265. [Google Scholar] [CrossRef]
- Williams, T.A.; Verhovez, A.; Milan, A.; Veglio, F.; Mulatero, P. Protective effect of spironolactone on endothelial cell apoptosis. Endocrinology 2006, 147, 2496–2505. [Google Scholar] [CrossRef] [Green Version]
- Hung, C.S.; Chang, Y.Y.; Tsai, C.H.; Liao, C.W.; Peng, S.Y.; Lee, B.C.; Pan, C.T.; Wu, X.M.; Chen, Z.W.; Wu, V.C.; et al. Aldosterone suppresses cardiac mitochondria. Transl. Res. J. Lab. Clin. Med. 2022, 239, 58–70. [Google Scholar] [CrossRef]
- Shao, Q.; Meng, L.; Lee, S.; Tse, G.; Gong, M.; Zhang, Z.; Zhao, J.; Zhao, Y.; Li, G.; Liu, T. Empagliflozin, a sodium glucose co-transporter-2 inhibitor, alleviates atrial remodeling and improves mitochondrial function in high-fat diet/streptozotocin-induced diabetic rats. Cardiovasc. Diabetol. 2019, 18, 165. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, M.; Kuno, A.; Yano, T.; Miki, T.; Oshima, H.; Sato, T.; Nakata, K.; Kimura, Y.; Tanno, M.; Miura, T. Empagliflozin normalizes the size and number of mitochondria and prevents reduction in mitochondrial size after myocardial infarction in diabetic hearts. Physiol. Rep. 2018, 6, e13741. [Google Scholar] [CrossRef]
- Yurista, S.R.; Silljé, H.H.W.; Oberdorf-Maass, S.U.; Schouten, E.M.; Pavez Giani, M.G.; Hillebrands, J.L.; van Goor, H.; van Veldhuisen, D.J.; de Boer, R.A.; Westenbrink, B.D. Sodium-glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Eur. J. Heart Fail. 2019, 21, 862–873. [Google Scholar] [CrossRef] [Green Version]
- Seefeldt, J.M.; Lassen, T.R.; Hjortbak, M.V.; Jespersen, N.R.; Kvist, F.; Hansen, J.; Bøtker, H.E. Cardioprotective effects of empagliflozin after ischemia and reperfusion in rats. Sci. Rep. 2021, 11, 9544. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Huang, C.; Sin, J.; Germano, J.F.; Taylor, D.J.R.; Thakur, R.; Gottlieb, R.A.; Mentzer, R.M., Jr.; Andres, A.M. Attenuation of Adverse Postinfarction Left Ventricular Remodeling with Empagliflozin Enhances Mitochondria-Linked Cellular Energetics and Mitochondrial Biogenesis. Int. J. Mol. Sci. 2021, 23, 437. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Xu, C.; Liu, X.; Li, X.; Li, T.; Yu, X.; Xue, M.; Yang, J.; Kosmas, C.E.; Moris, D.; et al. Empagliflozin Induces White Adipocyte Browning and Modulates Mitochondrial Dynamics in KK Cg-Ay/J Mice and Mouse Adipocytes. Front. Physiol. 2021, 12, 745058. [Google Scholar] [CrossRef]
- Yu, X.; Hao, M.; Liu, Y.; Ma, X.; Lin, W.; Xu, Q.; Zhou, H.; Shao, N.; Kuang, H. Liraglutide ameliorates non-alcoholic steatohepatitis by inhibiting NLRP3 inflammasome and pyroptosis activation via mitophagy. Eur. J. Pharmacol. 2019, 864, 172715. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; Ren, H.; Du, H.; Zhang, M.; Xiong, X.; Lv, R. Liraglutide repairs the infarcted heart: The role of the SIRT1/Parkin/mitophagy pathway. Mol. Med. Rep. 2018, 17, 3722–3734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Li, N.; Yan, S.; Lu, Y.; Miao, X.; Gu, Z.; Shao, Y. Liraglutide protects renal mesangial cells against hyperglycemia-mediated mitochondrial apoptosis by activating the ERK-Yap signaling pathway and upregulating Sirt3 expression. Mol. Med. Rep. 2019, 19, 2849–2860. [Google Scholar] [CrossRef] [PubMed]
- Durak, A.; Akkus, E.; Canpolat, A.G.; Tuncay, E.; Corapcioglu, D.; Turan, B. Glucagon-like peptide-1 receptor agonist treatment of high carbohydrate intake-induced metabolic syndrome provides pleiotropic effects on cardiac dysfunction through alleviations in electrical and intracellular Ca(2+) abnormalities and mitochondrial dysfunction. Clin. Exp. Pharmacol. Physiol. 2022, 49, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.K.; Lin, K.J.; Lin, H.Y.; Lin, K.L.; Lan, M.Y.; Wang, P.W.; Wang, T.J.; Wang, F.S.; Tsai, P.C.; Liou, C.W.; et al. Glucagon-Like Peptide-1 Receptor Agonist Ameliorates 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine (MPTP) Neurotoxicity Through Enhancing Mitophagy Flux and Reducing α-Synuclein and Oxidative Stress. Front. Mol. Neurosci. 2021, 14, 697440. [Google Scholar] [CrossRef]
- Chang, G.; Liu, J.; Qin, S.; Jiang, Y.; Zhang, P.; Yu, H.; Lu, K.; Zhang, N.; Cao, L.; Wang, Y.; et al. Cardioprotection by exenatide: A novel mechanism via improving mitochondrial function involving the GLP-1 receptor/cAMP/PKA pathway. Int. J. Mol. Med. 2018, 41, 1693–1703. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.H.; Ha, S.J.; Woo, J.S.; Lee, G.J.; Lee, S.R.; Kim, J.W.; Park, H.K.; Kim, W. Exenatide Prevents Morphological and Structural Changes of Mitochondria Following Ischaemia-Reperfusion Injury. Heart Lung Circ. 2017, 26, 519–523. [Google Scholar] [CrossRef]
- Zheng, W.; Pan, H.; Wei, L.; Gao, F.; Lin, X. Dulaglutide mitigates inflammatory response in fibroblast-like synoviocytes. Int. Immunopharmacol. 2019, 74, 105649. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Tuo, X.; Li, B.; Deng, Z.; Qiu, Y.; Xie, H. Semaglutide attenuates excessive exercise-induced myocardial injury through inhibiting oxidative stress and inflammation in rats. Life Sci. 2020, 250, 117531. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, L.; Li, L.; Hölscher, C. Semaglutide is Neuroprotective and Reduces α-Synuclein Levels in the Chronic MPTP Mouse Model of Parkinson’s Disease. J. Park. Dis. 2019, 9, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Pu, Y. Lixisenatide enhances mitochondrial biogenesis and function through regulating the CREB/PGC-1α pathway. Biochem. Biophys. Res. Commun. 2019, 508, 1120–1125. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Zhang, H.; Zhang, W.; Wang, Q.; Wang, W.; Ge, G.; Bai, J.; Guo, X.; Zhang, Y.; Jiang, X.; et al. The protective effects of lixisenatide against inflammatory response in human rheumatoid arthritis fibroblast-like synoviocytes. Int. Immunopharmacol. 2019, 75, 105732. [Google Scholar] [CrossRef]
- Krestinina, O.; Baburina, Y.; Krestinin, R.; Odinokova, I.; Fadeeva, I.; Sotnikova, L. Astaxanthin Prevents Mitochondrial Impairment Induced by Isoproterenol in Isolated Rat Heart Mitochondria. Antioxidants 2020, 9, 262. [Google Scholar] [CrossRef] [Green Version]
- Krestinin, R.; Baburina, Y.; Odinokova, I.; Kruglov, A.; Fadeeva, I.; Zvyagina, A.; Sotnikova, L.; Krestinina, O. Isoproterenol-Induced Permeability Transition Pore-Related Dysfunction of Heart Mitochondria Is Attenuated by Astaxanthin. Biomedicines 2020, 8, 437. [Google Scholar] [CrossRef]
- Stanely Mainzen Prince, P.; Dey, P.; Roy, S.J. Sinapic acid safeguards cardiac mitochondria from damage in isoproterenol-induced myocardial infarcted rats. J. Biochem. Mol. Toxicol. 2020, 34, e22556. [Google Scholar] [CrossRef]
- Sharmila Queenthy, S.; Stanely Mainzen Prince, P.; John, B. Diosmin Prevents Isoproterenol-Induced Heart Mitochondrial Oxidative Stress in Rats. Cardiovasc. Toxicol. 2018, 18, 120–130. [Google Scholar] [CrossRef]
- Kumaran, K.S.; Prince, P.S. Caffeic acid protects rat heart mitochondria against isoproterenol-induced oxidative damage. Cell Stress Chaperones 2010, 15, 791–806. [Google Scholar] [CrossRef] [Green Version]
- Thangaiyan, R.; Robert, B.M.; Arjunan, S.; Govindasamy, K.; Nagarajan, R.P. Preventive effect of apigenin against isoproterenol-induced apoptosis in cardiomyoblasts. J. Biochem. Mol. Toxicol. 2018, 32, e22213. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Morioka, S.; Kanazawa, H.; Yamawaki, H. Canstatin inhibits isoproterenol-induced apoptosis through preserving mitochondrial morphology in differentiated H9c2 cardiomyoblasts. Apoptosis Int. J. Program. Cell Death 2016, 21, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yang, J.; Lyu, Q.; Wu, G.; Lin, S.; Yang, Q.; Hu, J. Taurine attenuates isoproterenol-induced H9c2 cardiomyocytes hypertrophy by improving antioxidative ability and inhibiting calpain-1-mediated apoptosis. Mol. Cell. Biochem. 2020, 469, 119–132. [Google Scholar] [CrossRef]
- De Lacerda Alexandre, J.V.; Viana, Y.I.P.; David, C.E.B.; Cunha, P.L.O.; Albuquerque, A.C.; Varela, A.L.N.; Kowaltowski, A.J.; Facundo, H.T. Quercetin treatment increases H(2)O(2) removal by restoration of endogenous antioxidant activity and blocks isoproterenol-induced cardiac hypertrophy. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2021, 394, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Spasov, A.A.; Gurova, N.A.; Popova, T.A.; Perfilova, V.N.; Vishnevskaya, V.V.; Kustova, M.V.; Ovsyankina, N.V.; Ozerov, A.A. Effects of Zoniporide and BMA-1321 Compound on the Rate of Oxygen Absorption by Cardiomyocyte Mitochondria in Rats with Experimental Chronic Heart Failure. Bull. Exp. Biol. Med. 2021, 170, 316–320. [Google Scholar] [CrossRef]
- Malik, C.; Ghosh, S. Quinidine partially blocks mitochondrial voltage-dependent anion channel (VDAC). Eur. Biophys. J. EBJ 2020, 49, 193–205. [Google Scholar] [CrossRef]
- Finsterer, J.; Zarrouk-Mahjoub, S. Mitochondrial toxicity of cardiac drugs and its relevance to mitochondrial disorders. Expert Opin. Drug. Metab. Toxicol. 2015, 11, 15–24. [Google Scholar] [CrossRef]
- Zheng, W.B.; Li, Y.J.; Wang, Y.; Yang, J.; Zheng, C.C.; Huang, X.H.; Li, B.; He, Q.Y. Propafenone suppresses esophageal cancer proliferation through inducing mitochondrial dysfunction. Am. J. Cancer Res. 2017, 7, 2245–2256. [Google Scholar]
- Seydi, E.; Tabbati, Y.; Pourahmad, J. Toxicity of Atenolol and Propranolol on Rat Heart Mitochondria. Drug Res. 2020, 70, 151–157. [Google Scholar] [CrossRef]
- Brohée, L.; Peulen, O.; Nusgens, B.; Castronovo, V.; Thiry, M.; Colige, A.C.; Deroanne, C.F. Propranolol sensitizes prostate cancer cells to glucose metabolism inhibition and prevents cancer progression. Sci. Rep. 2018, 8, 7050. [Google Scholar] [CrossRef] [Green Version]
- Iwai, T.; Tanonaka, K.; Kasahara, S.; Inoue, R.; Takeo, S. Protective effect of propranolol on mitochondrial function in the ischaemic heart. Br. J. Pharmacol. 2002, 136, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Kaasik, A.; Kuum, M.; Joubert, F.; Wilding, J.; Ventura-Clapier, R.; Veksler, V. Mitochondria as a source of mechanical signals in cardiomyocytes. Cardiovasc. Res. 2010, 87, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Montoya, A.; Varela-Ramirez, A.; Dickerson, E.; Pasquier, E.; Torabi, A.; Aguilera, R.; Nahleh, Z.; Bryan, B. The beta adrenergic receptor antagonist propranolol alters mitogenic and apoptotic signaling in late stage breast cancer. Biomed. J. 2019, 42, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, H.; Wang, F.; Xu, R.; Wang, P.; Tang, F.; Zhang, X.; Zhu, Z.; Lv, H.; Han, T. Propranolol suppresses the proliferation and induces the apoptosis of liver cancer cells. Mol. Med. Rep. 2018, 17, 5213–5221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fromenty, B.; Fisch, C.; Berson, A.; Letteron, P.; Larrey, D.; Pessayre, D. Dual effect of amiodarone on mitochondrial respiration. Initial protonophoric uncoupling effect followed by inhibition of the respiratory chain at the levels of complex I and complex II. J. Pharmacol. Exp. Ther. 1990, 255, 1377–1384. [Google Scholar]
- Fromenty, B.; Fisch, C.; Labbe, G.; Degott, C.; Deschamps, D.; Berson, A.; Letteron, P.; Pessayre, D. Amiodarone inhibits the mitochondrial beta-oxidation of fatty acids and produces microvesicular steatosis of the liver in mice. J. Pharmacol. Exp. Ther. 1990, 255, 1371–1376. [Google Scholar] [PubMed]
- Kaufmann, P.; Török, M.; Hänni, A.; Roberts, P.; Gasser, R.; Krähenbühl, S. Mechanisms of benzarone and benzbromarone-induced hepatic toxicity. Hepatology 2005, 41, 925–935. [Google Scholar] [CrossRef]
- Spaniol, M.; Bracher, R.; Ha, H.R.; Follath, F.; Krähenbühl, S. Toxicity of amiodarone and amiodarone analogues on isolated rat liver mitochondria. J. Hepatol. 2001, 35, 628–636. [Google Scholar] [CrossRef]
- Serviddio, G.; Bellanti, F.; Giudetti, A.M.; Gnoni, G.V.; Capitanio, N.; Tamborra, R.; Romano, A.D.; Quinto, M.; Blonda, M.; Vendemiale, G.; et al. Mitochondrial oxidative stress and respiratory chain dysfunction account for liver toxicity during amiodarone but not dronedarone administration. Free. Radic. Biol. Med. 2011, 51, 2234–2242. [Google Scholar] [CrossRef]
- Felser, A.; Blum, K.; Lindinger, P.W.; Bouitbir, J.; Krähenbühl, S. Mechanisms of hepatocellular toxicity associated with dronedarone—A comparison to amiodarone. Toxicol. Sci. 2013, 131, 480–490. [Google Scholar] [CrossRef] [Green Version]
- Karkhanis, A.; Leow, J.W.H.; Hagen, T.; Chan, E.C.Y. Dronedarone-Induced Cardiac Mitochondrial Dysfunction and Its Mitigation by Epoxyeicosatrienoic Acids. Toxicol. Sci. 2018, 163, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felser, A.; Stoller, A.; Morand, R.; Schnell, D.; Donzelli, M.; Terracciano, L.; Bouitbir, J.; Krähenbühl, S. Hepatic toxicity of dronedarone in mice: Role of mitochondrial β-oxidation. Toxicology 2014, 323, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Nulton-Persson, A.C.; Szweda, L.I.; Sadek, H.A. Inhibition of cardiac mitochondrial respiration by salicylic acid and acetylsalicylate. J. Cardiovasc. Pharmacol. 2004, 44, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.S.; Stefanovich, V. Influence of anti-inflammatory agents on rat liver mitochondrial ATPase. Arzneim. Forsch. 1976, 26, 499–502. [Google Scholar]
- Al-Nasser, I.A. Salicylate-induced kidney mitochondrial permeability transition is prevented by cyclosporin A. Toxicol. Lett. 1999, 105, 1–8. [Google Scholar] [CrossRef]
- Tai, Y.K.; Cheong, Y.M.; Almsherqi, Z.A.; Chia, S.H.; Deng, Y.; McLachlan, C.S. High dose clopidogrel decreases mice liver mitochondrial respiration function in vitro. Int. J. Cardiol. 2009, 133, 250–252. [Google Scholar] [CrossRef]
- Zahno, A.; Bouitbir, J.; Maseneni, S.; Lindinger, P.W.; Brecht, K.; Krähenbühl, S. Hepatocellular toxicity of clopidogrel: Mechanisms and risk factors. Free Radic. Biol. Med. 2013, 65, 208–216. [Google Scholar] [CrossRef]
- Maseneni, S.; Donzelli, M.; Brecht, K.; Krähenbühl, S. Toxicity of thienopyridines on human neutrophil granulocytes and lymphocytes. Toxicology 2013, 308, 11–19. [Google Scholar] [CrossRef]
- Gjerde, H.; Helgeland, L. Effect of warfarin on ATP content, viability, glycosylation and protein synthesis in isolated rat hepatocytes. Acta Pharmacol. Toxicol. 1984, 54, 385–388. [Google Scholar] [CrossRef]
- Kurokawa, H.; Taninaka, A.; Shigekawa, H.; Matsui, H. Dabigatran Etexilate Induces Cytotoxicity in Rat Gastric Epithelial Cell Line via Mitochondrial Reactive Oxygen Species Production. Cells 2021, 10, 2508. [Google Scholar] [CrossRef]
- Manuel, M.A.; Weiner, M.W. Effects of ethacrynic acid and furosemide on isolated rat kidney mitochondria: Inhibition of electron transport in the region of phosphorylation site II. J. Pharmacol. Exp. Ther. 1976, 198, 209–221. [Google Scholar] [PubMed]
- Orita, Y.; Fukuhara, Y.; Yanase, M.; Ando, A.; Okada, N.; Abe, H. Effect of furosemide on mitochondrial electron transport system and oxidative phosphorylation. Arzneim. Forsch. 1983, 33, 1446–1450. [Google Scholar]
- Daiber, A.; Münzel, T. Organic Nitrate Therapy, Nitrate Tolerance, and Nitrate-Induced Endothelial Dysfunction: Emphasis on Redox Biology and Oxidative Stress. Antioxid Redox Signal 2015, 23, 899–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daiber, A.; Wenzel, P.; Oelze, M.; Münzel, T. New insights into bioactivation of organic nitrates, nitrate tolerance and cross-tolerance. Clin. Res. Cardiol. Off. J. Ger. Card. Soc. 2008, 97, 12–20. [Google Scholar] [CrossRef]
- Albuck, A.L.; Sakamuri, S.; Sperling, J.A.; Evans, W.R.; Kolli, L.; Sure, V.N.; Mostany, R.; Katakam, P.V.G. Peroxynitrite decomposition catalyst enhances respiratory function in isolated brain mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H630–H641. [Google Scholar] [CrossRef]
- Singh, I.N.; Sullivan, P.G.; Hall, E.D. Peroxynitrite-mediated oxidative damage to brain mitochondria: Protective effects of peroxynitrite scavengers. J. Neurosci. Res. 2007, 85, 2216–2223. [Google Scholar] [CrossRef]
- Xiong, Y.; Singh, I.N.; Hall, E.D. Tempol protection of spinal cord mitochondria from peroxynitrite-induced oxidative damage. Free. Radic. Res. 2009, 43, 604–612. [Google Scholar] [CrossRef]
- Uribe, P.; Treulen, F.; Boguen, R.; Sánchez, R.; Villegas, J.V. Nitrosative stress by peroxynitrite impairs ATP production in human spermatozoa. Andrologia 2017, 49, e12615. [Google Scholar] [CrossRef]
- Esselun, C.; Bruns, B.; Hagl, S.; Grewal, R.; Eckert, G.P. Differential Effects of Silibinin A on Mitochondrial Function in Neuronal PC12 and HepG2 Liver Cells. Oxidative Med. Cell. Longev. 2019, 2019, 1652609. [Google Scholar] [CrossRef]
- Lin, J.; Wu, G.; Chen, J.; Fu, C.; Hong, X.; Li, L.; Liu, X.; Wu, M. Electroacupuncture inhibits sodium nitroprusside-mediated chondrocyte apoptosis through the mitochondrial pathway. Mol. Med. Rep. 2018, 18, 4922–4930. [Google Scholar] [CrossRef]
- Fukushiro-Lopes, D.; Hegel, A.D.; Russo, A.; Senyuk, V.; Liotta, M.; Beeson, G.C.; Beeson, C.C.; Burdette, J.; Potkul, R.K.; Gentile, S. Repurposing Kir6/SUR2 Channel Activator Minoxidil to Arrests Growth of Gynecologic Cancers. Front. Pharmacol. 2020, 11, 577. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, A.; Tanaka, M.; Sumi, C.; Oku, K.; Kusunoki, M.; Nishi, K.; Matsuo, Y.; Takenaga, K.; Shingu, K.; Hirota, K. The antioxidant N-acetyl cysteine suppresses lidocaine-induced intracellular reactive oxygen species production and cell death in neuronal SH-SY5Y cells. BMC Anesthesiol. 2016, 16, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, C.; Kawasaki, T.; Ogata, M.; Sata, T.; Chaudry, I.H. Lidocaine enhances apoptosis and suppresses mitochondrial functions of human neutrophil in vitro. J. Trauma 2010, 68, 401–408. [Google Scholar] [CrossRef]
- Li, J.; Zhu, X.; Yang, S.; Xu, H.; Guo, M.; Yao, Y.; Huang, Z.; Lin, D. Lidocaine Attenuates Cognitive Impairment After Isoflurane Anesthesia by Reducing Mitochondrial Damage. Neurochem. Res. 2019, 44, 1703–1714. [Google Scholar] [CrossRef] [PubMed]
- Eghbal, M.A.; Taziki, S.; Sattari, M.R. Mechanisms of phenytoin-induced toxicity in freshly isolated rat hepatocytes and the protective effects of taurine and/or melatonin. J. Biochem. Mol. Toxicol. 2014, 28, 111–118. [Google Scholar] [CrossRef]
- Finsterer, J.; Scorza, F.A. Effects of antiepileptic drugs on mitochondrial functions, morphology, kinetics, biogenesis, and survival. Epilepsy Res. 2017, 136, 5–11. [Google Scholar] [CrossRef]
- Finsterer, J. Toxicity of Antiepileptic Drugs to Mitochondria. Handb. Exp. Pharmacol. 2017, 240, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Carreira, R.S.; Monteiro, P.; Gon Alves, L.M.; Providência, L.A. Carvedilol: Just another Beta-blocker or a powerful cardioprotector? Cardiovasc. Hematol. Disord. Drug Targets 2006, 6, 257–266. [Google Scholar] [CrossRef]
- Oliveira, P.J.; Marques, M.P.; Batista de Carvalho, L.A.; Moreno, A.J. Effects of carvedilol on isolated heart mitochondria: Evidence for a protonophoretic mechanism. Biochem. Biophys. Res. Commun. 2000, 276, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, P.J.; Gonçalves, L.; Monteiro, P.; Providencia, L.A.; Moreno, A.J. Are the antioxidant properties of carvedilol important for the protection of cardiac mitochondria? Curr. Vasc. Pharmacol. 2005, 3, 147–158. [Google Scholar] [CrossRef]
- Erguven, M.; Yazihan, N.; Aktas, E.; Sabanci, A.; Li, C.J.; Oktem, G.; Bilir, A. Carvedilol in glioma treatment alone and with imatinib in vitro. Int. J. Oncol. 2010, 36, 857–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhadri, N.; Razdan, R.; Goswami, S.K. Nebivolol, a β-blocker abrogates streptozotocin-induced behavioral, biochemical, and neurophysiological deficit by attenuating oxidative-nitrosative stress: A possible target for the prevention of diabetic neuropathy. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2018, 391, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Nuevo-Tapioles, C.; Santacatterina, F.; Stamatakis, K.; Núñez de Arenas, C.; Gómez de Cedrón, M.; Formentini, L.; Cuezva, J.M. Coordinate β-adrenergic inhibition of mitochondrial activity and angiogenesis arrest tumor growth. Nat. Commun. 2020, 11, 3606. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Jiang, H.; Wang, Z.; Cai, L.Y.; Jiang, Y.C.; Xie, L.; Zhou, Y.; Zeng, X.; Ji, N.; Shen, Y.Q.; et al. Adrenergic Blockade by Nebivolol to Suppress Oral Squamous Cell Carcinoma Growth via Endoplasmic Reticulum Stress and Mitochondria Dysfunction. Front. Pharmacol. 2021, 12, 691998. [Google Scholar] [CrossRef] [PubMed]
- Kotsinas, A.; Gorgoulis, V.; Zacharatos, P.; Zioris, H.; Triposkiadis, F.; Donta, I.; Kyriakidis, M.; Karayannacos, P.; Kittas, C. Antioxidant agent nimesulid and beta-blocker metoprolol do not exert protective effects against rat mitochondrial DNA alterations in adriamycin-induced cardiotoxicity. Biochem. Biophys. Res. Commun. 1999, 254, 651–656. [Google Scholar] [CrossRef]
- Lysko, P.G.; Webb, C.L.; Gu, J.L.; Ohlstein, E.H.; Ruffolo, R.R., Jr.; Yue, T.L. A comparison of carvedilol and metoprolol antioxidant activities in vitro. J. Cardiovasc. Pharmacol. 2000, 36, 277–281. [Google Scholar] [CrossRef]
- Zhu, B.Q.; Simonis, U.; Cecchini, G.; Zhou, H.Z.; Li, L.; Teerlink, J.R.; Karliner, J.S. Comparison of pyrroloquinoline quinone and/or metoprolol on myocardial infarct size and mitochondrial damage in a rat model of ischemia/reperfusion injury. J. Cardiovasc. Pharmacol. Ther. 2006, 11, 119–128. [Google Scholar] [CrossRef]
- Wang, P.; Zaragoza, C.; Holman, W. Sodium-hydrogen exchange inhibition and beta-blockade additively decrease infarct size. Ann. Thorac. Surg. 2007, 83, 1121–1127. [Google Scholar] [CrossRef]
- Sanchez-Roman, I.; Gomez, A.; Naudí, A.; Jove, M.; Gómez, J.; Lopez-Torres, M.; Pamplona, R.; Barja, G. Independent and additive effects of atenolol and methionine restriction on lowering rat heart mitochondria oxidative stress. J. Bioenergy Biomembr. 2014, 46, 159–172. [Google Scholar] [CrossRef]
- Gómez, A.; Sánchez-Roman, I.; Gomez, J.; Cruces, J.; Mate, I.; Lopez-Torres, M.; Naudi, A.; Portero-Otin, M.; Pamplona, R.; De la Fuente, M.; et al. Lifelong treatment with atenolol decreases membrane fatty acid unsaturation and oxidative stress in heart and skeletal muscle mitochondria and improves immunity and behavior, without changing mice longevity. Aging Cell 2014, 13, 551–560. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Roman, I.; Gomez, J.; Naudi, A.; Ayala, V.; Portero-Otín, M.; Lopez-Torres, M.; Pamplona, R.; Barja, G. The β-blocker atenolol lowers the longevity-related degree of fatty acid unsaturation, decreases protein oxidative damage, and increases extracellular signal-regulated kinase signaling in the heart of C57BL/6 mice. Rejuvenation Res. 2010, 13, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Quintana-Villamandos, B.; Delgado-Martos, M.J.; Delgado-Baeza, E. Early reversal cardiac with esmolol in hypertensive rats: The role of subcellular organelle phenotype. Pharmacol. Rep. PR 2019, 71, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Kavakcıoğlu Yardımcı, B.; Geyikoglu, F.; Aysin, F.; Koc, K.; Simsek Ozek, N.; Küçükatay, V. The cytotoxic and apoptotic effects of beta-blockers with different selectivity on cancerous and healthy lung cell lines. Mol. Biol. Rep. 2021, 48, 4009–4019. [Google Scholar] [CrossRef]
- Lu, Y.; Yang, Y.; He, X.; Dong, S.; Wang, W.; Wang, D.; Zhang, P. Esmolol reduces apoptosis and inflammation in early sepsis rats with abdominal infection. Am. J. Emerg. Med. 2017, 35, 1480–1484. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, H.; Zhou, G.; Wang, Y. [Changes of endoplasmic reticulum stress- and apoptosis-related factors in rat cerebral cortex following controlled hypotension]. Nan Fang Yi Ke Da Xue Xue Bao J. South. Med. Univ. 2014, 34, 1804–1808. [Google Scholar]
- Valdez, L.B.; Zaobornyj, T.; Bombicino, S.; Iglesias, D.E.; Boveris, A.; Donato, M.; D’Annunzio, V.; Buchholz, B.; Gelpi, R.J. Complex I syndrome in myocardial stunning and the effect of adenosine. Free. Radic. Biol. Med. 2011, 51, 1203–1212. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, J.; Zhang, Q.; Chen, P.; Song, J.; Yu, S.; Liu, H.; Liu, F.; Song, C.; Yang, D.; et al. Adenosine induces apoptosis in human liver cancer cells through ROS production and mitochondrial dysfunction. Biochem. Biophys. Res. Commun. 2014, 448, 8–14. [Google Scholar] [CrossRef]
- Tamura, K.; Kanno, T.; Fujita, Y.; Gotoh, A.; Nakano, T.; Nishizaki, T. A(2a) adenosine receptor mediates HepG2 cell apoptosis by downregulating Bcl-X(L) expression and upregulating Bid expression. J. Cell. Biochem. 2012, 113, 1766–1775. [Google Scholar] [CrossRef]
- Lu, Q.; Sakhatskyy, P.; Newton, J.; Shamirian, P.; Hsiao, V.; Curren, S.; Gabino Miranda, G.A.; Pedroza, M.; Blackburn, M.R.; Rounds, S. Sustained adenosine exposure causes lung endothelial apoptosis: A possible contributor to cigarette smoke-induced endothelial apoptosis and lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L361–L370. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; O’Rourke, B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ. Res. 2008, 103, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Brown, D.A.; O’Rourke, B. Role of mitochondrial dysfunction in cardiac glycoside toxicity. J. Mol. Cell. Cardiol. 2010, 49, 728–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campia, I.; Sala, V.; Kopecka, J.; Leo, C.; Mitro, N.; Costamagna, C.; Caruso, D.; Pescarmona, G.; Crepaldi, T.; Ghigo, D.; et al. Digoxin and ouabain induce the efflux of cholesterol via liver X receptor signalling and the synthesis of ATP in cardiomyocytes. Biochem. J. 2012, 447, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Hou, Y.; Hou, L.; Wang, W.; Li, K.; Zhang, Z.; Du, B.; Kong, D. Digoxin exerts anticancer activity on human nonsmall cell lung cancer cells by blocking PI3K/Akt pathway. Biosci. Rep. 2021, 41, BSR20211056. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.T.; Yan, J.Y.; Han, X.C.; Niu, F.L.; Zhang, J.H.; Hu, W.N. Anti-proliferative effect of digoxin on breast cancer cells via inducing apoptosis. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 5837–5842. [Google Scholar] [CrossRef] [PubMed]
- Alonso, E.; Cano-Abad, M.F.; Moreno-Ortega, A.J.; Novalbos, J.; Milla, J.; García, A.G.; Ruiz-Nuño, A. Nanomolar ouabain elicits apoptosis through a direct action on HeLa cell mitochondria. Steroids 2013, 78, 1110–1118. [Google Scholar] [CrossRef]
- Safari, F.; Bayat, G.; Shekarforoush, S.; Hekmatimoghaddam, S.; Anvari, Z.; Moghadam, M.F.; Hajizadeh, S. Expressional profile of cardiac uncoupling protein-2 following myocardial ischemia reperfusion in losartan- and ramiprilat-treated rats. J. Renin-Angiotensin-Aldosterone Syst. JRAAS 2014, 15, 209–217. [Google Scholar] [CrossRef]
- Shi, Q.; Abusarah, J.; Baroudi, G.; Fernandes, J.C.; Fahmi, H.; Benderdour, M. Ramipril attenuates lipid peroxidation and cardiac fibrosis in an experimental model of rheumatoid arthritis. Arthritis Res. Ther. 2012, 14, R223. [Google Scholar] [CrossRef] [Green Version]
- Taskin, E.; Guven, C.; Sahin, L.; Dursun, N. The Cooperative Effect of Local Angiotensin-II in Liver with Adriamycin Hepatotoxicity on Mitochondria. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2016, 22, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Taskin, E.; Ozdogan, K.; Kunduz Kindap, E.; Dursun, N. The restoration of kidney mitochondria function by inhibition of angiotensin-II production in rats with acute adriamycin-induced nephrotoxicity. Ren. Fail. 2014, 36, 606–612. [Google Scholar] [CrossRef] [Green Version]
- Taskin, E.; Kindap, E.K.; Ozdogan, K.; Aycan, M.B.Y.; Dursun, N. Acute adriamycin-induced cardiotoxicity is exacerbated by angiotension II. Cytotechnology 2016, 68, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Kojic, Z.; Gopcevic, K.; Marinkovic, D.; Tasic, G. Effect of captopril on serum lipid levels and cardiac mitochondrial oxygen consumption in experimentally-induced hypercholesterolemia in rabbits. Physiol. Res. 2011, 60 (Suppl. 1), S177–S184. [Google Scholar] [CrossRef] [PubMed]
- Ghazi-Khansari, M.; Mohammadi-Bardbori, A. Captopril ameliorates toxicity induced by paraquat in mitochondria isolated from the rat liver. Toxicol. Vitr. 2007, 21, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Mehrvar, S.; la Cour, M.F.; Medhora, M.; Camara, A.K.S.; Ranji, M. Optical Metabolic Imaging for Assessment of Radiation-Induced Injury to Rat Kidney and Mitigation by Lisinopril. Ann. Biomed. Eng. 2019, 47, 1564–1574. [Google Scholar] [CrossRef]
- Ederer, K.A.; Jin, K.; Bouslog, S.; Wang, L.; Gorman, G.S.; Rowe, G.C.; Abadir, P.; Raftery, D.; Moellering, D.; Promislow, D.; et al. Age- and Genotype-Specific Effects of the Angiotensin-Converting Enzyme Inhibitor Lisinopril on Mitochondrial and Metabolic Parameters in Drosophila melanogaster. Int. J. Mol. Sci. 2018, 19, 3351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samiei, F.; Sajjadi, H.; Jamshidzadeh, A.; Seydi, E.; Pourahmad, J. Contrasting Role of Concentration in Rivaroxaban Induced Toxicity and Oxidative Stress in Isolated Kidney Mitochondria. Drug Res. 2019, 69, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, Y.; Matsui, T.; Fukami, K.; Ueda, S.; Okuda, S.; Yamagishi, S. Rivaroxaban inhibits oxidative and inflammatory reactions in advanced glycation end product-exposed tubular cells by blocking thrombin/protease-activated receptor-2 system. Thromb. Res. 2015, 135, 770–773. [Google Scholar] [CrossRef]
- Imano, H.; Kato, R.; Tanikawa, S.; Yoshimura, F.; Nomura, A.; Ijiri, Y.; Yamaguchi, T.; Izumi, Y.; Yoshiyama, M.; Hayashi, T. Factor Xa inhibition by rivaroxaban attenuates cardiac remodeling due to intermittent hypoxia. J. Pharmacol. Sci. 2018, 137, 274–282. [Google Scholar] [CrossRef]
- Nakamaru-Ogiso, E.; Seo, B.B.; Yagi, T.; Matsuno-Yagi, A. Amiloride inhibition of the proton-translocating NADH-quinone oxidoreductase of mammals and bacteria. FEBS Lett. 2003, 549, 43–46. [Google Scholar] [CrossRef] [Green Version]
- Manoli, S.S.; Kisor, K.; Webb, B.A.; Barber, D.L. Ethyl isopropyl amiloride decreases oxidative phosphorylation and increases mitochondrial fusion in clonal untransformed and cancer cells. Am. J. Physiol. Cell Physiol. 2021, 321, C147–C157. [Google Scholar] [CrossRef]
- Rong, C.; Chen, F.H.; Jiang, S.; Hu, W.; Wu, F.R.; Chen, T.Y.; Yuan, F.L. Inhibition of acid-sensing ion channels by amiloride protects rat articular chondrocytes from acid-induced apoptosis via a mitochondrial-mediated pathway. Cell Biol. Int. 2012, 36, 635–641. [Google Scholar] [CrossRef]
- Mollazadeh, H.; Tavana, E.; Fanni, G.; Bo, S.; Banach, M.; Pirro, M.; von Haehling, S.; Jamialahmadi, T.; Sahebkar, A. Effects of statins on mitochondrial pathways. J. Cachexia Sarcopenia Muscle 2021, 12, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Muntean, D.M.; Thompson, P.D.; Catapano, A.L.; Stasiolek, M.; Fabis, J.; Muntner, P.; Serban, M.C.; Banach, M. Statin-associated myopathy and the quest for biomarkers: Can we effectively predict statin-associated muscle symptoms? Drug Discov. Today 2017, 22, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin Toxicity. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulou, M.; Corsini, A.; Roden, M. The role of mitochondria in statin-induced myopathy. Eur. J. Clin. Investig. 2015, 45, 745–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufmann, P.; Török, M.; Zahno, A.; Waldhauser, K.M.; Brecht, K.; Krähenbühl, S. Toxicity of statins on rat skeletal muscle mitochondria. Cell. Mol. Life Sci. CMLS 2006, 63, 2415–2425. [Google Scholar] [CrossRef] [Green Version]
- Broniarek, I.; Jarmuszkiewicz, W. Atorvastatin affects negatively respiratory function of isolated endothelial mitochondria. Arch. Biochem. Biophys. 2018, 637, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Durhuus, J.A.; Hansson, S.; Morville, T.; Kuhlman, A.B.; Dohlmann, T.L.; Larsen, S.; Helge, J.W.; Angleys, M.; Muniesa-Vargas, A.; Bundgaard, J.R.; et al. Simvastatin improves mitochondrial respiration in peripheral blood cells. Sci. Rep. 2020, 10, 17012. [Google Scholar] [CrossRef]
- Li, C.; Su, Z.; Ge, L.; Chen, Y.; Chen, X.; Li, Y. Cardioprotection of hydralazine against myocardial ischemia/reperfusion injury in rats. Eur. J. Pharmacol. 2020, 869, 172850. [Google Scholar] [CrossRef]
- Daiber, A.; Mülsch, A.; Hink, U.; Mollnau, H.; Warnholtz, A.; Oelze, M.; Münzel, T. The oxidative stress concept of nitrate tolerance and the antioxidant properties of hydralazine. Am. J. Cardiol. 2005, 96, 25i–36i. [Google Scholar] [CrossRef]
- Kalkhoran, S.B.; Kriston-Vizi, J.; Hernandez-Resendiz, S.; Crespo-Avilan, G.E.; Rosdah, A.A.; Lees, J.G.; Costa, J.; Ling, N.X.Y.; Holien, J.K.; Samangouei, P.; et al. Hydralazine protects the heart against acute ischaemia/reperfusion injury by inhibiting Drp1-mediated mitochondrial fission. Cardiovasc. Res. 2022, 118, 282–294. [Google Scholar] [CrossRef]
- Dehghan, E.; Goodarzi, M.; Saremi, B.; Lin, R.; Mirzaei, H. Hydralazine targets cAMP-dependent protein kinase leading to sirtuin1/5 activation and lifespan extension in C. elegans. Nat. Commun. 2019, 10, 4905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Magaña, M.J.; Martínez-Aguilar, R.; Lucendo, E.; Campillo-Davo, D.; Schulze-Osthoff, K.; Ruiz-Ruiz, C. The antihypertensive drug hydralazine activates the intrinsic pathway of apoptosis and causes DNA damage in leukemic T cells. Oncotarget 2016, 7, 21875–21886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belosludtseva, N.V.; Starinets, V.S.; Belosludtsev, K.N. Effect of Dapagliflozin on the Functioning of Rat Liver Mitochondria In Vitro. Bull. Exp. Biol. Med. 2021, 171, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Zaibi, N.; Li, P.; Xu, S.Z. Protective effects of dapagliflozin against oxidative stress-induced cell injury in human proximal tubular cells. PLoS ONE 2021, 16, e0247234. [Google Scholar] [CrossRef] [PubMed]
- Lahnwong, S.; Palee, S.; Apaijai, N.; Sriwichaiin, S.; Kerdphoo, S.; Jaiwongkam, T.; Chattipakorn, S.C.; Chattipakorn, N. Acute dapagliflozin administration exerts cardioprotective effects in rats with cardiac ischemia/reperfusion injury. Cardiovasc. Diabetol. 2020, 19, 91. [Google Scholar] [CrossRef] [PubMed]
- Tanajak, P.; Sa-Nguanmoo, P.; Sivasinprasasn, S.; Thummasorn, S.; Siri-Angkul, N.; Chattipakorn, S.C.; Chattipakorn, N. Cardioprotection of dapagliflozin and vildagliptin in rats with cardiac ischemia-reperfusion injury. J. Endocrinol. 2018, 236, 69–84. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Starinets, V.S.; Belosludtsev, M.N.; Mikheeva, I.B.; Dubinin, M.V.; Belosludtseva, N.V. Chronic treatment with dapagliflozin protects against mitochondrial dysfunction in the liver of C57BL/6NCrl mice with high-fat diet/streptozotocin-induced diabetes mellitus. Mitochondrion 2021, 59, 246–254. [Google Scholar] [CrossRef]
- Secker, P.F.; Beneke, S.; Schlichenmaier, N.; Delp, J.; Gutbier, S.; Leist, M.; Dietrich, D.R. Canagliflozin mediated dual inhibition of mitochondrial glutamate dehydrogenase and complex I: An off-target adverse effect. Cell Death Dis. 2018, 9, 226. [Google Scholar] [CrossRef] [Green Version]
- Papadopoli, D.; Uchenunu, O.; Palia, R.; Chekkal, N.; Hulea, L.; Topisirovic, I.; Pollak, M.; St-Pierre, J. Perturbations of cancer cell metabolism by the antidiabetic drug canagliflozin. Neoplasia 2021, 23, 391–399. [Google Scholar] [CrossRef]
- Wei, D.; Liao, L.; Wang, H.; Zhang, W.; Wang, T.; Xu, Z. Canagliflozin ameliorates obesity by improving mitochondrial function and fatty acid oxidation via PPARα in vivo and in vitro. Life Sci. 2020, 247, 117414. [Google Scholar] [CrossRef]
- Feng, C.C.; Liao, P.H.; Tsai, H.I.; Cheng, S.M.; Yang, L.Y.; PadmaViswanadha, V.; Pan, L.F.; Chen, R.J.; Lo, J.F.; Huang, C.Y. Tumorous imaginal disc 1 (TID1) inhibits isoproterenol-induced cardiac hypertrophy and apoptosis by regulating c-terminus of hsc70-interacting protein (CHIP) mediated degradation of Gαs. Int. J. Med. Sci. 2018, 15, 1537–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitali Serdoz, L.; Rittger, H.; Furlanello, F.; Bastian, D. Quinidine-A legacy within the modern era of antiarrhythmic therapy. Pharmacol. Res. 2019, 144, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Sheets, M.F.; Fozzard, H.A.; Lipkind, G.M.; Hanck, D.A. Sodium channel molecular conformations and antiarrhythmic drug affinity. Trends Cardiovasc. Med. 2010, 20, 16–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beecham, G.B.; Bansal, P.; Nessel, T.A.; Goyal, A. Lidocaine. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Okamoto, A.; Sumi, C.; Tanaka, H.; Kusunoki, M.; Iwai, T.; Nishi, K.; Matsuo, Y.; Harada, H.; Takenaga, K.; Bono, H.; et al. HIF-1-mediated suppression of mitochondria electron transport chain function confers resistance to lidocaine-induced cell death. Sci. Rep. 2017, 7, 3816. [Google Scholar] [CrossRef] [Green Version]
- Patocka, J.; Wu, Q.; Nepovimova, E.; Kuca, K. Phenytoin—An anti-seizure drug: Overview of its chemistry, pharmacology and toxicology. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2020, 142, 111393. [Google Scholar] [CrossRef]
- Alsaad, A.A.; Ortiz Gonzalez, Y.; Austin, C.O.; Kusumoto, F. Revisiting propafenone toxicity. BMJ Case Rep. 2017, 2017, bcr2017219270. [Google Scholar] [CrossRef]
- Dézsi, C.A.; Szentes, V. The Real Role of β-Blockers in Daily Cardiovascular Therapy. Am. J. Cardiovasc. Drugs Drugs Devices Other Interv. 2017, 17, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Santos, D.J.; Moreno, A.J. Inhibition of heart mitochondrial lipid peroxidation by non-toxic concentrations of carvedilol and its analog BM-910228. Biochem. Pharmacol. 2001, 61, 155–164. [Google Scholar] [CrossRef]
- Diogo, C.V.; Deus, C.M.; Lebiedzinska-Arciszewska, M.; Wojtala, A.; Wieckowski, M.R.; Oliveira, P.J. Carvedilol and antioxidant proteins in a type I diabetes animal model. Eur. J. Clin. Investig. 2017, 47, 19–29. [Google Scholar] [CrossRef]
- Cheng, J.; Kamiya, K.; Kodama, I. Carvedilol: Molecular and cellular basis for its multifaceted therapeutic potential. Cardiovasc. Drug Rev. 2001, 19, 152–171. [Google Scholar] [CrossRef] [Green Version]
- Seleme, V.B.; Marques, G.L.; Mendes, A.E.M.; Rotta, I.; Pereira, M.; Júnior, E.L.; da Cunha, C.L.P. Nebivolol for the Treatment of Essential Systemic Arterial Hypertension: A Systematic Review and Meta-Analysis. Am. J. Cardiovasc. Drugs Drugs Devices Other Interv. 2021, 21, 165–180. [Google Scholar] [CrossRef]
- Power, A.S.; Norman, R.; Jones, T.L.M.; Hickey, A.J.; Ward, M.L. Mitochondrial function remains impaired in the hypertrophied right ventricle of pulmonary hypertensive rats following short duration metoprolol treatment. PLoS ONE 2019, 14, e0214740. [Google Scholar] [CrossRef]
- DeLima, L.G.; Kharasch, E.D.; Butler, S. Successful pharmacologic treatment of massive atenolol overdose: Sequential hemodynamics and plasma atenolol concentrations. Anesthesiology 1995, 83, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.; Nation, R.L.; Chiou, W.L.; Mehta, P.K. Plasma concentrations of propranolol and 4-hydroxypropranolol during chronic oral propranolol therapy. Br. J. Clin. Pharmacol. 1979, 8, 163–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazreg, S.; Merad, Z.; Nouri, M.T.; Garout, R.; Derdour, A.; Ghroud, N.; Kherroubi, R.; Meziane, M.; Belkacem, S.; Ouhadj, O.; et al. Efficacy and safety of preservative-free timolol 0.1% gel in open-angle glaucoma and ocular hypertension in treatment-naïve patients and patients intolerant to other hypotensive medications. J. Fr. D’ophtalmologie 2018, 41, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Cruickshank, J.M. The Role of Beta-Blockers in the Treatment of Hypertension. Adv. Exp. Med. Biol. 2017, 956, 149–166. [Google Scholar] [CrossRef]
- Cicek, F.A.; Toy, A.; Tuncay, E.; Can, B.; Turan, B. Beta-blocker timolol alleviates hyperglycemia-induced cardiac damage via inhibition of endoplasmic reticulum stress. J. Bioenerg. Biomembr. 2014, 46, 377–387. [Google Scholar] [CrossRef]
- Garnock-Jones, K.P. Esmolol: A review of its use in the short-term treatment of tachyarrhythmias and the short-term control of tachycardia and hypertension. Drugs 2012, 72, 109–132. [Google Scholar] [CrossRef]
- Soar, J.; Perkins, G.D.; Maconochie, I.; Böttiger, B.W.; Deakin, C.D.; Sandroni, C.; Olasveengen, T.M.; Wyllie, J.; Greif, R.; Lockey, A.; et al. European Resuscitation Council Guidelines for Resuscitation: 2018 Update—Antiarrhythmic drugs for cardiac arrest. Resuscitation 2019, 134, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Ren, Z.; Yu, D.; Ning, B.; Guo, L. DNA damage-induced apoptosis and mitogen-activated protein kinase pathway contribute to the toxicity of dronedarone in hepatic cells. Environ. Mol. Mutagen. 2018, 59, 278–289. [Google Scholar] [CrossRef]
- Lüker, J.; Sultan, A.; Sehner, S.; Hoffmann, B.; Servatius, H.; Willems, S.; Steven, D. Use of antiarrhythmic drugs during ablation of persistent atrial fibrillation: Observations from a large single-centre cohort. Heart Vessel. 2016, 31, 1669–1675. [Google Scholar] [CrossRef] [PubMed]
- Samanta, R.; Thiagalingam, A.; Turner, C.; Lakkireddy, D.J.; Kovoor, P. The Use of Intravenous Sotalol in Cardiac Arrhythmias. Heart Lung Circ. 2018, 27, 1318–1326. [Google Scholar] [CrossRef]
- Shenasa, F.; Shenasa, M. Dofetilide: Electrophysiologic Effect, Efficacy, and Safety in Patients with Cardiac Arrhythmias. Card. Electrophysiol. Clin. 2016, 8, 423–436. [Google Scholar] [CrossRef]
- Ponne, S.; Kumar, C.R.; Boopathy, R. Verapamil attenuates scopolamine induced cognitive deficits by averting oxidative stress and mitochondrial injury—A potential therapeutic agent for Alzheimer’s Disease. Metab. Brain Dis. 2020, 35, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Xiao, C.; Zhang, K.; Jia, C.; Ding, X.; Zhang, B.; Wang, Y.; Li, M. The calcium channel blocker verapamil inhibits oxidative stress response in Candida albicans. Mycopathologia 2014, 177, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez Padial, L.; Barón-Esquivias, G.; Hernández Madrid, A.; Marzal Martín, D.; Pallarés-Carratalá, V.; de la Sierra, A. Clinical Experience with Diltiazem in the Treatment of Cardiovascular Diseases. Cardiol. Ther. 2016, 5, 75–82. [Google Scholar] [CrossRef]
- Faulds, D.; Chrisp, P.; Buckley, M.M. Adenosine. An evaluation of its use in cardiac diagnostic procedures, and in the treatment of paroxysmal supraventricular tachycardia. Drugs 1991, 41, 596–624. [Google Scholar] [CrossRef]
- Xu, Z.; Park, S.S.; Mueller, R.A.; Bagnell, R.C.; Patterson, C.; Boysen, P.G. Adenosine produces nitric oxide and prevents mitochondrial oxidant damage in rat cardiomyocytes. Cardiovasc. Res. 2005, 65, 803–812. [Google Scholar] [CrossRef] [Green Version]
- Kalogeris, T.J.; Baines, C.; Korthuis, R.J. Adenosine prevents TNFα-induced decrease in endothelial mitochondrial mass via activation of eNOS-PGC-1α regulatory axis. PLoS ONE 2014, 9, e98459. [Google Scholar] [CrossRef] [Green Version]
- Malik, A.; Masson, R.; Singh, S.; Wu, W.C.; Packer, M.; Pitt, B.; Waagstein, F.; Morgan, C.J.; Allman, R.M.; Fonarow, G.C.; et al. Digoxin Discontinuation and Outcomes in Patients With Heart Failure With Reduced Ejection Fraction. J. Am. Coll. Cardiol. 2019, 74, 617–627. [Google Scholar] [CrossRef]
- Zeng, W.T.; Liu, Z.H.; Li, Z.Y.; Zhang, M.; Cheng, Y.J. Digoxin Use and Adverse Outcomes in Patients With Atrial Fibrillation. Medicine 2016, 95, e2949. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, N.R.; Masi, S.; Taddei, S. The renin-angiotensin-aldosterone system: A crossroad from arterial hypertension to heart failure. Heart Fail. Rev. 2020, 25, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Mancia, G.; Rea, F.; Ludergnani, M.; Apolone, G.; Corrao, G. Renin-Angiotensin-Aldosterone System Blockers and the Risk of COVID-19. N. Engl. J. Med. 2020, 382, 2431–2440. [Google Scholar] [CrossRef] [PubMed]
- Marampon, F.; Gravina, G.L.; Scarsella, L.; Festuccia, C.; Lovat, F.; Ciccarelli, C.; Zani, B.M.; Polidoro, L.; Grassi, D.; Desideri, G.; et al. Angiotensin-converting-enzyme inhibition counteracts angiotensin II-mediated endothelial cell dysfunction by modulating the p38/SirT1 axis. J. Hypertens. 2013, 31, 1972–1983. [Google Scholar] [CrossRef] [PubMed]
- Zoll, J.; Monassier, L.; Garnier, A.; N’Guessan, B.; Mettauer, B.; Veksler, V.; Piquard, F.; Ventura-Clapier, R.; Geny, B. ACE inhibition prevents myocardial infarction-induced skeletal muscle mitochondrial dysfunction. J. Appl. Physiol. 2006, 101, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Mujkosová, J.; Ulicná, O.; Waczulíková, I.; Vlkovicová, J.; Vancová, O.; Ferko, M.; Polák, S.; Ziegelhöffer, A. Mitochondrial function in heart and kidney of spontaneously hypertensive rats: Influence of captopril treatment. Gen. Physiol. Biophys. 2010, 29, 203–207. [Google Scholar] [CrossRef]
- Ziegelhöffer, A.; Mujkošová, J.; Ferko, M.; Vrbjar, N.; Ravingerová, T.; Uličná, O.; Waczulíková, I.; Ziegelhöffer, B. Dual influence of spontaneous hypertension on membrane properties and ATP production in heart and kidney mitochondria in rat: Effect of captopril and nifedipine, adaptation and dysadaptation. Can. J. Physiol. Pharmacol. 2012, 90, 1311–1323. [Google Scholar] [CrossRef]
- Kancirová, I.; Jašová, M.; Waczulíková, I.; Ravingerová, T.; Ziegelhöffer, A.; Ferko, M. Effect of antihypertensive agents—Captopril and nifedipine—On the functional properties of rat heart mitochondria. Iran. J. Basic Med. Sci. 2016, 19, 615–623. [Google Scholar]
- Van Ginkel, S.; Ruoss, S.; Valdivieso, P.; Degens, H.; Waldron, S.; de Haan, A.; Flück, M. ACE inhibition modifies exercise-induced pro-angiogenic and mitochondrial gene transcript expression. Scand. J. Med. Sci. Sport. 2016, 26, 1180–1187. [Google Scholar] [CrossRef] [Green Version]
- Costa, L.E.; La-Padula, P.; Lores-Arnaiz, S.; D’Amico, G.; Boveris, A.; Kurnjek, M.L.; Basso, N. Long-term angiotensin II inhibition increases mitochondrial nitric oxide synthase and not antioxidant enzyme activities in rat heart. J. Hypertens. 2002, 20, 2487–2494. [Google Scholar] [CrossRef]
- Cole, B.K.; Keller, S.R.; Wu, R.; Carter, J.D.; Nadler, J.L.; Nunemaker, C.S. Valsartan protects pancreatic islets and adipose tissue from the inflammatory and metabolic consequences of a high-fat diet in mice. Hypertension 2010, 55, 715–721. [Google Scholar] [CrossRef] [PubMed]
- De Cavanagh, E.M.; Ferder, L.; Toblli, J.E.; Piotrkowski, B.; Stella, I.; Fraga, C.G.; Inserra, F. Renal mitochondrial impairment is attenuated by AT1 blockade in experimental Type I diabetes. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H456–H465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cavanagh, E.M.; Piotrkowski, B.; Basso, N.; Stella, I.; Inserra, F.; Ferder, L.; Fraga, C.G. Enalapril and losartan attenuate mitochondrial dysfunction in aged rats. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 1096–1098. [Google Scholar] [CrossRef]
- De Cavanagh, E.M.; Toblli, J.E.; Ferder, L.; Piotrkowski, B.; Stella, I.; Fraga, C.G.; Inserra, F. Angiotensin II blockade improves mitochondrial function in spontaneously hypertensive rats. Cell. Mol. Biol. 2005, 51, 573–578. [Google Scholar] [PubMed]
- Kurokawa, H.; Sugiyama, S.; Nozaki, T.; Sugamura, K.; Toyama, K.; Matsubara, J.; Fujisue, K.; Ohba, K.; Maeda, H.; Konishi, M.; et al. Telmisartan enhances mitochondrial activity and alters cellular functions in human coronary artery endothelial cells via AMP-activated protein kinase pathway. Atherosclerosis 2015, 239, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Al-Harbi, N.O.; Imam, F.; Al-Harbi, M.M.; Iqbal, M.; Nadeem, A.; Sayed-Ahmed, M.M.; Alabidy, A.D.; Almukhallafi, A.F. Olmesartan attenuates tacrolimus-induced biochemical and ultrastructural changes in rat kidney tissue. BioMed Res. Int. 2014, 2014, 607246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Yeh, J.N.; Yue, Y.; Chu, Y.C.; Huang, C.R.; Yang, C.C.; Chiang, J.Y.; Yip, H.K.; Guo, J. Entresto protected the cardiomyocytes and preserved heart function in cardiorenal syndrome rat fed with high-protein diet through regulating the oxidative stress and Mfn2-mediated mitochondrial functional integrity. Biomed. Pharmacother. Biomed. Pharmacother. 2021, 144, 112244. [Google Scholar] [CrossRef]
- Fares, H.; DiNicolantonio, J.J.; O’Keefe, J.H.; Lavie, C.J. Amlodipine in hypertension: A first-line agent with efficacy for improving blood pressure and patient outcomes. Open Heart 2016, 3, e000473. [Google Scholar] [CrossRef] [Green Version]
- Patti, G.; Micieli, G.; Cimminiello, C.; Bolognese, L. The Role of Clopidogrel in 2020: A Reappraisal. Cardiovasc. Ther. 2020, 2020, 8703627. [Google Scholar] [CrossRef] [Green Version]
- Kubisa, M.J.; Jezewski, M.P.; Gasecka, A.; Siller-Matula, J.M.; Postuła, M. Ticagrelor—Toward more efficient platelet inhibition and beyond. Ther. Clin. Risk Manag. 2018, 14, 129–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawarskas, J.J.; Montoya, T.N. Switching From Ticagrelor or Prasugrel to Clopidogrel. Cardiol. Rev. 2018, 26, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W. Dual antiplatelet therapy for coronary artery disease. Circ. J. Off. J. Jpn. Circ. Soc. 2015, 79, 255–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witt, D.M.; Clark, N.P.; Kaatz, S.; Schnurr, T.; Ansell, J.E. Guidance for the practical management of warfarin therapy in the treatment of venous thromboembolism. J. Thromb. Thrombolysis 2016, 41, 187–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkarda, B.; Arda, O.; Tasyurekli, M.; Derman, U. Mitochondria-lytic action of warfarin in lymphocytes. Int. J. Clin. Pharmacol. Ther. Toxicol. 1992, 30, 277–279. [Google Scholar] [PubMed]
- Chen, A.; Stecker, E.; Warden, B.A. Direct Oral Anticoagulant Use: A Practical Guide to Common Clinical Challenges. J. Am. Heart Assoc. 2020, 9, e017559. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.; Chen, J.; Guo, Y.; Ji, T.; Xie, P. Rivaroxaban ameliorates angiotensin II-induced cardiac remodeling by attenuating TXNIP/Trx2 interaction in KKAy mice. Thromb. Res. 2020, 193, 45–52. [Google Scholar] [CrossRef]
- Zamorano-Leon, J.J.; Serna-Soto, M.; Moñux, G.; Freixer, G.; Zekri-Nechar, K.; Cabrero-Fernandez, M.; Segura, A.; Gonzalez-Cantalapiedra, A.; Serrano, J.; Farré, A.L. Factor Xa Inhibition by Rivaroxaban Modified Mitochondrial-Associated Proteins in Human Abdominal Aortic Aneurysms. Ann. Vasc. Surg. 2020, 67, 482–489. [Google Scholar] [CrossRef]
- Ellison, D.H. Clinical Pharmacology in Diuretic Use. Clin. J. Am. Soc. Nephrol. CJASN 2019, 14, 1248–1257. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.G.; Card, J.W.; Racz, W.J. The role of mitochondrial injury in bromobenzene and furosemide induced hepatotoxicity. Toxicol. Lett. 2000, 116, 171–181. [Google Scholar] [CrossRef]
- Church, R.J.; Schomaker, S.J.; Eaddy, J.S.; Boucher, G.G.; Kreeger, J.M.; Aubrecht, J.; Watkins, P.B. Glutamate dehydrogenase as a biomarker for mitotoxicity; insights from furosemide hepatotoxicity in the mouse. PLoS ONE 2020, 15, e0240562. [Google Scholar] [CrossRef] [PubMed]
- Roush, G.C.; Kaur, R.; Ernst, M.E. Diuretics: A review and update. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Gong, W.; He, M.; Liu, Y.; Yang, Y.; Wang, M.; Wu, M.; Guo, S.; Yu, Y.; Wang, X.; et al. Spironolactone Protects against Diabetic Cardiomyopathy in Streptozotocin-Induced Diabetic Rats. J. Diabetes Res. 2018, 2018, 9232065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Sever, P. Amiloride: A review. J. Renin-Angiotensin-Aldosterone Syst. JRAAS 2020, 21, 1470320320975893. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [Green Version]
- Broniarek, I.; Dominiak, K.; Galganski, L.; Jarmuszkiewicz, W. The Influence of Statins on the Aerobic Metabolism of Endothelial Cells. Int. J. Mol. Sci. 2020, 21, 1485. [Google Scholar] [CrossRef] [Green Version]
- Abdoli, N.; Heidari, R.; Azarmi, Y.; Eghbal, M.A. Mechanisms of the statins cytotoxicity in freshly isolated rat hepatocytes. J. Biochem. Mol. Toxicol. 2013, 27, 287–294. [Google Scholar] [CrossRef]
- Sadighara, M.; Amirsheardost, Z.; Minaiyan, M.; Hajhashemi, V.; Naserzadeh, P.; Salimi, A.; Seydi, E.; Pourahmad, J. Toxicity of Atorvastatin on Pancreas Mitochondria: A Justification for Increased Risk of Diabetes Mellitus. Basic Clin. Pharmacol. Toxicol. 2017, 120, 131–137. [Google Scholar] [CrossRef]
- Avram, V.F.; Chamkha, I.; Åsander-Frostner, E.; Ehinger, J.K.; Timar, R.Z.; Hansson, M.J.; Muntean, D.M.; Elmér, E. Cell-Permeable Succinate Rescues Mitochondrial Respiration in Cellular Models of Statin Toxicity. Int. J. Mol. Sci. 2021, 22, 424. [Google Scholar] [CrossRef]
- Björkhem-Bergman, L.; Lindh, J.D.; Bergman, P. What is a relevant statin concentration in cell experiments claiming pleiotropic effects? Br. J. Clin. Pharmacol. 2011, 72, 164–165. [Google Scholar] [CrossRef] [Green Version]
- Vevera, J.; Fišar, Z.; Nekovářová, T.; Vrablík, M.; Zlatohlávek, L.; Hroudová, J.; Singh, N.; Raboch, J.; Valeš, K. Statin-induced changes in mitochondrial respiration in blood platelets in rats and human with dyslipidemia. Physiol. Res. 2016, 65, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Avram, V.F.; Bîna, A.M.; Sima, A.; Aburel, O.M.; Sturza, A.; Burlacu, O.; Timar, R.Z.; Muntean, D.M.; Elmér, E.; Crețu, O.M. Improvement of Platelet Respiration by Cell-Permeable Succinate in Diabetic Patients Treated with Statins. Life 2021, 11, 288. [Google Scholar] [CrossRef] [PubMed]
- Gvozdjakova, A.; Sumbalova, Z.; Kucharska, J.; Chladekova, A.; Rausova, Z.; Vancova, O.; Komlosi, M.; Ulicna, O.; Mojto, V. Platelet mitochondrial bioenergetic analysis in patients with nephropathies and non-communicable diseases: A new method. Bratisl. Lek. Listy 2019, 120, 630–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohlmann, T.L.; Morville, T.; Kuhlman, A.B.; Chrøis, K.M.; Helge, J.W.; Dela, F.; Larsen, S. Statin Treatment Decreases Mitochondrial Respiration But Muscle Coenzyme Q10 Levels Are Unaltered: The LIFESTAT Study. J. Clin. Endocrinol. Metab. 2019, 104, 2501–2508. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, P.; Hink, U.; Oelze, M.; Schuppan, S.; Schaeuble, K.; Schildknecht, S.; Ho, K.K.; Weiner, H.; Bachschmid, M.; Münzel, T.; et al. Role of reduced lipoic acid in the redox regulation of mitochondrial aldehyde dehydrogenase (ALDH-2) activity. Implications for mitochondrial oxidative stress and nitrate tolerance. J. Biol. Chem. 2007, 282, 792–799. [Google Scholar] [CrossRef] [Green Version]
- Daiber, A.; Oelze, M.; Coldewey, M.; Bachschmid, M.; Wenzel, P.; Sydow, K.; Wendt, M.; Kleschyov, A.L.; Stalleicken, D.; Ullrich, V.; et al. Oxidative stress and mitochondrial aldehyde dehydrogenase activity: A comparison of pentaerythritol tetranitrate with other organic nitrates. Mol. Pharmacol. 2004, 66, 1372–1382. [Google Scholar] [CrossRef]
- Bogaert, M.G. Pharmacokinetics of organic nitrates in man: An overview. Eur. Heart J. 1988, 9 (Suppl. A), 33–37. [Google Scholar] [CrossRef]
- Blanco Garcia, J.; Aldinucci, C.; Maiorca, S.M.; Palmi, M.; Valoti, M.; Buonocore, G.; Pessina, G.P. Physiopathological effects of the NO donor 3-morpholinosydnonimine on rat cortical synaptosomes. Neurochem. Res. 2009, 34, 931–941. [Google Scholar] [CrossRef]
- Dudek, J.; Maack, C. Grandfather’s moonlighting: Hydralazine’s novel liaison with mitochondria. Cardiovasc. Res. 2022, 118, 13–15. [Google Scholar] [CrossRef]
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E., Jr.; Collins, K.J.; Dennison Himmelfarb, C.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2018, 71, e13–e115. [Google Scholar] [CrossRef]
- Chiusa, M.; Timolati, F.; Perriard, J.C.; Suter, T.M.; Zuppinger, C. Sodium nitroprusside induces cell death and cytoskeleton degradation in adult rat cardiomyocytes in vitro: Implications for anthracycline-induced cardiotoxicity. Eur. J. Histochem. EJH 2012, 56, e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.R.; Kwak, J.H.; Noh, S.J.; Pronto, J.R.; Ko, K.S.; Rhee, B.D.; Xu, Z.; Kim, N.; Han, J. Kobophenol A inhibits sodium nitroprusside-induced cardiac H9c2 cell death through suppressing activation of JNK and preserving mitochondrial anti-apoptotic Bcl-2 and Mcl-1. Chem. Pharm. Bull. 2014, 62, 713–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shorter, K.; Farjo, N.P.; Picksley, S.M.; Randall, V.A. Human hair follicles contain two forms of ATP-sensitive potassium channels, only one of which is sensitive to minoxidil. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2008, 22, 1725–1736. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Wang, X.; Ye, X.; Ares, I.; Lopez-Torres, B.; Martínez, M.; Martínez-Larrañaga, M.-R.; Wang, X.; Anadón, A.; Martínez, M.-A. Mitochondria as an important target of metformin: The mechanism of action, toxic and side effects, and new therapeutic applications. Pharmacol. Res. 2022, 177, 106114. [Google Scholar] [CrossRef] [PubMed]
- Merce, A.P.; Ionică, L.N.; Bînă, A.M.; Popescu, S.; Lighezan, R.; Petrescu, L.; Borza, C.; Sturza, A.; Muntean, D.M.; Creţu, O.M. Monoamine oxidase is a source of cardiac oxidative stress in obese rats: The beneficial role of metformin. Mol. Cell. Biochem. 2022. [Google Scholar] [CrossRef] [PubMed]
- Ionică, L.N.; Gaiță, L.; Bînă, A.M.; Soșdean, R.; Lighezan, R.; Sima, A.; Malița, D.; Crețu, O.M.; Burlacu, O.; Muntean, D.M.; et al. Metformin alleviates monoamine oxidase-related vascular oxidative stress and endothelial dysfunction in rats with diet-induced obesity. Mol. Cell. Biochem. 2021, 476, 4019–4029. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Khan, M.S.; Marx, N.; Lam, C.S.P.; Schnaidt, S.; Ofstad, A.P.; Brueckmann, M.; Jamal, W.; et al. Effect of Empagliflozin on Cardiovascular and Renal Outcomes in Patients With Heart Failure by Baseline Diabetes Status: Results From the EMPEROR-Reduced Trial. Circulation 2021, 143, 337–349. [Google Scholar] [CrossRef]
- Tsampasian, V.; Baral, R.; Chattopadhyay, R.; Debski, M.; Joshi, S.S.; Reinhold, J.; Dweck, M.R.; Garg, P.; Vassiliou, V.S. The Role of SGLT2 Inhibitors in Heart Failure: A Systematic Review and Meta-Analysis. Cardiol. Res. Pract. 2021, 2021, 9927533. [Google Scholar] [CrossRef]
- Vaduganathan, M.; Januzzi, J.L., Jr. Preventing and Treating Heart Failure with Sodium-Glucose Co-Transporter 2 Inhibitors. Am. J. Cardiol. 2019, 124 (Suppl. 1), S20–S27. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.C.; Chau, Y.Y.; Ng, H.Y.; Chen, C.H.; Wang, P.W.; Liou, C.W.; Lin, T.K.; Chen, J.B. Empagliflozin Protects HK-2 Cells from High Glucose-Mediated Injuries via a Mitochondrial Mechanism. Cells 2019, 8, 1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.H.; Kim, S.H.; Kang, J.M.; Heo, J.H.; Kim, D.J.; Park, S.H.; Sung, M.; Kim, J.; Oh, J.; Yang, D.H.; et al. Empagliflozin attenuates diabetic tubulopathy by improving mitochondrial fragmentation and autophagy. Am. J. Physiol. Ren. Physiol. 2019, 317, F767–F780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Xu, C.; Xu, L.; Li, X.; Sun, H.; Xue, M.; Li, T.; Yu, X.; Sun, B.; Chen, L. Empagliflozin improves diabetic renal tubular injury by alleviating mitochondrial fission via AMPK/SP1/PGAM5 pathway. Metab. Clin. Exp. 2020, 111, 154334. [Google Scholar] [CrossRef] [PubMed]
- Durak, A.; Olgar, Y.; Degirmenci, S.; Akkus, E.; Tuncay, E.; Turan, B. A SGLT2 inhibitor dapagliflozin suppresses prolonged ventricular-repolarization through augmentation of mitochondrial function in insulin-resistant metabolic syndrome rats. Cardiovasc. Diabetol. 2018, 17, 144. [Google Scholar] [CrossRef]
- Hawley, S.A.; Ford, R.J.; Smith, B.K.; Gowans, G.J.; Mancini, S.J.; Pitt, R.D.; Day, E.A.; Salt, I.P.; Steinberg, G.R.; Hardie, D.G. The Na+/Glucose Cotransporter Inhibitor Canagliflozin Activates AMPK by Inhibiting Mitochondrial Function and Increasing Cellular AMP Levels. Diabetes 2016, 65, 2784–2794. [Google Scholar] [CrossRef] [Green Version]
- Villani, L.A.; Smith, B.K.; Marcinko, K.; Ford, R.J.; Broadfield, L.A.; Green, A.E.; Houde, V.P.; Muti, P.; Tsakiridis, T.; Steinberg, G.R. The diabetes medication Canagliflozin reduces cancer cell proliferation by inhibiting mitochondrial complex-I supported respiration. Mol. Metab. 2016, 5, 1048–1056. [Google Scholar] [CrossRef]
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; Grobbee, D.E.; Hansen, T.B.; et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Heart J. 2020, 41, 255–323. [Google Scholar] [CrossRef]
- Sheahan, K.H.; Wahlberg, E.A.; Gilbert, M.P. An overview of GLP-1 agonists and recent cardiovascular outcomes trials. Postgrad. Med. J. 2020, 96, 156–161. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.C.; Wang, W.T.; Lee, S.S.; Kuo, Y.R.; Wang, Y.C.; Yen, S.J.; Lee, M.Y.; Yeh, J.L. Glucagon-Like Peptide-1 Receptor Agonist Attenuates Autophagy to Ameliorate Pulmonary Arterial Hypertension through Drp1/NOX- and Atg-5/Atg-7/Beclin-1/LC3β Pathways. Int. J. Mol. Sci. 2019, 20, 3435. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; He, W.; Wei, W.; Mei, X.; Yang, M.; Wang, Y. Exenatide Attenuates Obesity-Induced Mitochondrial Dysfunction by Activating SIRT1 in Renal Tubular Cells. Front. Endocrinol. 2021, 12, 622737. [Google Scholar] [CrossRef]
- Monji, A.; Mitsui, T.; Bando, Y.K.; Aoyama, M.; Shigeta, T.; Murohara, T. Glucagon-like peptide-1 receptor activation reverses cardiac remodeling via normalizing cardiac steatosis and oxidative stress in type 2 diabetes. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H295–H304. [Google Scholar] [CrossRef] [PubMed]
- Li, P.C.; Liu, L.F.; Jou, M.J.; Wang, H.K. The GLP-1 receptor agonists exendin-4 and liraglutide alleviate oxidative stress and cognitive and micturition deficits induced by middle cerebral artery occlusion in diabetic mice. BMC Neurosci. 2016, 17, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khin, P.P.; Hong, Y.; Yeon, M.; Lee, D.H.; Lee, J.H.; Jun, H.S. Dulaglutide improves muscle function by attenuating inflammation through OPA-1-TLR-9 signaling in aged mice. Aging 2021, 13, 21962–21974. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wang, Z.; Hu, S.; Zhou, B. Assessing Drug-Induced Mitochondrial Toxicity in Cardiomyocytes: Implications for Preclinical Cardiac Safety Evaluation. Pharmaceutics 2022, 14, 1313. [Google Scholar] [CrossRef]
- Fermini, B.; Coyne, S.T.; Coyne, K.P. Clinical Trials in a Dish: A Perspective on the Coming Revolution in Drug Development. SLAS Discov. 2018, 23, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Fialová, D.; Laffon, B.; Marinković, V.; Tasić, L.; Doro, P.; Sόos, G.; Mota, J.; Dogan, S.; Brkić, J.; Teixeira, J.P.; et al. Medication use in older patients and age-blind approach: Narrative literature review (insufficient evidence on the efficacy and safety of drugs in older age, frequent use of PIMs and polypharmacy, and underuse of highly beneficial nonpharmacological strategies). Eur. J. Clin. Pharmacol. 2019, 75, 451–466. [Google Scholar] [CrossRef] [Green Version]
- Madla, C.M.; Gavins, F.K.H.; Merchant, H.A.; Orlu, M.; Murdan, S.; Basit, A.W. Let’s talk about sex: Differences in drug therapy in males and females. Adv. Drug. Deliv. Rev. 2021, 175, 113804. [Google Scholar] [CrossRef]
- Zucker, I.; Prendergast, B.J. Sex differences in pharmacokinetics predict adverse drug reactions in women. Biol. Sex Differ. 2020, 11, 32. [Google Scholar] [CrossRef]
- Merce, A.P.M.; Bînă, A.M.B.; Avram, V.F.A.; Buriman, D.G.B.; Lascu, A.L.; Feier, H.B.F.; Petrescu, L.P.; Muntean, D.M.M.; Elmér, E.E.; Crețu, O.M.C. Cell-Permeable Succinate Improves Platelet Respiration in Patients Undergoing Cardiopulmonary Bypass: A Pilot Study. Timis. Med. J. 2022, 2022, 2. [Google Scholar] [CrossRef]
- Bonora, M.; Wieckowski, M.R.; Sinclair, D.A.; Kroemer, G.; Pinton, P.; Galluzzi, L. Targeting mitochondria for cardiovascular disorders: Therapeutic potential and obstacles. Nat. Rev. Cardiol. 2019, 16, 33–55. [Google Scholar] [CrossRef]


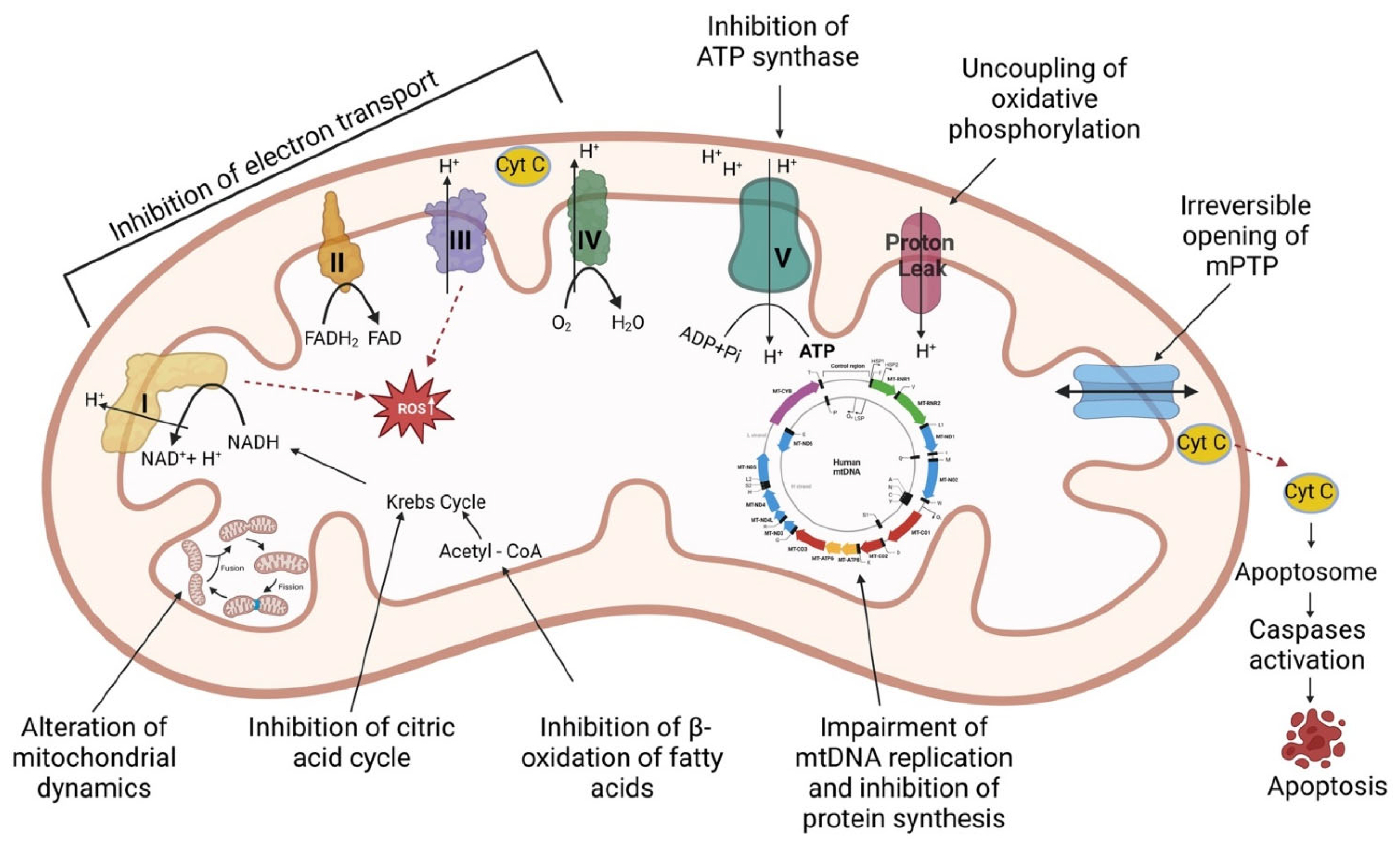{kind=link}
| Class of Drugs | Drug Name | Mitochondrial Effects | Experimental Model | References |
|---|---|---|---|---|
| Antiarrhytmics | ||||
| Class II (β-blockers) | Timolol | Prevention of oxidative damage (25–5000 µM) Prevention of lipid peroxidation (5 mg/kg body weight, 9 months) | in vitro in vivo (female rat model of aging-related altered left ventricular function) | [28,29,30,31] |
| Class III (K-channel blockers) | Ibutilide | Attenuation of oxidative stress (10−8 to 10−3 mol/L) Inhibition of mitochondrial-related apoptosis Increase in glutathione peroxidase and superoxide dismutase levels (10−8 to 10−3 mol/L) | in vitro (rat cardiomyocytes) | [32] |
| Sotalol | No mitochondrial dysfunction (15–240 µM) | in vitro (human platelets) | [33] | |
| Dofetilide | Correction of the calcium handling (2 mg/kg, 3 days; 10−6–10−8 mol/L) Correction of NADPH oxidase (2 mg/kg, 3 days; 10−6–10−8 mol/L) | in vivo (heart failure rat model) in vitro (primary neonatal cardiomyocytes) | [34] | |
| Class IV (Ca-channel blockers) | Verapamil | Inhibition of lipid peroxidation (7 mg/kg) Antioxidant enzyme activity (10 mg/kg twice) Reduction in apoptosis (10 mg/kg twice) Reduction in ROS formation and cytochrome c release (10 mg/kg twice) Increase the ATP concentration (10 mg/kg twice) Reduction in mitochondrial swelling (10 mg/kg twice) Inhibition of mitochondrial membrane potential decrease (10 mg/kg twice) | in vivo (streptozotocin-induced diabetic rats) in vivo (rat model of forebrain ischemia/reperfusion) | [35,36] |
| Diltiazem | Protection of mitochondrial integrity (0.1–0.5 µmol/L) Conservation of high-energy phosphate levels (200 µg/kg bolus + 15 µg/kg/min continuous iv infusion) Prevention of mitochondrial swelling (7.5 µM) Prevention of mitochondrial Ca2+ increase (7.5 µM) Reduction in lipid peroxidation (5 × 10−7M) Decrease apoptosis (10 µmol/L) | ex vivo (drug administered during reperfusion in a rabbit model of heart ischemia/reperfusion) in vivo (rabbit model of myocardium ischemia/reperfusion) ex vivo (drug added before ischemia in a rat model of heart ischemia/reperfusion) ex vivo (reperfused isolated rabbit hearts) in vitro (rat hepatocytes) | [37,38,39,40,41] | |
| Angiotensin-converting enzyme inhibitors (ACEI) | Zofenopril | Prevention of mitochondrial calcium overload (10−9–10−4 M) Maintenance of oxidative phosphorylation (10−9–10−4 M) Maintenance of ATP production (10−9–10−4 M) Preservation of membrane integrity (10−9–10−4 M) Decrease oxidative stress (10−9–10−4 M) | ex vivo (rabbit model of myocardium ischemia/reperfusion) | [42] |
| Perindopril | Decreased ROS synthesis (2 mg/kg/day, 6 weeks) Increased antioxidant enzymes (2 mg/kg/day, 6 weeks) Increased number of mitochondria (2 mg/kg/day, 6 weeks) Alleviation of mitochondrial ETS dysfunction (2 mg/kg/day, 6 weeks) Increase ATP production (2 mg/kg/day, 6 weeks) Reduction in apoptosis (2 mg/kg/day, 6 weeks) Increased calcium retention capacity (0.2 mg/kg) | in vivo (rat model of isoprotenerol-induced cardiomyopathy) in vivo (drug administered prior to ischemia in a pig model of heart ischemia/reperfusion) | [43,44] | |
| Trandolapril | Increase in ETS complexes I, II and IV activities (4–6 mg/kg/day, 12 days) Attenuation of oxidative stress (4–6 mg/kg/day, 12 days) Reduction in lipid peroxidation (4–6 mg/kg/day, 12 days) Improvement of mitochondrial oxygen consumption rates (3 mg/kg/day, 6 weeks) Increase in ATP production (3 mg/kg/day, 6 weeks) | in vivo (in a rat model of 3-nitropropionic acid induced brain lesions) in vivo (rat model of failing heart following acute myocardial infarction) | [45,46,47] | |
| Enalapril | Enhance antioxidant defenses (20 mg/L in drinking water, 11 weeks) Decreased ROS production (10 mg/kg/day, 14 days) Increased mitochondrial mass/biogenesis (20 mg/kg/day, 3 months) Promotion of mitochondrial fusion and autophagy (20 mg/kg/day, 3 months) Reduction in lipid peroxidation (10 mg/kg/day, 12 weeks) Improvement of mitochondrial respiratory efficiency (10 mg/kg/day, 10 weeks) | in vivo (mouse tissues) in vivo (rat kidney mitochondria) in vivo (aged rat hearts) in vivo (rat model of heart failure) in vivo (rat model of doxorubicin-induced cardiomyopathy) | [48,49,50,51,52] | |
| Angiotensin receptor blockers (ARBs) | Valsartan | Improvement of mitochondrial biogenesis and mitophagy (320 mg/day, 4 weeks) Increase mitochondrial respiration (15 mg/kg/day, 4 months) Reduction in mitochondrial oxidative stress (30 mg/kg/day in drinking water, 3 weeks) Increase in mitochondrial β-oxidation (30 mg/kg/day in drinking water, 3 weeks) | in vivo (pig model with renovascular hypertension) in vivo (rats with type 2 diabetes) in vivo (rats with elevated levels of angiotensin II) | [53,54,55] |
| Losartan | Reduction in oxidative stress (40 mg/kg/day, 6 months) Increased mitochondrial membrane potential (40 mg/kg/day, 6 months) Amelioration of mtDNA content decrease (30 mg/kg/day, 16.5 months) Improvement of mitochondrial biogenesis (100 mg/L in drinking water, 30 days) | in vivo (spontaneously hypertensive rats) in vivo (aged rats) in vivo (obese mice) | [56,57,58] | |
| Candesartan | Decreased ROS production (10 μmol/L) Regulation of mitochondrial dynamics (10 μmol/L) Improvement of mitochondrial structure and dynamics (2 mg/kg/day, 8 weeks) Increased mitochondrial membrane potential (2 mg/kg/day, 8 weeks) Alleviation of mitochondrial ETS dysfunction (Complex I, II, III, and IV) (0.1–0.3 mg/kg, 7 days) | in vitro (vascular smooth muscle cells) in vivo (spontaneously hypertensive rats) in vivo (rat model of cerebral ischemia) | [59,60,61] | |
| Irbesartan | Inhibition of mitochondrial apoptosis (50 mg/kg/day) Increase ATP production (10 nM) Increased mitochondrial membrane potential (10 nM) Decreased ROS production (10 nM) | in vivo (rat model of sleep apnea) in vitro (human and mouse model of non-alcoholic fatty liver disease) | [62,63] | |
| Telmisartan | Upregulation of mitochondria-specific genes expression (3–10 mg/kg) Increased mitochondrial membrane potential Decreased oxidative stress (5 mg/kg/day, 12 weeks) Modulation of mitochondrial Ca2+ homeostasis (5 mg/kg/day, 12 weeks) Enhancement of ATP synthesis (1–10 µM) Increase in mitochondrial complex II activity (1–10 µM) Reduction in apoptosis (1–10 µM) | in vivo (mouse model of Parkinsonism) in vitro (renal glomerular endothelial cells exposed to high glucose) in vivo (hypertensive rats) in vitro (human vascular smooth muscle cells) | [64,65,66,67] | |
| Olmesartan | Increase in mitochondrial ETS activities (complex I, II) (10 mg/kg/day, 6 weeks) Reduction in oxidative damage (3 mg/kg/day in drinking water, 8 weeks) Improvement of ADP-dependent mitochondrial respiration (3 mg/kg/day in drinking water, 8 weeks) | in vivo (obese insulin resistant rats exposed to an acute glucose load) in vivo (mice model of high-fat diet-induced diabetes) | [68,69] | |
| Azilsartan | Decreased ROS production (0.1–10 µM) Inhibition of lipid peroxidation (0.1–10 µM) Increased mitochondrial membrane potential (0.1–10 µM) Preservation of ATP production (0.1–10 µM) Reduction in mitochondrial swelling (0.1–10 µM) Alleviation of ETS complexes I, II and IV dysfunction Increased mitochondrial respiration (2–4 mg/kg) Inhibition of apoptosis (2–4 mg/kg) Increased glutathione level (2–4 mg/kg) | in vitro (murine brain endothelial cells) in vivo (rat model of cerebral ischemia) | [70,71] | |
| Angiotensin receptor neprilysin inhibitor (ARNi) | Sacubitril/Valsartan | Attenuation of oxidative stress (68 mg/kg/day, 10 weeks) Improvement of mitochondrial state-3 respiration (100 mg/day, 3 months) Increased mitochondrial membrane potential (100 mg/day, 3 months) Prevention of mitochondrial permeability transition pore opening (100 mg/day, 3 months) Increased ATP production (100 mg/day, 3 months) Normalization of complex-I and IV activities (100 mg/day, 3 months) Inhibition of apoptosis (100 mg/day, 3 months) | in vivo (rat model of pressure overloaded hearts) in vivo (dogs with experimental cardiorenal syndrome) | [72,73] |
| Calcium channel blockers-dihydropyridines | Amlodipine | Increased oxygen consumption in state 3 (0.4 mg/kg) Increased calcium retention capacity (0.4 mg/kg) Reduction in ROS production (0.4 mg/kg) Decrease in mitochondrial swelling (0.4 mg/kg) Antioxidant properties (5 mg/kg/day, 8 weeks) Increased glutathione peroxidase, catalase and superoxide dismutase activity (1 mg/kg, 7 days) Reduction in lipid peroxidation (1 mg/kg, 7 days) Inhibition of apoptosis (1 mg/kg, 7 days) Enhancement of mitochondrial biogenesis (0.1–1000 μM) | ex vivo (pig ischemia/reperfusion model) in vivo (cholesterol-induced rabbit model of atherosclerosis and a liver and a heart rat model of ischemia/reperfusion injury) in vitro (neural stem cells exposed to oxygen glucose deprivation) | [44,74,75,76,77,78] |
| Antithrombotic agents | Ticagrelor | Increased mitochondrial membrane potential (1 µM) Decreased ROS production (1 µM) Preservation of ATP synthesis (1 µM) Restoration of mitochondria ultrastructural changes (swelling and loss of crista) (1 µM) | in vitro (insulin-resistant H9 c2 cells) | [79,80] |
| Oral anticoagulants | ||||
| Direct oral anticoagulants | Apixaban | Antioxidant properties (60 ng/mL) Reduction in ROS production (60 ng/mL) | in vitro (model of endothelial dysfunction in uremia) | [81] |
| Edoxaban | Increase mitochondrial oxygen consumption (1 μmol/L) Improve mitochondrial ATP generation consumption (1 μmol/L) | in vitro (human alveolar epithelial cells) | [82] | |
| Diuretics | ||||
| Loop diuretics | Bumetanide | Attenuation of mitochondrial Ca2+ overload (5 μM) Attenuation of mitochondrial membrane potential dissipation (5 μM) Decreased cytochrome c release (5 μM) | in vitro (astrocytes following ischemia) | [83,84] |
| Antagonists of aldosterone | Spironolactone | Improvement of mitochondrial membrane potential (0.01–1 µM) Increase in ATP synthesis (0.01–1 µM) Inhibition of ROS production (0.01–1 µM) Inhibition of apoptosis (1–10 µM) | in vitro (methylglyoxal exposed osteoblastic cells) | [85,86] |
| Eplerenone | Increased number of cardiac mitochondria (100 mg/kg/day, 6 weeks) Increase in mitochondrial DNA copy number (100 mg/kg/day, 6 weeks) | in vivo (aldosterone-infused mice) | [87] | |
| Sodium-glucose cotransporter 2 (SGLT2) inhibitors | Empagliflozin | Improvement of mitochondrial biogenesis (10–30 mg/kg/day, 8 weeks) Increased state 3 respiratory rate (10–30 mg/kg/day, 8 weeks) Increased mitochondrial membrane potential (10–30 mg/kg/day, 8 weeks) Suppression of ROS generation (10 mg/kg/day, 2 weeks) Reduction in mitochondrial DNA damage (30 mg/kg/day, 10 weeks) Reduction in oxidative stress (30 mg/kg/day, 10 weeks) Restoration of fatty acid oxidation (30 mg/kg/day, 10 weeks) Enhancement of mitochondrial fusion (3.8 mg/kg/day, 8 weeks) | in vivo (rat model of high-fat diet/streptozocin-induced diabetes) in vivo (rat diabetic hearts after myocardial infarction) in vivo (rats with left ventricular dysfunction after myocardial infarction) | [88,89,90,91,92,93] |
| Glucagon-like peptide-1 receptor agonists (GLP-1 RAs) | Liraglutide | Decreased ROS production (50–500 nM) Increased mitophagy (50–500 nM) Alleviation of mitochondrial membrane potential decrease (1–20 nM) Inhibition of mitochondrial permeability transition pore opening (1–20 nM) Inhibition of apoptosis (1–20 nM) Attenuation of Ca2+ abnormalities (0.3 mg/kg, 4 weeks) Normalization of mitochondrial dynamics (0.15 mg/kg) | in vitro (HepG2 cell model of non-alcoholic steatohepatitis) in vitro (human renal mesangial cells exposed to hyperglycemia) in vivo (rat model of high-carbohydrate induced metabolic syndrome) in vivo (acute mouse model of Parkinson’s disease) | [94,95,96,97,98] |
| Exenatide | Decreased oxidative stress (0.05–0.6 μM) Increased ATP production (0.05–0.6 μM) Increased mitochondrial ATPase activity (0.05–0.6 μM) Increased mitochondrial membrane potential (0.05–0.6 μM) Decreased mitochondrial calcium overload (0.05–0.6 μM) Inhibition of mitochondrial permeability transition pore opening (0.05–0.6 μM) Improvement of morphological and structural changes of mitochondria (10 mg/kg or 0.3 nM) | in vitro (H9c2 cardiomyocytes subjected to hypoxia/reoxygenation) in vivo (rat model of ischemia/reperfusion injury) and ex vivo (Langendorff model) | [99,100] | |
| Dulaglutide | Increased mitochondrial membrane potential (50–100 nM) Decreased ROS generation (50–100 nM) Increased glutathione level (50–100 nM) | in vitro (human fibroblast-like synoviocytes exposed to TNF-α) | [101] | |
| Semaglutide | Decreased ROS production (1–5 mmol/L) Improvement of autophagy (1–5 mmol/L) Decreased lipid peroxidation (25 nmol/kg, 30 days) | in vitro (lipopolysaccharides treated H9c2 cardiomyocytes) in vivo (aged mice) | [102,103] | |
| Lixisenatide | Promotion of mitochondrial biogenesis (5–20 nM) Increased mitochondrial respiration (5–20 nM) Enhancement of ATP generation (5–20 nM) Inhibition of oxidative stress (10–20 nM) Increased mitochondrial membrane potential (10–20 nM) | in vitro (human umbilical vein endothelial cells) in vitro (human rheumatoid arthritis fibroblast-like synoviocytes) | [104,105] | |
| Class of Drugs | Drug Name | Mitochondrial Effects/Dosage | Experimental Model | References |
|---|---|---|---|---|
| β1 and 2 receptor agonists | Isoprotenerol | Inhibition of respiration (85 mg/kg of body weight) Inhibition of ETS complexes I, II, IV and ATP synthase (85 mg/kg of body weight) Stimulation of the mPTP opening (100 mg/kg body weight) Increase in lipid peroxidation (100 mg/kg body weight) Alteration of glutathione status (100 mg/kg body weight) Induction of antioxidant depletion (≅30 μM) DNA damage and apoptotic signaling (≅30 μM) Induction of oxidative stress (30 mg/kg/day, 8 days) Uncoupling of oxidative phosphorylation (1 mg/kg, 10 days) Decrease ATP levels (1 mg/kg, 10 days) Increased expression of inflammatory markers | in vivo (rats) in vivo (rats) in vitro (cardiomyoblasts) in vivo (mice) in vivo (rat model of experimental chronic heart failure) | [106,107,108,109,110,111,112,113,114,115] |
| Antiarrhytmics | ||||
| Class I (Na-channel blockers) | Quinidine | Uncoupling of oxidative phosphorylation (50 mg/kg/day, 5 days/week for 4 weeks) Reduction in mitochondrial creatine phosphate kinase activity (50 mg/kg/day, 5 days/week for 4 weeks) Decrease ATP production (50 mg/kg/day, 5 days/week for 4 weeks) Inhibition of the protein synthesis in heart mitochondria (50 mg/kg/day, 5 days/week for 4 weeks) Inhibition of respiration (1–4 mM in vitro; 75 mg/kg twice a day for 4 days in vivo) | in vivo (rats) in vitro (isolated kidney cortex mitochondria); in vivo (male rats) | [116,117] |
| Propafenone | Reduction in mitochondrial membrane potential (10–20 µM) Decrease the expression of apoptotic inhibitors Bcl-xL and Bcl-2 (10–20 µM) | in vitro (esophageal squamous cell carcinoma) | [118] | |
| Class II (β-blockers) | Propanolol | Induction of mitochondrial swelling and cytochrome c release (2.5–20 µg/mL) Activation of caspase cascade and apoptotic cell death (2.5–20 µg/mL) Inhibition of the ETS complex II (2.5–20 µg/mL) Increased ROS formation (2.5–20 µg/mL) Decreased mitochondrial membrane potential (2.5–20 µg/mL) Depletion of the ATP level (2.5–20 µg/mL) | in vitro (rat cardiomyocytes) | [119,120,121,122,123,124] |
| Class III (K-channel blockers) | Amiodarone | Inhibition of respiration (20–400 µM) Uncoupling of oxidative phosphorylation (20–400 µM) Inhibition of the mitochondrial complexes I and II (20–400 µM) Inhibition of fatty acid ß-oxidation (20–400 µM) Depletion of ATP content (20–400 µM) | in vitro (isolated rat liver mitochondria, human hepatocytes, rat cardiomyocytes, human platelets, peripheral blood mononuclear cells, HepG2 cells) | [125,126,127,128,129,130,131] |
| Dronedarone | Inhibition of fatty acid β-oxidation (1–50 µM) Dissipation of the mitochondrial membrane potential Inhibition of respiration (1–50 µM) Inhibition of mitochondrial complex I (1–50 µM) Uncoupling of oxidative phosphorylation (1–50 µM) Decrease in the intracellular ATP content (1–50 µM) | in vitro (rat liver mitochondria, primary human hepatocytes, HepG2 cells, rat cardiomyocytes) | [129,130,131,132] | |
| Antithrombotic agents | Acetyl-salicylic acid | Inhibition of respiration (2–10 mM) Inhibition of ATP synthesis (2–10 mM) Uncoupling of oxidative phosphorylation (2–10 mM) Inhibition of the respiratory chain ATPase Opening of the mitochondrial transition pore (400 µM) Reduction in mitochondrial membrane potential (400 µM) Increase Ca2+ release from the mitochondrion (400 µM) | in vitro (isolated rat cardiac mitochondria) in vitro (rat liver mitochondria) in vitro (rat kidney mitochondria) | [133,134,135] |
| Clopidogrel | Inhibition of mitochondrial respiratory state 3 and state 4 respiration—in high doses (10 µg/mL) Reduction in glutathione content (10–100 μM) Decreased mitochondrial membrane potential (10–100 μM) Increased ROS production (10–100 μM) Induction of apoptosis (10–100 μM) | in vitro (isolated mice liver mitochondria) in vitro (primary human hepatocytes and HepG2 cells) | [136,137,138] | |
| Prasugrel and Ticlopidine | Decreased mitochondrial membrane potential (10–100 μM) Increased ROS production (10–100 μM) Induction of apoptosis (10–100 μM) | in vitro (human neutrophil granulocytes and lymphocytes) | [138] | |
| Oral anticoagulants | ||||
| Coumarin derivatives | Warfarin | Reduction in ATP content (0.5–1 mM) | in vitro (isolated rat hepatocytes) | [139] |
| Direct Oral Anticoagulants | Dabigatran | Increased ROS generation (1–100 µM) Decreased mitochondrial membrane potential (1–100 µM) Increased lipid peroxidation (1–100 µM) | in vitro (rat gastric epithelial cell line) | [140] |
| Diuretics | ||||
| Loop diuretics | Furosemide | Inhibition of ETS complex II Inhibition of state 3 (ADP-dependent) respiration (2 × 10−3 mol/L) | in vitro (rat kidney mitochondria, rat liver mitochondria) | [141,142] |
| Direct vasodilators | Organic nitrates | Increase in ROS production (50–5000 μM) Induction of lipid peroxidation (50–5000 μM) Decreased mitochondrial membrane potential (50–5000 μM) Induction of mitochondrial swelling (50–5000 μM) | in vitro (rat heart mitochondria) | [143,144] |
| Molsidomine/ Lisindomine | Induction of oxidative stress (1–20 µM) Inhibition of mitochondrial respiration (50 µmol/L) Inhibition of complex I (1–20 µM) Decreased mitochondrial membrane potential (0.2–0.8 mmol/L) Decrease in ATP synthesis (0.2–0.8 mmol/L) | in vitro (isolated rat brain mitochondria) in vitro (human spermatozoa) | [145,146,147,148] | |
| Sodium nitroprusside | Decreased mitochondrial membrane potential (0.5–5 mM) Inhibition of ATP generation (0.5–5 mM) Induction of apoptosis (1 mM) | in vitro (neuronal PC12 cells and HepG2 liver cells) in vitro (rat chondrocytes) | [149,150] | |
| Minoxidil | Induction of mitochondrial morphological abnormalities (50 µg/mL) Increased ROS production (50 µg/mL) | in vitro (ovarian cancer cells) | [151] | |
| Class of Drugs | Drug Name | Beneficial Effects | Experimental Model | Deleterious Effects | Experimental Model | References |
|---|---|---|---|---|---|---|
| Antiarrhytmics | ||||||
| Lidocaine | Alleviation of isoflurane-induced mitochondrial structure damage and the decline in mitochondrial membrane potential 40–100 μg/mL) Reversal of isoflurane-induced mitochondrial ETS dysfunction (40–100 μg/mL) Inhibition of isoflurane-induced apoptosis (40–100 μg/mL) | in vitro (H4 cells exposed to isoflurane) | Suppression of the mitochondrial ETS (0.1–10 mM) Decreased mitochondrial membrane potential (0.1–10 mM) Increased ROS production (0.1–10 mM) Inhibition of ATP synthesis (0.1–10 mM) Induction of mitochondrial structural changes and apoptosis (4–4000 µM) | in vitro (neuronal SH-SY5Y cells) in vitro (human neutrophils) | [152,153,154] | |
| Phenytoin | Decreased cerebral malondialdehyde as marker of oxidative stress Decreased monoamine oxidase A + B activity in an animal model of epilepsy | in vivo | Increased oxidative stress (200–600 µM) Depletion of glutathione (200–600 µM) Increased lipid peroxidation (200–600 µM) Inhibition of respiration (0.025–1 mM) Decreased ATP synthesis (0.025–1 mM) Decreased mitochondrial membrane potential (0.025–1 mM) | in vitro (rat hepatocytes) in vitro (murine hepatic microsomal system) | [155,156,157] | |
| Class II (β-blockers) | Carvedilol | Antioxidant effects (10 µM) Inhibition of lipid peroxidation (1–50 µM) Mild uncoupling of mitochondrial oxidative phosphorylation (10–100 µM) Decrease in ROS production (10–20 µM) Prevention of calcium overload (10–20 µM) Inhibition of NADH dehydrogenase and prevention of oxidative damage (10–20 µM) Inhibition of mPTP (5–20 µM) | in vitro (swine ventricular membranes, rat brain homogenates, human LDL, bovine and human endothelial cells, rat heart mitochondria) | Induction of severe mitochondria damage—mitochondrial swelling, crista damage and formation of myelin figures inside the mitochondria (10 µM) | in vitro (rat C6 glioma cells) | [158,159,160,161] |
| Nebivolol | Antioxidant activity (1–2 mg/kg, 8 weeks) Inhibition of NADPH oxidase activity (1–2 mg/kg, 8 weeks) | in vivo (streptozocin treated diabetic rats) | Inhibition of complex I and V (1 µM) Inhibition of respiration (1 µM) Depletion of ATP levels (10 µM) Induction of mitochondrial morphology changes (10 µM) Increased ROS production (10 µM) | in vitro (breast, colon and oral squamous carcinoma cells) | [162,163,164] | |
| Metoprolol | Increased mitochondrial respiratory control ratio (1 mg/kg -bolus infusion) | in vivo (rat model of ischemia/reperfusion injury) | No protective effect against adriamycin-induced mitochondrial DNA impairment (3 mg/kg/12 h, 12 days) | in vivo (rat model of adriamycin-induced cardiotoxicity) | [165,166,167,168] | |
| Atenolol | Decrease in membrane fatty acid unsaturation degree of mitochondria (0.1 g/L of atenolol drinking water solution) Reduction in mitochondrial protein oxidative, glycoxidative, and lipoxidative modification (0.1 g/L in drinking water) Reduction in oxidative damage in heart mitochondrial DNA (0.1 g/L in drinking water) | in vivo (rats) | Increased ROS production (2.5–20 μg/mL) Decrease in mitochondrial succinate dehydrogenase activity (2.5–20 μg/mL) Decreased mitochondrial membrane potential (2.5–20 μg/mL) Induction of mitochondrial swelling (2.5–20 μg/mL) Decreased ATP content (2.5–20 μg/mL) | in vitro (isolated rat heart mitochondria) | [119,169,170,171] | |
| Esmolol | Improvement of mitochondrial morphology (300 μg/kg/min, 48 h) Prevention of apoptosis by decreasing the Bax/Bcl-2 levels (1.75–3.5 mg/Kg/h) | in vivo (spontaneously hypertensive rats) in vivo (early sepsis rats with abdominal infection) | Increased ROS level (5–250 μM) Decreased mitochondrial membrane potential (5–250 μM) | in vitro (human lung fibroblast cells) | [172,173,174,175] | |
| Others | Adenosine | Attenuation of the decline of complex I and mitochondrial NO synthase activities (0.03 μg/kg/min, 65 min) Reduction in mitochondrial phospholipid oxidation (0.03 μg/kg/min, 65 min) | ex vivo (experimental model of rabbit heart ischemia/reperfusion) | Induction of apoptosis (0.1–10 mM) Increased ROS production (0.1–10 mM) Reduction in Bcl-X(L) expression (3 mM) Disruption of mitochondrial membrane potential (3 mM) | in vitro (liver cancer cells) in vitro (HepG2 cells) | [176,177,178,179] |
| Digitalis | Enhancement of the efficiency of mitochondrial electron transport and ATP synthesis (1–100 nM in vitro; 1 mg/kg, 5–8 days in vivo) | in vitro (rat cardiomyocytes) in vivo (mice) | Reduction in mitochondrial Ca2+ accumulation (1 µM) Reduction in the NADH/NAD+ redox potential (1 µM) Increased ROS production (1 µM) Decrease mitochondrial membrane potential (0.025–0.2 µM) Induction of mitochondrial related apoptosis (0.025–0.2 µM) Increase Bax/Bcl-2 proportion (50–200 nM) Depletion of ATP (0.03–100 µM) | in vitro (guinea pig ventricular myocytes) in vitro (human non-small cell lung cancer cells A549) in vitro (breast cancer cells) in vitro (HeLa cell line) | [180,181,182,183,184,185] | |
| Angiotensin-converting enzyme inhibitors (ACEI) | Ramipril | Attenuation of lipid peroxidation (10 mg/kg/day, 28 days in vivo; 10 µM in vitro) | in vivo (rat model of rheumatoid arthritis) and in vitro (rat cardiomyocytes) | Inhibition of cardiac uncoupling protein-2 expression (50 µg/kg/day, 4 weeks) | in vivo (rat model of ischemia/reperfusion) | [186,187] |
| Captopril | Attenuation of mitochondrial membrane potential dissipation (10 mg/kg, 7–8 days) Increase ATP production (10 mg/kg, 7–8 days) Restoration of mitochondrial oxygen consumption (5 mg/kg, 12 weeks) Antioxidant effect (0.08 mM) | in vivo (rat model of adriamycin toxicity) in vivo (rabbits with experimentally induces hypercholesterolemia) in vitro (rat liver mitochondria) | Decrease in respiration rates (5 mg/kg, 12 weeks) Inhibition of ATP synthase activity (0.1–0.5 mmol/L) | in vivo (rabbits with experimentally induces hypercholesterolemia) in vitro (rat heart mitochondria) | [188,189,190,191,192] | |
| Lisinopril | Attenuation of oxidative stress (40 mg/L in drinking water) Increase mitochondrial content (40 mg/L in drinking water) | in vivo (rat model of irradiation-induced kidney damage) | Reduction in mitochondrial respiration (50–10,000 ng/mL) | in vitro (Drosophila melanogaster strains) | [193,194] | |
| Direct Oral Anticoagulants | Rivaroxaban | Reduction in ROS generation (300 nM in vitro; 12 mg/kg/day, 28 days in vivo) Antioxidant effects (5.6 mM) | in vitro (advanced glycation end products-exposed proximal tubular cells); in vivo (intermittent hypoxia exposed mice) in vitro (rat kidney mitochondria) | Decrease in mitochondrial succinate dehydrogenase activity (1.4–2.8 mM) Increase ROS production (1.4–2.8 mM) Induction of mitochondrial swelling (1.4–2.8 mM) Reduction in mitochondrial membrane potential (1.4–2.8 mM) | in vitro (rat kidney mitochondria) | [195,196,197] |
| Diuretics | ||||||
| Epithelial sodium channel blockers | Amiloride | Attenuation of the mitochondrial membrane potential dissipation (50–200 μM) Inhibition of apoptosis (50–200 μM) | in vitro (rat articular chondrocytes) | Inhibition of mitochondrial NADH-quinone oxidoreductase (complex I) (5–100 µM) Inhibition of oxidative phosphorylation (10 µM) Increased mitochondrial fusion (10 µM) | in vitro (in bovine submitochondrial particles and in bacterial membranes) in vitro (clonal untransformed and cancer cells) | [198,199,200] |
| Statins | Enhancement of mitochondrial respiration (2.5–10 µM Simva) Increase in complex I and IV activity (2.5–10 µM Simva) | in vitro (peripheral blood mononuclear cells and platelets) | Reduction in coenzyme Q10 level (1–100 µM) Increase in ROS generation (25–700 µM) Inhibition of respiration (1–1000 µmol/L) Inhibition of respiratory chain complexes (1–1000 µmol/L) Uncoupling of oxidative phosphorylation (1–1000 µmol/L) Reduction in ATP production (1–1000 µmol/L) Induction of mitochondrial membrane depolarization (1–1000 µmol/L) Induction of mitochondrial apoptosis (1–1000 µmol/L) Induction of mitochondrial swelling (1–1000 µmol/L) Dysregulation of calcium metabolism (25–700 µM) Induction of fatty acid oxidation (Simva, 80 mg/day, 12 weeks) | in vitro (rat myoblasts, isolated rat skeletal muscle mitochondria, isolated endothelial mitochondria, rat hepatocytes, pancreas mitochondria, human platelets) in vivo | [201,202,203,204,205,206,207] | |
| Direct vasodilators | Hydralazine | Antioxidant properties (10–30 mg/kg) Inhibition of apoptosis (10–30 mg/kg) Inhibition of mitochondrial fission (1 µM) Preservation of mitochondrial fusion (1 µM) Promotion of mitochondrial biogenesis (5–20 µM) Increase in ETS complexes activity (5–20 µM) Increase in ATP production (5–20 µM) Enhancement of mitochondrial membrane potential (5–20 µM) Increase in mtDNA/nDNA ratio (5–20 µM) Increase in mitochondrial mass (5–20 µM) | in vivo (rat model of ischemia/reperfusion) in vitro (isolated murine cardiomyocytes subjected to ischemia/reperfusion injury) in vitro (human neuroblastoma SH-SY5Y and mouse myoblast C2C12 cells) | Inhibition of respiration (10–100 µM) Induction of apoptosis (200–600 µM) Increase ROS production (200–600 µM) Reduction in mitochondrial membrane potential (200–600 µM) | in vitro (human neuroblastoma SH-SY5Y and mouse myoblast C2C12 cells) in vitro (leukemic T cells) | [208,209,210] [211,212] |
| Sodium-glucose cotransporter 2 (SGLT2) inhibitors | Dapagliflozin | Antioxidant properties (10 µM) Reduction in ROS production (0.1–10 μM) Alteration of Ca2+ dynamics (0.01–10 μM) Decrease in mitochondrial swelling (1 mg/kg) Reduction in mitochondrial fission (1 mg/kg/day, 28 days) Increase in mitochondrial fusion (1 mg/kg/day, 28 days) Normalization of respiratory control ratio (1 mg/kg/day, 20 days) Decrease lipid peroxidation (1 mg/kg/day, 20 days) | in vitro (rat liver mitochondria) in vitro (human proximal tubular cells) in vivo (rat model of ischemia/reperfusion injury) in vivo (metabolic syndrome rats subjected to ischemia/reperfusion) in vivo (mice model of streptozocin induced diabetes) | Inhibition of mitochondrial respiration (20–50 µM) Reduction in calcium retention capacity (20–50 µM) | in vitro (rat liver mitochondria) | [213,214,215,216,217] |
| Canagliflozin | Improvement of mitochondrial biogenesis (60 mg/kg/day, 14 weeks) Improvement of fatty acid oxidation (60 mg/kg/day, 14 weeks) | in vivo (mice model of high-fat diet induced obesity) | Inhibition of the ETS complex I (10–50 µM) Inhibition of the ETS complex II (50 µM) | in vitro (human renal proximal tubule epithelial cell model system) in vitro (breast cancer cells) | [218,219] [220] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bețiu, A.M.; Noveanu, L.; Hâncu, I.M.; Lascu, A.; Petrescu, L.; Maack, C.; Elmér, E.; Muntean, D.M. Mitochondrial Effects of Common Cardiovascular Medications: The Good, the Bad and the Mixed. Int. J. Mol. Sci. 2022, 23, 13653. https://doi.org/10.3390/ijms232113653
Bețiu AM, Noveanu L, Hâncu IM, Lascu A, Petrescu L, Maack C, Elmér E, Muntean DM. Mitochondrial Effects of Common Cardiovascular Medications: The Good, the Bad and the Mixed. International Journal of Molecular Sciences. 2022; 23(21):13653. https://doi.org/10.3390/ijms232113653
Chicago/Turabian StyleBețiu, Alina M., Lavinia Noveanu, Iasmina M. Hâncu, Ana Lascu, Lucian Petrescu, Christoph Maack, Eskil Elmér, and Danina M. Muntean. 2022. "Mitochondrial Effects of Common Cardiovascular Medications: The Good, the Bad and the Mixed" International Journal of Molecular Sciences 23, no. 21: 13653. https://doi.org/10.3390/ijms232113653