
Density Functional Theory Study of Low-Dimensional (2D, 1D, 0D) Boron Nitride Nanomaterials Catalyzing Acetylene Acetate Reaction

{kind=link}
{kind=link}
{kind=link}
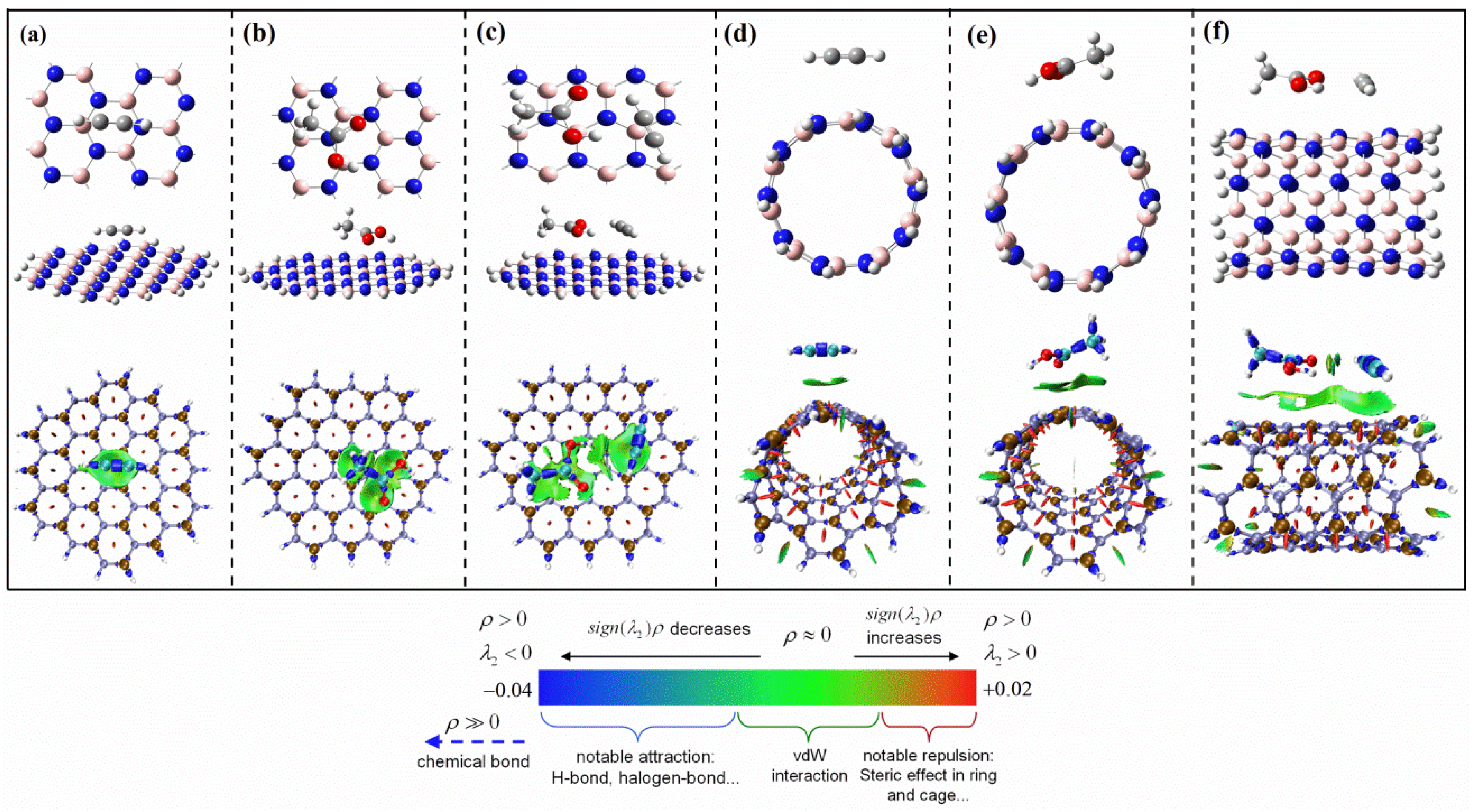{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Analysis of Structure and Properties of Catalysts
2.2. Adsorption of C2H2 and CH3COOH on BN Nanomaterials with Different Structures
2.3. Reaction Mechanism of Acetylene Acetotization Catalyzed by BN Nanomaterials with Different Structures
2.3.1. Reaction Process of Acetylene Acetylation on BN Nanosheets
2.3.2. Reaction Process of Acetylene Acetylation on BN Nanotubes
2.3.3. Reaction Process of Acetylene Acetylation on B12N12 Nanocages
2.4. Source Analysis of Catalytic Activity Differences of Different Boron Nitride Nanomaterials
2.4.1. Effects of Different Geometric Structures (Dimension) on Catalytic Properties of BN Nanomaterials
2.4.2. Effects of Different Reaction Sites on Catalytic Properties of BN Nanomaterials
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- García-Mota, M.; López, N. Template Effects in Vinyl Acetate Synthesis on PdAu Surface Alloys: A Density Functional Theory Study. J. Am. Chem. Soc. 2008, 130, 14406–14407. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Chen, M.S.; Goodman, D.W. Synthesis of vinyl acetate on Pd-based catalysts. Catal. Today 2007, 123, 77–85. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, Z.; Song, Z.; Zhou, J.; Ning, X.; Wu, L. Simulation and improvement of the separation process of synthesizing vinyl acetate by acetylene gas-phase method. Green Process. Synth. 2021, 10, 912–922. [Google Scholar] [CrossRef]
- Cui, X.M. Supply and Demand Status of Vinyl Acetate at Home and Abroad and its Development Prospect. Technol. Econ. Petrochem. 2021, 37, 14–20. [Google Scholar]
- Guo, X.; Chen, G.; Wang, W.; Li, J. Research progress of catalysts for synthesis of vinyl acetate from acetylene and acetic acid. Chem. Ind. Eng. Prog. 2017, 9, 3293–3299. [Google Scholar]
- Temkin, O.N.; Abanto-Chavez, H.I.; Hoang, K.B. Kinetic models of vinyl acetate synthesis on new-generation zinc acetate catalysts. React. Kinet. Catal. Lett. 2000, 41, 638–654. [Google Scholar] [CrossRef]
- Xu, H.; Yu, T.; Li, M. Zinc Acetate Immobilized on Mesoporous Materials by Acetate Ionic Liquids as Catalysts for Vinyl Acetate Synthesis. J. Chem. 2015, 2015, 1–5. [Google Scholar] [CrossRef]
- Yan, F.W.; Guo, C.Y.; Yan, F.; Li, F.B.; Qian, Q.L.; Yuan, G.Q. Vinyl acetate formation in the reaction of acetylene with acetic acid catalyzed by zinc acetate supported on porous carbon spheres. Russ. J. Phys. Chem. A 2010, 84, 796–801. [Google Scholar] [CrossRef]
- Fang, H.; Banjade, H.; Deepika; Jena, P. Realization of the Zn3+ oxidation state. Nanoscale 2021, 13, 14041–14048. [Google Scholar] [CrossRef]
- Norsko, J.K. Chemisorption on metal surfaces. Rep. Prog. Phys. 1999, 53, 1253–1295. [Google Scholar] [CrossRef]
- Zhou, S.; Pei, W.; Zhao, Y.; Yang, X.; Zhao, J. Low-dimensional non-metal catalysts: Principles for regulating p-orbital-dominated reactivity. NPJ Comput. Mater. 2021, 7, 186. [Google Scholar] [CrossRef]
- Vyskoil, J.; Mayorga-Martinez, C.C.; Szklová, K.; Sofer, Z.; Pumera, M. Hexagonal and Cubic Boron Nitride in Bulk and Nanosized Forms and Their Capacitive Behavior. ChemElectroChem 2020, 7, 74–77. [Google Scholar] [CrossRef] [Green Version]
- Esrafili, M.D. NO reduction by CO molecule over Si-doped boron nitride nanosheet: A dispersion-corrected DFT study. Chem. Phys. Lett. 2018, 695, 131–137. [Google Scholar] [CrossRef]
- He, X.; Gui, Y.; Xie, J.; Liu, X.; Wang, Q.; Tang, C. A DFT study of dissolved gas (C2H2, H2, CH4) detection in oil on CuO-modified BNNT. Appl. Surf. Sci. 2020, 500, 144030. [Google Scholar] [CrossRef]
- Oh, J.H.; Kim, M.; Lee, Y.H.; Yong, H.; Hong, S.H.; Park, S.S.; Kim, T.H.; Choi, S. Synthesis of cobalt boride nanoparticles and h-BN nanocage encapsulation by thermal plasma. Ceram. Int. 2020, 46, 28792–28799. [Google Scholar] [CrossRef]
- Li, L.H.; Chen, Y. Atomically Thin Boron Nitride: Unique Properties and Applications. Adv. Funct. Mater. 2016, 26, 2594–2608. [Google Scholar] [CrossRef]
- Elham, T.L.; Alireza, S.; Mohammad, T.B.; Masoud, B.J.; Sahar, M.R. Theoretical study on pure and doped B12N12 fullerenes as thiophene sensor. Adsorption 2018, 24, 585–593. [Google Scholar]
- Pino-Rios, R.; Chigo-Anota, E.; Shakerzadeh, E.; Cárdenas-Jirón, G. B12N12 cluster as a collector of noble gases: A quantum chemical study. Physica E 2020, 115, 113697. [Google Scholar] [CrossRef]
- Singh, K.; Kaur, M.; Chauhan, I.; Singh, H.; Awasthi, A.; Kumar, M.; Thakur, A.; Kumar, A. Effect of ammonia gas on electrical properties of boron nitride/nickel oxide (BN80/NiO20) nanocomposite. J. Mater. Sci.-Mater. Electron. 2021, 32, 5556–5566. [Google Scholar] [CrossRef]
- Singh, B.; Singh, K.; Kumar, M.; Thakur, S.; Kumar, A. Insights of preferred growth, elemental and morphological properties of BN/SnO2 composite for photocatalytic applications towards organic pollutants. Chem. Phys. 2020, 531, 110659. [Google Scholar] [CrossRef]
- Cortese, R.; Campisi, D.; Prestianni, A.; Duca, D. Alkane dehydrogenation on defective BN quasi-molecular nanoflakes: DFT studies. Mol. Catal. 2020, 493, 110891. [Google Scholar] [CrossRef]
- Li, P.; Li, H.; Pan, X.; Tie, K.; Cui, T.T.; Ding, M.Z.; Bao, X.H. Catalytically Active Boron Nitride in Acetylene Hydrochlorination. ACS Catal. 2017, 7, 8572–8577. [Google Scholar] [CrossRef]
- Mananghaya, M.R. Hydrogen saturation limit of Ti-doped BN nanotube with B-N defects: An insight from DFT calculations. Int. J. Hydrogen Energy 2018, 43, 10368–10375. [Google Scholar] [CrossRef]
- Sainsbury, T.; Satti, A.; May, P.; Wang, Z.; McGovern, I.; Gun, Y.K.; Coleman, J. Oxygen radical functionalization of boron nitride nanosheets. J. Am. Chem. Soc. 2012, 134, 18758. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Guan, J.; Zgierski, M.Z.; Kim, K.S.; Kingston, C.T.; Simard, B. Covalent Functionalization of Boron Nitride Nanotubes via Reduction Chemistry. ACS Nano 2015, 9, 12573–12582. [Google Scholar] [CrossRef]
- Nejati, K.; Hosseinian, A.; Vessally, E.; Bekhradnia, A.; Edjlali, L. A comparative DFT study on the interaction of cathinone drug with BN nanotubes, nanocages, and nanosheets. Appl. Surf. Sci. 2017, 422, 763–768. [Google Scholar] [CrossRef]
- Esmaeili, S.; Samadizadeh, M.; Khaleghian, M.; Shiraz, N.Z. DFT Study of Adsorption of (4E,6E)-4-(4-Hydroxyphenyldiazenyl) -N-((furan-2-yl)methylene)benzenamine on BN(6,6-8) Nanotube. Russ. J. Phys. Chem. 2020, 94, 778–788. [Google Scholar] [CrossRef]
- Sun, C.; Zhao, J.; Zhang, D.; Guo, H.; Wang, X.; Hu, H. Covalent functionalization of boron nitride nanosheets via reductive activation. Nanoscale 2020, 35, 1–29. [Google Scholar] [CrossRef]
- Nishiwaki, A.; Oku, T. Atomic structures of multi-walled boron nitride nanohorns. J. Electron Microsc. 2005, 54, 9–14. [Google Scholar] [CrossRef]
- Nishiwaki, A.; Oku, T.; Narita, I. Formation and atomic structures of boron nitride nanohorns. Sci. Technol. Adv. Mater. 2004, 5, 629–634. [Google Scholar] [CrossRef]
- Oku, T.; Kuno, M.; Kitahara, H.; Narita, I. Formation, atomic structures and properties of boron nitride and carbon nanocage fullerene materials. Int. J. Inorg. Mater. 2001, 3, 597–612. [Google Scholar] [CrossRef]
- Mamusi, F.; Farmanzadeh, D. Mechanism of ethanol steam reforming on B12N12 and Al12N12 nano-cages: A theoretical study. Mater. Today Commun. 2021, 30, 103014. [Google Scholar] [CrossRef]
- Xie, W.; Wang, J.; Wang, J.P.; Wu, X.C.; Wang, Z.G. High Angular Momentum Orbitals and Superatomic Characteristics of Boron-Nitrogen Cages. J. Phys. Chem. C 2020, 6, 3881–3885. [Google Scholar] [CrossRef]
- Strout, D.L. Structure and Stability of Boron Nitrides: Isomers of B12N12. J. Phys. Chem. A 2000, 104, 3364–3366. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Interaction Region Indicator: A Simple Real Space Function Clearly Revealing Both Chemical Bonds and Weak Interactions. Chem. Methods 2021, 1, 231–239. [Google Scholar] [CrossRef]
- Murdoch, J.R. What is the rate-limiting step of a multistep reaction? J. Chem. Educ. 1981, 58, 32–36. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Newton, M.D. Analysis of Koopmans’ Theorem. J. Chem. Phys. 1968, 48, 2825–2826. [Google Scholar] [CrossRef]
- Tian, L.; Chen, Q. Revealing Molecular Electronic Structure via Analysis of Valence Electron Density. Acta Mech. Solida Sin. 2017, 34, 503–513. [Google Scholar]
- Petersson, G.A.; Al-Laham, M.A. A complete basis set model chemistry. II. Open-shell systems and the total energies of the first-row atoms. J. Chem. Phys. 1991, 94, 6081–6090. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, J.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Schlegel, H.B.; Scalmani, G.; Barone, V.; Mennucci, B.; et al. Gaussian 09, Revision B.01; Gaussian Inc.: Wallingford, UK, 2010. [Google Scholar]
- Rhodes, C.J. Chapter 7. Electron spin resonance. Annu. Rep. Prog. Chem. Sect. C Phys. Chem. 1999, 95, 199–234. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Shermo: A General Code for Calculating Molecular Thermochemistry Properties. Comput. Theor. Chem. 2020, 1200, 113249. [Google Scholar] [CrossRef]
- Schlegel, H.B. Some thoughts on reaction-path following. J. Chem. Soc. Faraday Trans. 1994, 90, 1569–1574. [Google Scholar] [CrossRef]
- Stacho, L.L.; Domotor, G.; Ban, M.I. On the Elber-Karplus reaction path-following method and related procedures. Chem. Phys. Lett. 1999, 311, 328–334. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Kang, L.; Zhu, M. Density Functional Theory Study of Low-Dimensional (2D, 1D, 0D) Boron Nitride Nanomaterials Catalyzing Acetylene Acetate Reaction. Int. J. Mol. Sci. 2022, 23, 9997. https://doi.org/10.3390/ijms23179997
Zhang X, Kang L, Zhu M. Density Functional Theory Study of Low-Dimensional (2D, 1D, 0D) Boron Nitride Nanomaterials Catalyzing Acetylene Acetate Reaction. International Journal of Molecular Sciences. 2022; 23(17):9997. https://doi.org/10.3390/ijms23179997
Chicago/Turabian StyleZhang, Xunchao, Lihua Kang, and Mingyuan Zhu. 2022. "Density Functional Theory Study of Low-Dimensional (2D, 1D, 0D) Boron Nitride Nanomaterials Catalyzing Acetylene Acetate Reaction" International Journal of Molecular Sciences 23, no. 17: 9997. https://doi.org/10.3390/ijms23179997