
The Kinesin Gene KIF26B Modulates the Severity of Post-Traumatic Heterotopic Ossification

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
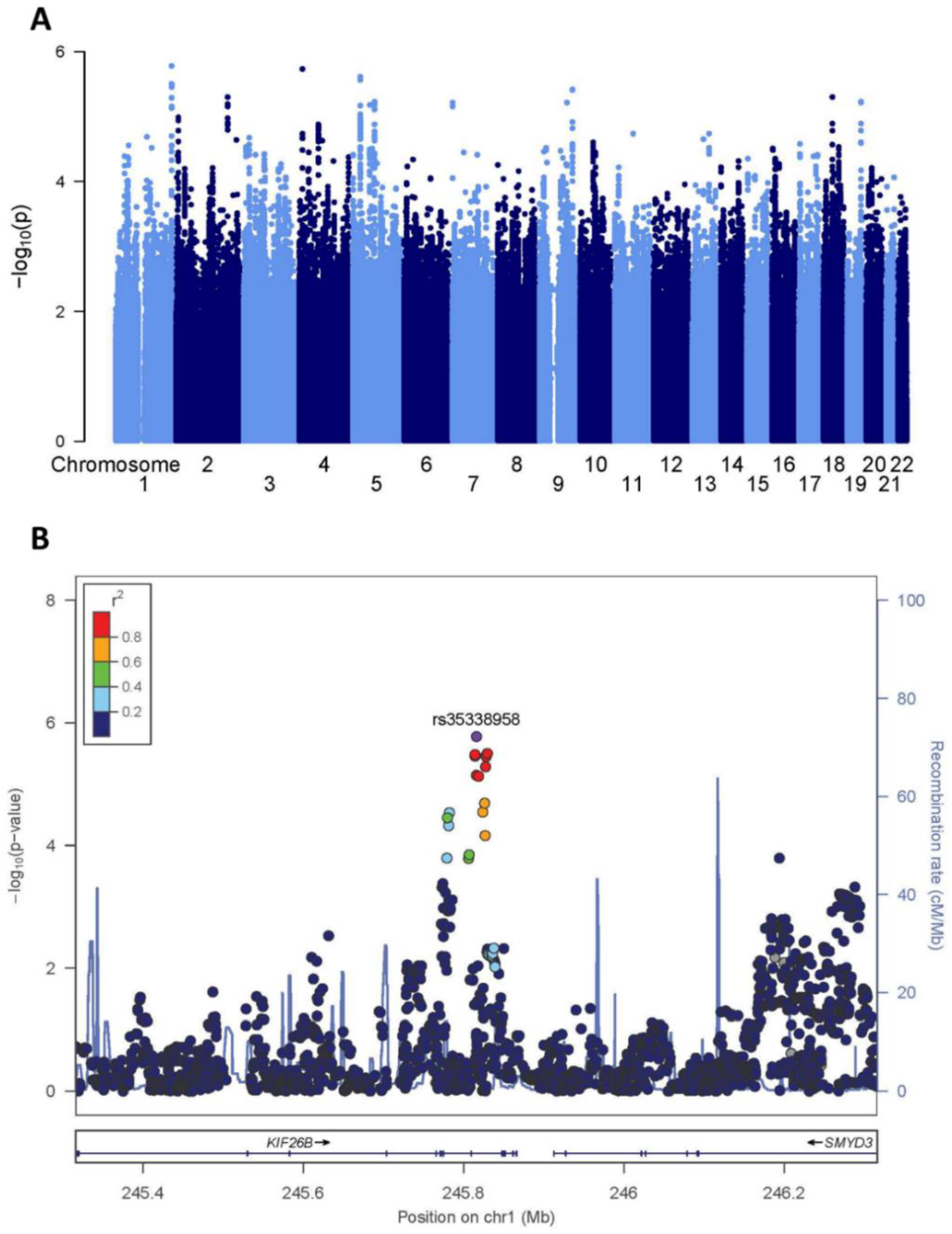
2.1. Screening GWAS
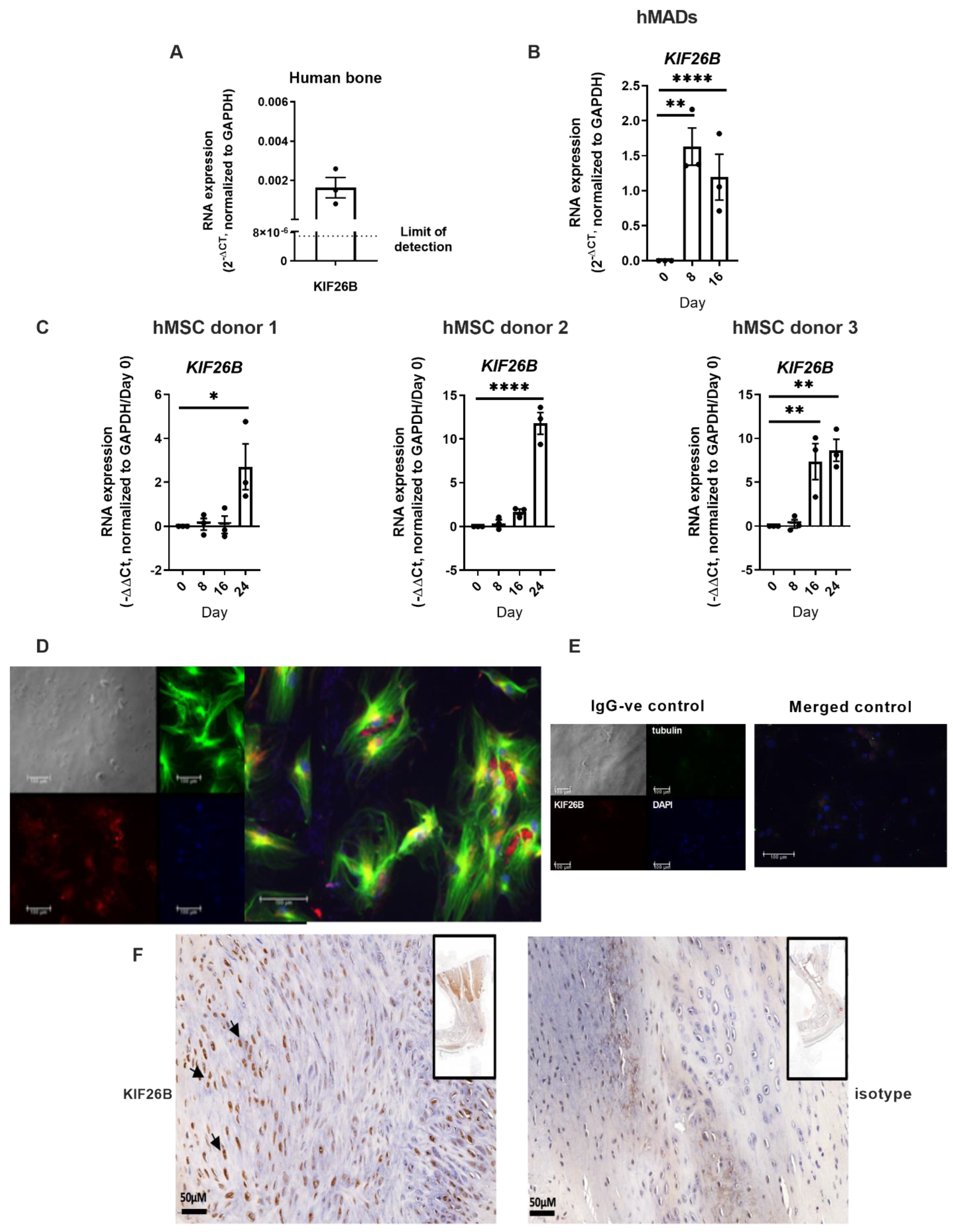
2.2. KIF26B Is Expressed in Human Bone, in BMP2 Stimulated Human Mesenchymal Stem Cells, Co-Localises with Microtubules in the Cytoskeleton, and Is Induced in a Mammalian Post-traumatic HO Model
2.3. Knockout of Murine Kif26b Prevents BMP2-Mediated Mineralisation of C2C12 Myoblasts
2.4. Kif26b Knockout Disrupts the Mineralisation and Trans-Differentiation of C2C12 Myoblasts by Modulating the Expression of Osteogenic Genes
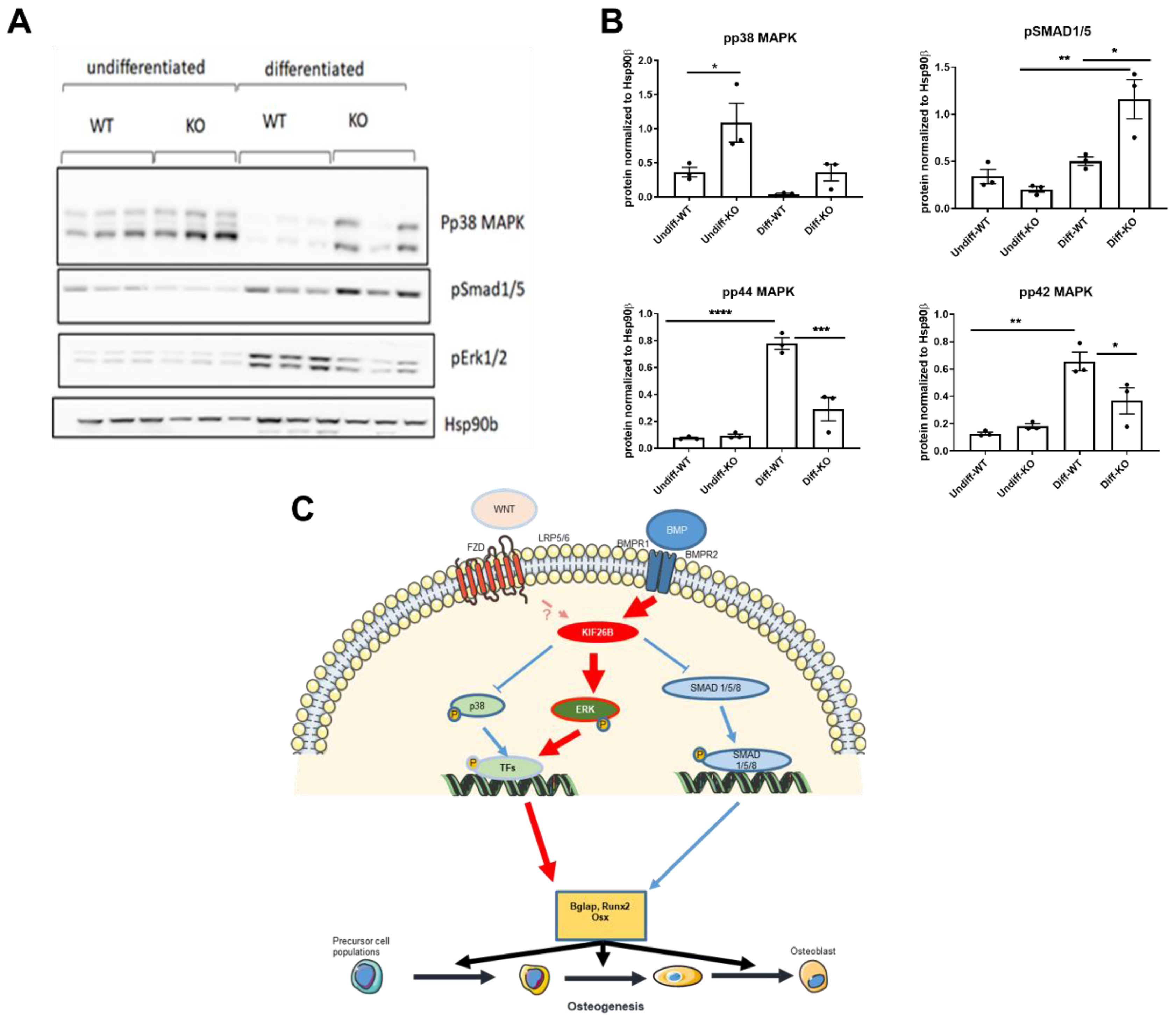
2.5. KIF26B Deficiency Inhibits ERK MAP Kinase Activation during Osteogenesis, Whilst Augmenting p38 and SMAD 1/5/8 Phosphorylation
3. Discussion
4. Materials and Methods
4.1. Study Oversight and Populations
4.2. Screening GWAS
4.3. Cell Culture
4.4. CRISPR-Cas9 Knockout
4.5. Osteogenic Differentiation
4.6. RNA Isolation and RT-qPCR
4.7. Protein Isolation and Western Blotting
4.8. Alizarin Red S Staining
4.9. Immunocytochemistry
4.10. Immunohistochemistry
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Forsberg, J.A.; Pepek, J.M.; Wagner, S.; Wilson, K.; Flint, J.; Andersen, R.C.; Tadaki, D.; Gage, F.A.; Stojadinovic, A.; Elster, E.A. Heterotopic ossification in high-energy wartime extremity injuries: Prevalence and risk factors. J. Bone Jt. Surg. Am. 2009, 91, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Potter, B.K.; Burns, T.C.; Lacap, A.P.; Granville, R.R.; Gajewski, D.A. Heterotopic ossification following traumatic and combat-related amputations. Prevalence, risk factors, and preliminary results of excision. J. Bone Jt. Surg. Am. 2007, 89, 476–486. [Google Scholar] [CrossRef] [PubMed]
- DeLee, J.; Ferrari, A.; Charnley, J. Ectopic bone formation following low friction arthroplasty of the hip. Clin. Orthop. Relat. Res. 1976, 121, 53–59. [Google Scholar] [CrossRef]
- Neal, B.; Gray, H.; MacMahon, S.; Dunn, L. Incidence of heterotopic bone formation after major hip surgery. ANZ J. Surg. 2002, 72, 808–821. [Google Scholar] [CrossRef]
- Garland, D.E. Clinical observations on fractures and heterotopic ossification in the spinal cord and traumatic brain injured populations. Clin. Orthop. Relat. Res. 1988, 233, 86–101. [Google Scholar] [CrossRef]
- Richards, A.M.; Klaassen, M.F. Heterotopic ossification after severe burns: A report of three cases and review of the literature. Burns 1997, 23, 64–68. [Google Scholar] [CrossRef]
- Edwards, D.S.; Clasper, J.C. Heterotopic ossification: A systematic review. J. R. Army Med. Corps 2015, 161, 315–321. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, F.; Chen, W.; Zhang, Q.; Liu, S.; Zhang, Y. Incidence and risk factors for heterotopic ossification after total hip arthroplasty: A meta-analysis. Arch. Orthop. Trauma Surg. 2015, 135, 1307–1314. [Google Scholar] [CrossRef]
- Davis, G.; Patel, R.P.; Tan, T.L.; Alijanipour, P.; Naik, T.U.; Parvizi, J. Ethnic differences in heterotopic ossification following total hip arthroplasty. Bone Jt. J. 2016, 98-B, 761–766. [Google Scholar] [CrossRef]
- Felix-Ilemhenbhio, F.; Pickering, G.A.E.; Kiss-Toth, E.; Wilkinson, J.M. Pathophysiology and Emerging Molecular Therapeutic Targets in Heterotopic Ossification. Int. J. Mol. Sci. 2022, 23, 6983. [Google Scholar] [CrossRef]
- Eisenstein, N.; Stapley, S.; Grover, L. Post-Traumatic Heterotopic Ossification: An Old Problem in Need of New Solutions. J. Orthop. Res. 2018, 36, 1061–1068. [Google Scholar] [CrossRef]
- Joice, M.; Vasileiadis, G.I.; Amanatullah, D.F. Non-steroidal anti-inflammatory drugs for heterotopic ossification prophylaxis after total hip arthroplasty: A systematic review and meta-analysis. Bone Jt. J. 2018, 100-B, 915–922. [Google Scholar] [CrossRef]
- Hoyt, B.W.; Pavey, G.J.; Potter, B.K.; Forsberg, J.A. Heterotopic ossification and lessons learned from fifteen years at war: A review of therapy, novel research, and future directions for military and civilian orthopaedic trauma. Bone 2018, 109, 3–11. [Google Scholar] [CrossRef]
- Lees-Shepard, J.B.; Nicholas, S.E.; Stoessel, S.J.; Devarakonda, P.M.; Schneider, M.J.; Yamamoto, M.; Goldhamer, D.J. Palovarotene reduces heterotopic ossification in juvenile FOP mice but exhibits pronounced skeletal toxicity. Elife 2018, 7, e40814. [Google Scholar] [CrossRef]
- Hatzikotoulas, K.; Pickering, G.A.E.; Clark, M.J.; Felix-Ilemhenbhio, F.; Kocsy, K.; Simpson, J.; MacInnes, S.J.; Koprulu, M.; Southam, L.; Bellantuono, I.; et al. Genome-wide association and functional analyses identify CASC20 and KIF26B as target loci in heterotopic ossification. bioRxiv 2019, 845958. [Google Scholar] [CrossRef]
- Susman, M.W.; Karuna, E.P.; Kunz, R.C.; Gujral, T.S.; Cantu, A.V.; Choi, S.S.; Jong, B.Y.; Okada, K.; Scales, M.K.; Hum, J.; et al. Kinesin superfamily protein Kif26b links Wnt5a-Ror signaling to the control of cell and tissue behaviors in vertebrates. Elife 2017, 6, e26509. [Google Scholar] [CrossRef]
- Karuna, E.P.; Choi, S.S.; Scales, M.K.; Hum, J.; Cohen, M.; Fierro, F.A.; Ho, H.H. Identification of a WNT5A-Responsive Degradation Domain in the Kinesin Superfamily Protein KIF26B. Genes 2018, 9, 196. [Google Scholar] [CrossRef]
- Rai, M.F.; Schmidt, E.J.; Hashimoto, S.; Cheverud, J.M.; Sandell, L.J. Genetic loci that regulate ectopic calcification in response to knee trauma in LG/J by SM/J advanced intercross mice. J. Orthop. Res. 2015, 33, 1412–1423. [Google Scholar] [CrossRef]
- Rodriguez, A.M.; Elabd, C.; Amri, E.Z.; Ailhaud, G.; Dani, C. The human adipose tissue is a source of multipotent stem cells. Biochimie 2005, 87, 125–128. [Google Scholar] [CrossRef]
- Baumgart, S.J.; Najafova, Z.; Hossan, T.; Xie, W.; Nagarajan, S.; Kari, V.; Ditzel, N.; Kassem, M.; Johnsen, S.A. CHD1 regulates cell fate determination by activation of differentiation-induced genes. Nucleic. Acids. Res. 2017, 45, 7722–7735. [Google Scholar] [CrossRef]
- Lin, L.; Shen, Q.; Xue, T.; Yu, C. Heterotopic ossification induced by Achilles tenotomy via endochondral bone formation: Expression of bone and cartilage related genes. Bone 2010, 46, 425–431. [Google Scholar] [CrossRef]
- Rooney, P.; Grant, M.E.; McClure, J. Endochondral ossification and de novo collagen synthesis during repair of the rat Achilles tendon. Matrix 1992, 12, 274–281. [Google Scholar] [CrossRef]
- Katagiri, T.; Yamaguchi, A.; Komaki, M.; Abe, E.; Takahashi, N.; Ikeda, T.; Rosen, V.; Wozney, J.M.; Fujisawa-Sehara, A.; Suda, T. Bone morphogenetic protein-2 converts the differentiation pathway of C2C12 myoblasts into the osteoblast lineage. J. Cell Biol. 1994, 127 Pt 1, 1755–1766. [Google Scholar] [CrossRef]
- Pohl, F.; Hassel, S.; Nohe, A.; Flentje, M.; Knaus, P.; Sebald, W.; Koelbl, O. Radiation-induced suppression of the Bmp2 signal transduction pathway in the pluripotent mesenchymal cell line C2C12: An in vitro model for prevention of heterotopic ossification by radiotherapy. Radiat. Res. 2003, 159, 345–350. [Google Scholar] [CrossRef]
- Matsushita, T.; Chan, Y.Y.; Kawanami, A.; Balmes, G.; Landreth, G.E.; Murakami, S. Extracellular signal-regulated kinase 1 (ERK1) and ERK2 play essential roles in osteoblast differentiation and in supporting osteoclastogenesis. Mol. Cell Biol. 2009, 29, 5843–5857. [Google Scholar] [CrossRef]
- Xiao, G.; Jiang, D.; Thomas, P.; Benson, M.D.; Guan, K.; Karsenty, G.; Franceschi, R.T. MAPK pathways activate and phosphorylate the osteoblast-specific transcription factor, Cbfa1. J. Biol. Chem. 2000, 275, 4453–4459. [Google Scholar] [CrossRef]
- Xiao, G.; Gopalakrishnan, R.; Jiang, D.; Reith, E.; Benson, M.D.; Franceschi, R.T. Bone morphogenetic proteins, extracellular matrix, and mitogen-activated protein kinase signaling pathways are required for osteoblast-specific gene expression and differentiation in MC3T3-E1 cells. J. Bone Miner Res. 2002, 17, 101–110. [Google Scholar] [CrossRef]
- Xiao, G.; Jiang, D.; Gopalakrishnan, R.; Franceschi, R.T. Fibroblast growth factor 2 induction of the osteocalcin gene requires MAPK activity and phosphorylation of the osteoblast transcription factor, Cbfa1/Runx2. J. Biol. Chem. 2002, 277, 36181–36187. [Google Scholar] [CrossRef]
- Afzal, F.; Pratap, J.; Ito, K.; Ito, Y.; Stein, J.L.; van Wijnen, A.J.; Stein, G.S.; Lian, J.B.; Javed, A. Smad function and intranuclear targeting share a Runx2 motif required for osteogenic lineage induction and BMP2 responsive transcription. J. Cell Physiol. 2005, 204, 63–72. [Google Scholar] [CrossRef]
- Hammond, R.K.; Pahl, M.C.; Su, C.; Cousminer, D.L.; Leonard, M.E.; Lu, S.; Doege, C.A.; Wagley, Y.; Hodge, K.M.; Lasconi, C.; et al. Biological constraints on GWAS SNPs at suggestive significance thresholds reveal additional BMI loci. Elife 2021, 10, e62206. [Google Scholar] [CrossRef]
- Panoutsopoulou, K.; Thiagarajah, S.; Zengini, E.; Day-Williams, A.G.; Ramos, Y.F.; Meessen, J.M.; Huetink, K.; Nelissen, R.G.; Southam, L.; Rayner, N.W.; et al. Radiographic endophenotyping in hip osteoarthritis improves the precision of genetic association analysis. Ann. Rheum. Dis. 2016, 76, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Govaere, O.; Petersen, S.K.; Martinez-Lopez, N.; Wouters, J.; Van Haele, M.; Mancina, R.M.; Jamialahmadi, O.; Bilkei-Gorzo, O.; Lassen, P.B.; Darlay, R.; et al. Macrophage scavenger receptor 1 mediates lipid-induced inflammation in non-alcoholic fatty liver disease. J. Hepatol. 2022, 76, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Gibson, J.; Russ, T.C.; Clarke, T.K.; Howard, D.M.; Hillary, R.F.; Evans, K.L.; Walker, R.M.; Bermingham, M.L.; Morris, S.W.; Campbell, A.; et al. A meta-analysis of genome-wide association studies of epigenetic age acceleration. PLoS Genet. 2019, 15, e1008104. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Duan, X.; Cai, L.; Zhang, W.; Silva, M.J.; Brophy, R.H.; Rai, M.F. KIF26B silencing prevents osseous transdifferentiation of progenitor/stem cells and attenuates ectopic calcification in a murine model. J. Bone Miner Res. 2022, 37, 349–368. [Google Scholar] [CrossRef]
- Toom, A.; Arend, A.; Gunnarsson, D.; Ulfsparre, R.; Suutre, S.; Haviko, T.; Selstam, G. Bone formation zones in heterotopic ossifications: Histologic findings and increased expression of bone morphogenetic protein 2 and transforming growth factors beta2 and beta3. Calcif. Tissue Int. 2007, 80, 259–267. [Google Scholar] [CrossRef]
- Suutre, S.; Toom, A.; Arend, A.; Selstam, G. Bone tissue content of TGF-beta2 changes with time in human heterotopic ossification after total hip arthroplasty. Growth Factors 2009, 27, 114–120. [Google Scholar] [CrossRef]
- Akhurst, R.J.; FitzPatrick, D.R.; Gatherer, D.; Lehnert, S.A.; Millan, F.A. Transforming growth factor betas in mammalian embryogenesis. Prog. Growth Factor Res. 1990, 2, 153–168. [Google Scholar] [CrossRef]
- Salazar, V.S.; Gamer, L.W.; Rosen, V. BMP signalling in skeletal development, disease and repair. Nat. Rev. Endocrinol. 2016, 12, 203–221. [Google Scholar] [CrossRef]
- Chen, G.; Deng, C.; Li, Y.P. TGF-beta and BMP signaling in osteoblast differentiation and bone formation. Int. J. Biol. Sci. 2012, 8, 272–288. [Google Scholar] [CrossRef]
- Evans, K.N.; Potter, B.K.; Brown, T.S.; Davis, T.A.; Elster, E.A.; Forsberg, J.A. Osteogenic gene expression correlates with development of heterotopic ossification in war wounds. Clin. Orthop. Relat. Res. 2014, 472, 396–404. [Google Scholar] [CrossRef]
- Ju, C.; Lv, Z.; Zhang, C.; Jiao, Y. Regulatory effect of miR-421 on humeral fracture and heterotopic ossification in elderly patients. Exp. Ther. Med. 2019, 17, 1903–1911. [Google Scholar] [CrossRef]
- Yu, P.B.; Deng, D.Y.; Lai, C.S.; Hong, C.C.; Cuny, G.D.; Bouxsein, M.L.; Hong, D.W.; McManus, P.M.; Katagiri, T.; Sachidanandan, C.; et al. BMP type I receptor inhibition reduces heterotopic [corrected] ossification. Nat. Med. 2008, 14, 1363–1369. [Google Scholar] [CrossRef]
- Gorrell, R.E.; Totten, M.H.; Schoerning, L.J.; Newby, J.B.; Geyman, L.J.; Lawless, W.G.; Hum, J.M.; Lowery, J.W. Identification of a bone morphogenetic protein type 2 receptor neutralizing antibody. BMC Res. Notes 2019, 12, 331. [Google Scholar] [CrossRef]
- Terabayashi, T.; Sakaguchi, M.; Shinmyozu, K.; Ohshima, T.; Johjima, A.; Ogura, T.; Miki, H.; Nishinakamura, R. Phosphorylation of Kif26b promotes its polyubiquitination and subsequent proteasomal degradation during kidney development. PLoS ONE 2012, 7, e39714. [Google Scholar] [CrossRef]
- Guillabert-Gourgues, A.; Jaspard-Vinassa, B.; Bats, M.L.; Sewduth, R.N.; Franzl, N.; Peghaire, C.; Jeanningros, S.; Moreau, C.; Roux, E.; Larrieu-Lahargue, F.; et al. Kif26b controls endothelial cell polarity through the Dishevelled/Daam1-dependent planar cell polarity-signaling pathway. Mol. Biol. Cell 2016, 27, 941–953. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Uehara, S.; Udagawa, N.; Takahashi, N. Regulation of bone metabolism by Wnt signals. J. Biochem. 2016, 159, 387–392. [Google Scholar] [CrossRef]
- Lee, K.S.; Kim, H.J.; Li, Q.L.; Chi, X.Z.; Ueta, C.; Komori, T.; Wozney, J.M.; Kim, E.G.; Choi, J.Y.; Ryoo, H.M.; et al. Runx2 is a common target of transforming growth factor beta1 and bone morphogenetic protein 2, and cooperation between Runx2 and Smad5 induces osteoblast-specific gene expression in the pluripotent mesenchymal precursor cell line C2C12. Mol. Cell Biol. 2000, 20, 8783–8792. [Google Scholar] [CrossRef]
- Li, Z.; Xu, Z.; Duan, C.; Liu, W.; Sun, J.; Han, B. Role of TCF/LEF Transcription Factors in Bone Development and Osteogenesis. Int. J. Med. Sci. 2018, 15, 1415–1422. [Google Scholar] [CrossRef]
- Gaur, T.; Lengner, C.J.; Hovhannisyan, H.; Bhat, R.A.; Bodine, P.V.; Komm, B.S.; Javed, A.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; et al. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J. Biol. Chem. 2005, 280, 33132–33140. [Google Scholar] [CrossRef]
- Guicheux, J.; Lemonnier, J.; Ghayor, C.; Suzuki, A.; Palmer, G.; Caverzasio, J. Activation of p38 mitogen-activated protein kinase and c-Jun-NH2-terminal kinase by BMP-2 and their implication in the stimulation of osteoblastic cell differentiation. J. Bone Miner Res. 2003, 18, 2060–2068. [Google Scholar] [CrossRef]
- Vinals, F.; Lopez-Rovira, T.; Rosa, J.L.; Ventura, F. Inhibition of PI3K/p70 S6K and p38 MAPK cascades increases osteoblastic differentiation induced by BMP-2. FEBS Lett. 2002, 510, 99–104. [Google Scholar] [CrossRef]
- Huang, R.L.; Yuan, Y.; Tu, J.; Zou, G.M.; Li, Q. Opposing TNF-alpha/IL-1beta- and BMP-2-activated MAPK signaling pathways converge on Runx2 to regulate BMP-2-induced osteoblastic differentiation. Cell Death Dis. 2014, 5, e1187. [Google Scholar] [CrossRef]
- Rosina, M.; Langone, F.; Giuliani, G.; Cerquone Perpetuini, A.; Reggio, A.; Calderone, A.; Fuoco, C.; Castagnoli, L.; Gargioli, C.; Cesareni, G. Osteogenic differentiation of skeletal muscle progenitor cells is activated by the DNA damage response. Sci. Rep. 2019, 9, 5447. [Google Scholar] [CrossRef]
- Yaffe, D.; Saxel, O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 1977, 270, 725–727. [Google Scholar] [CrossRef]
- Blau, H.M.; Pavlath, G.K.; Hardeman, E.C.; Chiu, C.P.; Silberstein, L.; Webster, S.G.; Miller, S.C.; Webster, C. Plasticity of the differentiated state. Science 1985, 230, 758–766. [Google Scholar] [CrossRef]
- Cocks, M.; Mohan, A.; Meyers, C.A.; Ding, C.; Levi, B.; McCarthy, E.; James, A.W. Vascular patterning in human heterotopic ossification. Hum. Pathol. 2017, 63, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Davies, O.G.; Liu, Y.; Player, D.J.; Martin, N.R.W.; Grover, L.M.; Lewis, M.P. Defining the Balance between Regeneration and Pathological Ossification in Skeletal Muscle Following Traumatic Injury. Front. Physiol. 2017, 8, 194. [Google Scholar] [CrossRef] [PubMed]
- Lammers, L.; Naujoks, C.; Berr, K.; Depprich, R.; Kubler, N.; Meyer, U.; Langenbach, F.; Luttenberg, B.; Kogler, G.; Wiesmann, H.P.; et al. Impact of DAG stimulation on mineral synthesis, mineral structure and osteogenic differentiation of human cord blood stem cells. Stem Cell Res. 2012, 8, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Langenbach, F.; Handschel, J. Effects of dexamethasone, ascorbic acid and beta-glycerophosphate on the osteogenic differentiation of stem cells in vitro. Stem Cell Res. Ther. 2013, 4, 117. [Google Scholar] [CrossRef]
- MacInnes, S.J.; Hatzikotoulas, K.; Fenstad, A.M.; Shah, K.; Southam, L.; Tachmazidou, I.; Hallan, G.; Dale, H.; Panoutsopoulou, K.; Furnes, O.; et al. The 2018 Otto Aufranc Award: How Does Genome-wide Variation Affect Osteolysis Risk After THA? Clin. Orthop. Relat. Res. 2019, 477, 297–309. [Google Scholar] [CrossRef]
- Brooker, A.F.; Bowerman, J.W.; Robinson, R.A.; Riley, L.H., Jr. Ectopic ossification following total hip arthroplasty. Incidence and method of classification. J. Bone Jt. Surg. 1973, 55-A, 1629–1632. [Google Scholar] [CrossRef]
- arcOGEN Consortium; arcOGEN Collaborators. Identification of new susceptibility loci for osteoarthritis (arcOGEN): A genome-wide association study. Lancet 2012, 380, 815–823. [Google Scholar] [CrossRef]
- Marchini, J.; Howie, B.; Myers, S.; McVean, G.; Donnelly, P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 2007, 39, 906–913. [Google Scholar] [CrossRef]
- Genomes Project, C.; Abecasis, G.R.; Altshuler, D.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Gibbs, R.A.; Hurles, M.E.; McVean, G.A. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef]
- Pavlou, G.; Salhab, M.; Murugesan, L.; Jallad, S.; Petsatodis, G.; West, R.; Tsiridis, E. Risk factors for heterotopic ossification in primary total hip arthroplasty. Hip Int. 2012, 22, 50–55. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Steinberg, J.; Ritchie, G.R.S.; Roumeliotis, T.I.; Jayasuriya, R.L.; Brooks, R.A.; Binch, A.L.A.; Shah, K.M.; Pardo, M.; LeMaitre, C.L.; Ramos, Y.F.; et al. Integrative epigenomics, transcriptomics and proteomics of patient chondrocytes reveal genes and pathways involved in osteoarthritis. biorxiv 2017, 7, 8935. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pickering, G.A.E.; Felix-Ilemhenbhio, F.; Clark, M.J.; Kocsy, K.; Simpson, J.; Bellantuono, I.; Gartland, A.; Wilkinson, J.M.; Hatzikotoulas, K.; Kiss-Toth, E. The Kinesin Gene KIF26B Modulates the Severity of Post-Traumatic Heterotopic Ossification. Int. J. Mol. Sci. 2022, 23, 9203. https://doi.org/10.3390/ijms23169203
Pickering GAE, Felix-Ilemhenbhio F, Clark MJ, Kocsy K, Simpson J, Bellantuono I, Gartland A, Wilkinson JM, Hatzikotoulas K, Kiss-Toth E. The Kinesin Gene KIF26B Modulates the Severity of Post-Traumatic Heterotopic Ossification. International Journal of Molecular Sciences. 2022; 23(16):9203. https://doi.org/10.3390/ijms23169203
Chicago/Turabian StylePickering, George A. E., Favour Felix-Ilemhenbhio, Matthew J. Clark, Klaudia Kocsy, Jonathan Simpson, Ilaria Bellantuono, Alison Gartland, Jeremy Mark Wilkinson, Konstantinos Hatzikotoulas, and Endre Kiss-Toth. 2022. "The Kinesin Gene KIF26B Modulates the Severity of Post-Traumatic Heterotopic Ossification" International Journal of Molecular Sciences 23, no. 16: 9203. https://doi.org/10.3390/ijms23169203