
The Common Cellular Events in the Neurodegenerative Diseases and the Associated Role of Endoplasmic Reticulum Stress


Abstract
:1. Introduction
2. The Neurodegenerative Diseases and Their Common Cellular Events
2.1. The Basic Etiologies of the Neurodegenerative Diseases
2.1.1. Alzheimer’s Disease
2.1.2. Parkinson’s Disease
2.1.3. Huntington’s Disease
2.1.4. Amyotrophic Lateral Sclerosis
2.1.5. Prion Disease
2.2. The Common Cellular Events in the Neurodegenerative Diseases
2.2.1. Calcium Dyshomeostasis
2.2.2. Neuroinflammation
2.2.3. Autophagy and Mitophagy
3. ER Protein Quality Control and ER Stress
3.1. Key Players in the UPR Pathway
3.2. ERAD Pathway and Autophagy-Lysosomal Pathway
3.3. ER stress, Inflammation, and Cell Death
3.4. ER Dysfunction and Neurodegenerative Diseases
4. Alzheimer’s Disease
4.1. ER UPR on Neuronal Pathophysiology in AD
4.2. The Age-Associated Decline of ER Capacity and AD
4.3. UPR Components and Their Role in Memory, Cognition, and Synaptic Plasticity in AD
5. Parkinson’s Disease
5.1. ER Stress and α-Synuclein-Related PD Pathology
5.2. Genetic Mutations in PD Related to ER Stress
5.3. Mitochondrial Dysfunction and Calcium Dyshomeostasis in ER Stress-Related PD Pathophysiology
6. Huntington’s Disease
6.1. Mutant Huntington Protein in the Pathogenesis of HD
6.2. Impact of Pathogenic Mutant Huntingtins on ER Stress
7. Amyotrophic Lateral Sclerosis
Proteostasis Disturbances and ER Stress in ALS Pathology
8. Prion Disease
ER Stress and UPS Impairment in Prion Diseases
9. Future Perspectives—Therapeutic Strategies for Targeting ER Stress and Neurodegenerative Diseases
9.1. Targeting Protein Misfolding and ER Stress
9.2. Targeting UPR Components
9.3. Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-PBA | 4-phenylbutyric acid |
| 6-OHDA | 6-hydroxy-dopamine |
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| AMPK | AMP-activated protein kinase |
| APP | Amyloid-β precursor protein |
| AR-JP | Autosomal recessive juvenile parkinsonism |
| ASK1 | Apoptotic-signaling kinase-1 |
| ATF4 | Activating transcription factor-4 |
| ATG | AuTophaGy(ATG)-related proteins |
| ATP13A2 | ATPase Cation Transporting 13A2 |
| Aβ | Amyloid-β |
| BAK | Bcl-2 homologous antagonist killer |
| BAX | Bcl-2-associated X protein |
| BDNF | Brain-derived growth factor |
| BSE | Bovine spongiform encephalopathy |
| BST1 | Bone Marrow Stromal Cell Antigen 1 |
| C/EBPa | CCAAT/enhancer binding protein a |
| C9ORF72 | Chromosome 9 open reading frame |
| CHIP | Carboxy-terminus of Hsc70 interacting protein |
| CHOP | C/EBP homologous protein |
| CJD | Creutzfeldt-Jakob disease |
| CREB | cAMP-response element binding protein |
| eIF2a | Eukaryotic translation initiation factor α |
| ER | Endoplasmic reticulum |
| ERAD | ER-associated protein degradation |
| ERK | Extracellular signal-regulated kinase |
| ERp57 | ER protein 57 |
| FKBP51 | FK506 binding protein 51 kDa |
| FoxO1 | Forkhead box protein O1 |
| FUS | Fused in sarcoma |
| GABA | γ-Aminobutyric acid |
| GADD153 | Growth arrest and DNA-damage-inducible 153 |
| GBA | Glucosylceramidase Beta |
| GCN2 | General control nonderepressible 2 |
| Gp78 | Glycoprotein 78 |
| GRP78/BiP | 78 KDa glucose-regulated protein/ |
| HD | Huntington’s disease |
| Herp | Homocysteine-induced ER protein |
| Hrd1 | HMG-CoA reductase degradation protein 1 |
| Hsp90 | Heat shock proteins 90 |
| HTT/mHtt | Huntington/mutant Huntington |
| IKK/NFκB | IkappaB kinase/Nuclear factor kappa B |
| IL- | Interleukin- |
| IP3R | Inositol 1,4,5-triphosphate receptor |
| IRE1 | Inositol requiring protein-1 |
| ISRIB | Integrated stress response inhibitor |
| ITPKB | Inositol-Trisphosphate 3-Kinase B |
| JAK/STAT | Janus kinase/Signal transducer and activator of transcription |
| JNK/c-Jun | c-Jun N-terminal kinase/c-Jun |
| LB | Lew bodies |
| LRRK2 | Leucine-rich repeat kinase 2 |
| MAM | Mitochondria-associated ER membrane |
| MAPK | Mitogen-activated protein kinase |
| MCU | Mitochondrial calcium uniporter |
| MICU | Mitochondrial Calcium Uptake |
| MPP+ | 1-methyl-4-phenylpyridinium |
| MPTP | N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| mTOR | Mechanistic target of rapamycin |
| NLRP3 | NLR Family Pyrin Domain Containing 3 |
| NMDAR | N-methyl-d-aspartate receptor |
| Npl4 | Nuclear protein localization protein 4 |
| Pael-R | Parkin-associated endothelin-like receptor |
| PD | Parkinson’s disease |
| PDI | Protein disulfide isomerase |
| PERK | Protein kinase RNA-like ER kinase |
| PI3K | Phosphoinositide 3-kinase |
| PINK1 | PTEN-induced kinase 1 |
| PKR | Protein kinase R |
| PLA2G6 | Phospholipase A2 Group VI |
| PolyQ | Polyglutamine |
| PrPC/PrPSc | Cellular α-helical prion proteins/Scrapie isoform of prion protein |
| PS1 | Presenilin1 |
| RIDD | Regulated IRE1-dependent decay |
| RyR | Ryanodine receptor |
| S1P/S2P | Site 1 protease/Site 2 protease |
| SNpc | Substantia nigra pars compacta |
| SOD1 | Superoxide dismutase |
| TARDBP/TDP-43 | TAR DNA binding protein |
| TFEB | Transcription factor EB |
| TNF- | Tumor necrosis factor- |
| TPR | Translocated promoter region |
| TRAF2 | Tumor necrosis factor receptor-associated factor 2 |
| TSEs | Transmissible spongiform encephalopathies |
| TUDCA | Tauroursodeoxycholic acid |
| TXNIP | Thioredoxin interacting protein |
| Ufd1 | Ubiquitin recognition factor in ER associated degradation 1 |
| ULK1 | Unc-51 like autophagy activating kinase 1 |
| uORFs | Upstream open reading frames |
| UPR | Unfolded protein response |
| UPS | Ubiquitin-proteasome system |
| VCP | Valosin-containing protein |
| VGCC | Voltage-gated calcium channel |
| XBP1 | X-box binding protein 1 |
| α-syn | α-synuclein |
References
- 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. 2021, 17, 327–406. [CrossRef] [PubMed]
- Citron, M. Alzheimer’s disease: Treatments in discovery and development. Nat. Neurosci. 2002, 5, 1055–1057. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Cell biology of protein misfolding: The examples of Alzheimer’s and Parkinson’s diseases. Nat. Cell. Biol. 2004, 6, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Ehrnhoefer, D.E.; Wong, B.K.; Hayden, M.R. Convergent pathogenic pathways in Alzheimer’s and Huntington’s diseases: Shared targets for drug development. Nat. Rev. Drug. Discov. 2011, 10, 853–867. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2012, 9, 13–24. [Google Scholar] [CrossRef]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The Function of α-Synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef] [Green Version]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [Green Version]
- Colla, E. Linking the Endoplasmic Reticulum to Parkinson’s Disease and Alpha-Synucleinopathy. Front. Neurosci. 2019, 13, 560. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Ross, C.A.; Margolis, R.L.; Rosenblatt, A.; Ranen, N.G.; Bêcher, M.W.; Aylward, E. Huntington Disease and the Related Disorder, Dentatorubral-Pallidoluysian Atrophy (DRPLA). Medicine 1997, 76, 305–338. [Google Scholar] [CrossRef]
- Kuhn, A.; Goldstein, D.R.; Hodges, A.; Strand, A.D.; Sengstag, T.; Kooperberg, C.; Becanovic, K.; Pouladi, M.A.; Sathasivam, K.; Cha, J.H.; et al. Mutant huntingtin’s effects on striatal gene expression in mice recapitulate changes observed in human Huntington’s disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum. Mol. Genet. 2007, 16, 1845–1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyama, F.; Miyazaki, H.; Sakamoto, N.; Becquet, C.; Machida, Y.; Kaneko, K.; Uchikawa, C.; Suzuki, T.; Kurosawa, M.; Ikeda, T.; et al. Sodium channel beta4 subunit: Down-regulation and possible involvement in neuritic degeneration in Huntington’s disease transgenic mice. J. Neurochem. 2006, 98, 518–529. [Google Scholar] [CrossRef]
- Hsu, Y.T.; Chang, Y.G.; Chern, Y. Insights into GABAAergic system alteration in Huntington’s disease. Open Biol. 2018, 8, 180165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdmanis, P.N.; Rouleau, G.A. Genetics of familial amyotrophic lateral sclerosis. Neurology 2008, 70, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Pasinelli, P.; Brown, R.H. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat. Rev. Neurosci. 2006, 7, 710–723. [Google Scholar] [CrossRef]
- Leblond, C.S.; Kaneb, H.M.; Dion, P.A.; Rouleau, G.A. Dissection of genetic factors associated with amyotrophic lateral sclerosis. Exp. Neurol. 2014, 262, 91–101. [Google Scholar] [CrossRef]
- Rosen, D.R. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 364, 362. [Google Scholar] [CrossRef]
- Ruegsegger, C.; Saxena, S. Proteostasis impairment in ALS. Brain Res. 2016, 1648, 571–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mays, C.E.; Soto, C. The stress of prion disease. Brain Res. 2016, 1648, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.A.; Soto, C.A.H.A.C. Stressing Out the ER: A Role of the Unfolded Protein Response in Prion-Related Disorders. Curr. Mol. Med. 2006, 6, 37–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.-B.; Shi, Q.; Xu, Y.; Xie, W.-L.; Zhang, J.; Tian, C.; Guo, Y.; Wang, K.; Zhang, B.-Y.; Chen, C.; et al. Protein Disulfide Isomerase Regulates Endoplasmic Reticulum Stress and the Apoptotic Process during Prion Infection and PrP Mutant-Induced Cytotoxicity. PLoS ONE 2012, 7, e38221. [Google Scholar] [CrossRef] [PubMed]
- Supnet, C.; Bezprozvanny, I. The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium 2010, 47, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Popugaeva, E.; Pchitskaya, E.; Bezprozvanny, I. Dysregulation of Intracellular Calcium Signaling in Alzheimer’s Disease. Antioxid. Redox. Signal 2018, 29, 1176–1188. [Google Scholar] [CrossRef]
- Costa, R.O.; Lacor, P.N.; Ferreira, I.L.; Resende, R.; Auberson, Y.P.; Klein, W.L.; Oliveira, C.R.; Rego, A.C.; Pereira, C.M. Endoplasmic reticulum stress occurs downstream of GluN2B subunit of N-methyl-d-aspartate receptor in mature hippocampal cultures treated with amyloid-beta oligomers. Aging Cell 2012, 11, 823–833. [Google Scholar] [CrossRef]
- Ferreiro, E.; Resende, R.; Costa, R.; Oliveira, C.R.; Pereira, C.M. An endoplasmic-reticulum-specific apoptotic pathway is involved in prion and amyloid-beta peptides neurotoxicity. Neurobiol. Dis. 2006, 23, 669–678. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zheng, W. Ca(2+) homeostasis dysregulation in Alzheimer’s disease: A focus on plasma membrane and cell organelles. FASEB J. 2019, 33, 6697–6712. [Google Scholar] [CrossRef]
- Chakroborty, S.; Goussakov, I.; Miller, M.B.; Stutzmann, G.E. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J. Neurosci. 2009, 29, 9458–9470. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Ludtmann, M.H.R.; Horrocks, M.H.; Negoda, A.; Cremades, N.; Klenerman, D.; Dobson, C.M.; Wood, N.W.; Pavlov, E.V.; Gandhi, S.; et al. Ca2+ is a key factor in α-synuclein-induced neurotoxicity. Development 2016, 143, e1.1. [Google Scholar] [CrossRef] [Green Version]
- Hurley, M.J.; Brandon, B.; Gentleman, S.M.; Dexter, D.T. Parkinson’s disease is associated with altered expression of CaV1 channels and calcium-binding proteins. Brain 2013, 136, 2077–2097. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.S.; Guzman, J.N.; Ilijic, E.; Mercer, J.N.; Rick, C.; Tkatch, T.; Meredith, G.E.; Surmeier, D.J. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 2007, 447, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Dryanovski, D.I.; Guzman, J.N.; Xie, Z.; Galteri, D.J.; Volpicelli-Daley, L.A.; Lee, V.M.-Y.; Miller, R.J.; Schumacker, P.T.; Surmeier, D.J. Calcium Entry and -Synuclein Inclusions Elevate Dendritic Mitochondrial Oxidant Stress in Dopaminergic Neurons. J. Neurosci. 2013, 33, 10154–10164. [Google Scholar] [CrossRef] [Green Version]
- Apicco, D.J.; Shlevkov, E.; Nezich, C.L.; Tran, D.T.; Guilmette, E.; Nicholatos, J.W.; Bantle, C.M.; Chen, Y.; Glajch, K.E.; Abraham, N.A.; et al. The Parkinson’s disease-associated gene ITPKB protects against α-synuclein aggregation by regulating ER-to-mitochondria calcium release. Proc. Natl. Acad. Sci. USA 2020, 118, e2006476118. [Google Scholar] [CrossRef]
- Saad, M.; Lesage, S.; Saint-Pierre, A.; Corvol, J.C.; Zelenika, D.; Lambert, J.C.; Vidailhet, M.; Mellick, G.D.; Lohmann, E.; Durif, F.; et al. Genome-wide association study confirms BST1 and suggests a locus on 12q24 as the risk loci for Parkinson’s disease in the European population. Hum. Mol. Genet. 2011, 20, 615–627. [Google Scholar] [CrossRef]
- Magrinelli, F.; Mehta, S.; Di Lazzaro, G.; Latorre, A.; Edwards, M.J.; Balint, B.; Basu, P.; Kobylecki, C.; Groppa, S.; Hegde, A.; et al. Dissecting the Phenotype and Genotype of PLA2G6-Related Parkinsonism. Mov. Disord. 2022, 37, 148–161. [Google Scholar] [CrossRef]
- Verma, M.; Callio, J.; Otero, P.A.; Sekler, I.; Wills, Z.P.; Chu, C.T. Mitochondrial Calcium Dysregulation Contributes to Dendrite Degeneration Mediated by PD/LBD-Associated LRRK2 Mutants. J. Neurosci. 2017, 37, 11151–11165. [Google Scholar] [CrossRef]
- Soman, S.; Keatinge, M.; Moein, M.; Da Costa, M.; Mortiboys, H.; Skupin, A.; Sugunan, S.; Bazala, M.; Kuznicki, J.; Bandmann, O. Inhibition of the mitochondrial calcium uniporter rescues dopaminergic neurons in pink1(-/-) zebrafish. Eur. J. Neurosci. 2017, 45, 528–535. [Google Scholar] [CrossRef]
- Torres, M.; Castillo, K.; Armisen, R.; Stutzin, A.; Soto, C.; Hetz, C. Prion protein misfolding affects calcium homeostasis and sensitizes cells to endoplasmic reticulum stress. PLoS ONE 2010, 5, e15658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czeredys, M. Dysregulation of Neuronal Calcium Signaling via Store-Operated Channels in Huntington’s Disease. Front. Cell Dev. Biol. 2020, 8, 611735. [Google Scholar] [CrossRef] [PubMed]
- Leal, S.S.; Gomes, C.M. Calcium dysregulation links ALS defective proteins and motor neuron selective vulnerability. Front. Cell Neurosci. 2015, 9, 225. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Barres, B.A. The Mystery and Magic of Glia: A Perspective on Their Roles in Health and Disease. Neuron 2008, 60, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The Role of Microglia and Astrocytes in Huntington’s Disease. Front. Mol. Neurosci. 2019, 12, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Chen, M.; Zhu, C. Neuroinflammation in Prion Disease. Int. J. Mol. Sci. 2021, 22, 2196. [Google Scholar] [CrossRef]
- Hong, S.; Dissing-Olesen, L.; Stevens, B. New insights on the role of microglia in synaptic pruning in health and disease. Curr. Opin. Neurobiol. 2015, 36, 128–134. [Google Scholar] [CrossRef] [Green Version]
- Baufeld, C.; O’Loughlin, E.; Calcagno, N.; Madore, C.; Butovsky, O. Differential contribution of microglia and monocytes in neurodegenerative diseases. J. Neural Transm. 2017, 125, 809–826. [Google Scholar] [CrossRef]
- Lee, J.-H.; Kim, J.-Y.; Noh, S.; Lee, H.; Lee, S.Y.; Mun, J.Y.; Park, H.; Chung, W.-S. Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis. Nature 2020, 590, 612–617. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, H.; Im, H.; Kang, Y.J.; Kim, Y.; Shin, J.H.; Won, W.; Lim, J.; Ju, Y.; Park, Y.M.; Kim, S.; et al. Severe reactive astrocytes precipitate pathological hallmarks of Alzheimer’s disease via H2O2− production. Nat. Neurosci. 2020, 23, 1555–1566. [Google Scholar] [CrossRef] [PubMed]
- Taipa, R.; das Neves, S.P.; Sousa, A.L.; Fernandes, J.; Pinto, C.; Correia, A.P.; Santos, E.; Pinto, P.S.; Carneiro, P.; Costa, P.; et al. Proinflammatory and anti-inflammatory cytokines in the CSF of patients with Alzheimer’s disease and their correlation with cognitive decline. Neurobiol. Aging 2019, 76, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Tarkowski, E.; Andreasen, N.; Tarkowski, A.; Blennow, K. Intrathecal inflammation precedes development of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1200–1205. [Google Scholar] [CrossRef]
- Collins, L.M.; Toulouse, A.; Connor, T.J.; Nolan, Y.M. Contributions of central and systemic inflammation to the pathophysiology of Parkinson’s disease. Neuropharmacology 2012, 62, 2154–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; Van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Fujikake, N.; Shin, M.; Shimizu, S. Association Between Autophagy and Neurodegenerative Diseases. Front. Neurosci. 2018, 12, 255. [Google Scholar] [CrossRef] [Green Version]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Sanchez, M.J.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [Green Version]
- Rubinsztein, D.C.; Bento, C.F.; Deretic, V. Therapeutic targeting of autophagy in neurodegenerative and infectious diseases. J. Exp. Med. 2015, 212, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Pedro, J.M.B.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive Involvement of Autophagy in Alzheimer Disease: An Immuno-Electron Microscopy Study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.H.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O. Macroautophagy--a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol 2005, 171, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H.V. Chaperone-Mediated Autophagy Markers in Parkinson Disease Brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [Green Version]
- Tanji, K.; Mori, F.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Alteration of autophagosomal proteins (LC3, GABARAP and GATE-16) in Lewy body disease. Neurobiol. Dis. 2011, 43, 690–697. [Google Scholar] [CrossRef]
- Moors, T.E.; Paciotti, S.; Ingrassia, A.; Quadri, M.; Breedveld, G.; Tasegian, A.; Chiasserini, D.; Eusebi, P.; Duran-Pacheco, G.; Kremer, T.; et al. Characterization of Brain Lysosomal Activities in GBA-Related and Sporadic Parkinson’s Disease and Dementia with Lewy Bodies. Mol. Neurobiol. 2018, 56, 1344–1355. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.R.; Maclean, G.D.; Foote, G.A.; Harvey, V.J. The Treatment of Advanced Recurrent Carcinoma of the Uterine Cervix with Platinum Based Cytotoxic Chemotherapy. Aust. N. Z. J. Obstet. Gynaecol. 1987, 27, 146–149. [Google Scholar] [CrossRef]
- Fernando, R.; Castro, J.P.; Flore, T.; Deubel, S.; Grune, T.; Ott, C. Age-Related Maintenance of the Autophagy-Lysosomal System Is Dependent on Skeletal Muscle Type. Oxidative Med. Cell. Longev. 2020, 2020, 4908162. [Google Scholar] [CrossRef]
- Carnio, S.; LoVerso, F.; Baraibar, M.A.; Longa, E.; Khan, M.M.; Maffei, M.; Reischl, M.; Canepari, M.; Löfler, S.; Kern, H.; et al. Autophagy Impairment in Muscle Induces Neuromuscular Junction Degeneration and Precocious Aging. Cell Rep. 2014, 8, 1509–1521. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Swerdlow, N.S.; Wilkins, H.M. Mitophagy and the Brain. Int. J. Mol. Sci. 2020, 21, 9661. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef]
- Frake, R.A.; Ricketts, T.; Menzies, F.M.; Rubinsztein, D.C. Autophagy and neurodegeneration. J. Clin. Investig. 2015, 125, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.-H.; Guo, F.; Shelburne, J.; Watkins, S.; Chu, C.T. Localization of Phosphorylated ERK/MAP Kinases to Mitochondria and Autophagosomes in Lewy Body Diseases. Brain Pathol. 2003, 13, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Franco-Iborra, S.; Plaza-Zabala, A.; Montpeyo, M.; Sebastian, D.; Vila, M.; Martinez-Vicente, M. Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy 2021, 17, 672–689. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Ozcan, U. Unfolded Protein Response Signaling and Metabolic Diseases. J. Biol. Chem. 2014, 289, 1203–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Kim, S.; Lee, Y.-I.; Lee, J. Cellular Stress-Modulating Drugs Can Potentially Be Identified by in Silico Screening with Connectivity Map (CMap). Int. J. Mol. Sci. 2019, 20, 5601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Grey, M.J.; Cloots, E.; Simpson, M.S.; LeDuc, N.; Serebrenik, Y.V.; De Luca, H.; De Sutter, D.; Luong, P.; Thiagarajah, J.R.; Paton, A.W.; et al. IRE1beta negatively regulates IRE1alpha signaling in response to endoplasmic reticulum stress. J. Cell Biol. 2020, 219, e201904048. [Google Scholar] [CrossRef] [Green Version]
- Gardner, B.M.; Walter, P. Unfolded Proteins Are Ire1-Activating Ligands That Directly Induce the Unfolded Protein Response. Science 2011, 333, 1891–1894. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Lee, J.; Reno, C.M.; Sun, C.; Park, S.W.; Chung, J.; Lee, J.; Fisher, S.J.; White, M.F.; Biddinger, S.B.; et al. Regulation of glucose homeostasis through a XBP-1–FoxO1 interaction. Nat. Med. 2011, 17, 356–365. [Google Scholar] [CrossRef] [Green Version]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef]
- Qi, L.; Tsai, B.; Arvan, P. New Insights into the Physiological Role of Endoplasmic Reticulum-Associated Degradation. Trends Cell Biol. 2017, 27, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Vembar, S.S.; Brodsky, J.L. One step at a time: Endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 2008, 9, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashid, H.-O.; Yadav, R.K.; Kim, H.-R.; Chae, H.-J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]
- Ogata, M.; Hino, S.-I.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy Is Activated for Cell Survival after Endoplasmic ReticulumStress. Mol. Cell. Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [Green Version]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [Green Version]
- Chino, H.; Mizushima, N. ER-Phagy: Quality Control and Turnover of Endoplasmic Reticulum. Trends Cell Biol. 2020, 30, 384–398. [Google Scholar] [CrossRef]
- Saez, I.; Vilchez, D. The Mechanistic Links Between Proteasome Activity, Aging and Agerelated Diseases. Curr. Genom. 2014, 15, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Vilchez, D.; Saez, I.; Dillin, A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 2014, 5, 5659. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.-H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Bernasconi, P.; Fisher, J.; Lee, A.-H.; Bassik, M.C.; Antonsson, B.; Brandt, G.S.; Iwakoshi, N.N.; Schinzel, A.; Glimcher, L.H.; et al. Proapoptotic BAX and BAK Modulate the Unfolded Protein Response by a Direct Interaction with IRE1α. Science 2006, 312, 572–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Ibi, D.; Taniguchi, K.; Lee, J.; Herrema, H.; Akosman, B.; Mucka, P.; Salazar Hernandez, M.A.; Uyar, M.F.; Park, S.W.; et al. Inflammation Improves Glucose Homeostasis through IKKbeta-XBP1s Interaction. Cell 2016, 167, 1052–1066.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Sun, C.; Zhou, Y.; Lee, J.; Gokalp, D.; Herrema, H.; Park, S.W.; Davis, R.J.; Ozcan, U. p38 MAPK–mediated regulation of Xbp1s is crucial for glucose homeostasis. Nat. Med. 2011, 17, 1251–1260. [Google Scholar] [CrossRef] [Green Version]
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T.; et al. Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab. 2012, 16, 265–273. [Google Scholar] [CrossRef] [Green Version]
- Meares, G.P.; Liu, Y.; Rajbhandari, R.; Qin, H.; Nozell, S.E.; Mobley, J.A.; Corbett, J.A.; Benveniste, E.N. PERK-Dependent Activation of JAK1 and STAT3 Contributes to Endoplasmic Reticulum Stress-Induced Inflammation. Mol. Cell. Biol. 2014, 34, 3911–3925. [Google Scholar] [CrossRef] [Green Version]
- Dahlmann, B. The Role of Proteasomes in Disease. BMC Biochem 2007, 8, S3. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Kanski, J. Brain protein oxidation in age-related neurodegenerative disorders that are associated with aggregated proteins. Mech. Ageing Dev. 2001, 122, 945–962. [Google Scholar] [CrossRef]
- Unterberger, U.; Höftberger, R.; Gelpi, E.; Flicker, H.; Budka, H.; Voigtländer, T. Endoplasmic Reticulum Stress Features Are Prominent in Alzheimer Disease but Not in Prion Diseases In Vivo. J. Neuropathol. Exp. Neurol. 2006, 65, 348–357. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-S.; Choi, Y.; Shin, K.-Y.; Joo, Y.; Lee, Y.-K.; Jung, S.Y.; Suh, Y.-H.; Kim, J.-H. Swedish amyloid precursor protein mutation increases phosphorylation of eIF2α in vitro and in vivo. J. Neurosci. Res. 2007, 85, 1528–1537. [Google Scholar] [CrossRef]
- Page, G.; Bilan, A.R.; Ingrand, S.; Lafay-Chebassier, C.; Pain, S.; Pochat, M.P.; Bouras, C.; Bayer, T.; Hugon, J. Activated double-stranded RNA-dependent protein kinase and neuronal death in models of Alzheimer’s disease. Neuroscience 2006, 139, 1343–1354. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.C.; Wong, A.K.; Ng, H.K.; Hugon, J. Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer’s disease. Neuroreport 2002, 13, 2429–2432. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.M.; Veerhuis, R.; Van Haastert, E.S.; Rozemuller, J.M.; Baas, F.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 2005, 110, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.; van Haastert, E.S.; Nijholt, D.A.; Rozemuller, A.J.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am. J. Pathol. 2009, 174, 1241–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheper, W.; Hoozemans, J.J.M. The unfolded protein response in neurodegenerative diseases: A neuropathological perspective. Acta Neuropathol. 2015, 130, 315–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baleriola, J.; Walker, C.A.; Jean, Y.Y.; Crary, J.F.; Troy, C.M.; Nagy, P.L.; Hengst, U. Axonally Synthesized ATF4 Transmits a Neurodegenerative Signal across Brain Regions. Cell 2014, 158, 1159–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijholt, D.A.; van Haastert, E.S.; Rozemuller, A.J.; Scheper, W.; Hoozemans, J.J. The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J. Pathol. 2012, 226, 693–702. [Google Scholar] [CrossRef]
- Hou, H.L.; Shen, Y.X.; Zhu, H.Y.; Sun, H.; Yan, X.B.; Fang, H.; Zhou, J.N. Alterations of hHrd1 expression are related to hyperphosphorylated tau in the hippocampus in Alzheimer’s disease. J. Neurosci. Res. 2006, 84, 1862–1870. [Google Scholar] [CrossRef]
- Nijholt, D.A.; de Graaf, T.R.; van Haastert, E.S.; Oliveira, A.O.; Berkers, C.R.; Zwart, R.; Ovaa, H.; Baas, F.; Hoozemans, J.J.; Scheper, W. Endoplasmic reticulum stress activates autophagy but not the proteasome in neuronal cells: Implications for Alzheimer’s disease. Cell Death Differ. 2011, 18, 1071–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.Y.; Lee, K.S.; Lee, H.J.; Kim, D.H.; Noh, Y.H.; Yu, K.; Jung, H.Y.; Lee, S.H.; Lee, J.Y.; Youn, Y.C.; et al. Activation of PERK signaling attenuates Abeta-mediated ER stress. PLoS ONE 2010, 5, e10489. [Google Scholar]
- Casas-Tinto, S.; Zhang, Y.; Sanchez-Garcia, J.; Gomez-Velazquez, M.; Rincon-Limas, D.E.; Fernandez-Funez, P. The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum. Mol. Genet. 2011, 20, 2144–2160. [Google Scholar] [CrossRef] [Green Version]
- Kraemer, B.C.; Burgess, J.K.; Chen, J.H.; Thomas, J.H.; Schellenberg, G.D. Molecular pathways that influence human tau-induced pathology in Caenorhabditis elegans. Hum. Mol. Genet. 2006, 15, 1483–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loewen, C.A.; Feany, M.B. The Unfolded Protein Response Protects from Tau Neurotoxicity In Vivo. PLoS ONE 2010, 5, e13084. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.; Wang, W.; Cai, Z.Y.; Yao, L.F.; Chen, Z.W.; Wang, C.Y.; Zhao, B.; Li, K.S. Polymorphism -116C/G of human X-box-binding protein 1 promoter is associated with risk of Alzheimer’s disease. CNS Neurosci. Ther. 2013, 19, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Iwakami, N.; Takeuchi, S.; Waragai, M.; Suzuki, M.; Kanazawa, I.; Lippa, C.F.; Ono, S.; Okazawa, H. JNK activation is associated with intracellular beta-amyloid accumulation. Brain Res. Mol. Brain Res. 2000, 85, 221–233. [Google Scholar] [CrossRef]
- Gourmaud, S.; Paquet, C.; Dumurgier, J.; Pace, C.; Bouras, C.; Gray, F.; Laplanche, J.-L.; Meurs, E.F.; Mouton-Liger, F.; Hugon, J. Increased levels of cerebrospinal fluid JNK3 associated with amyloid pathology: Links to cognitive decline. J. Psychiatry Neurosci. 2015, 40, 151–161. [Google Scholar] [CrossRef] [Green Version]
- Keller, J.N.; Hanni, K.B.; Markesbery, W.R. Possible involvement of proteasome inhibition in aging: Implications for oxidative stress. Mech. Ageing Dev. 2000, 113, 61–70. [Google Scholar] [CrossRef]
- Keller, J.N.; Gee, J.; Ding, Q. The proteasome in brain aging. Ageing Res. Rev. 2002, 1, 279–293. [Google Scholar] [CrossRef]
- Rabek, J.P.; Boylston, W.H., 3rd; Papaconstantinou, J. Carbonylation of ER chaperone proteins in aged mouse liver. Biochem. Biophys. Res. Commun. 2003, 305, 566–572. [Google Scholar] [CrossRef]
- Nuss, J.E.; Choksi, K.B.; DeFord, J.H.; Papaconstantinou, J. Decreased enzyme activities of chaperones PDI and BiP in aged mouse livers. Biochem. Biophys. Res. Commun. 2008, 365, 355–361. [Google Scholar] [CrossRef] [Green Version]
- Gavilan, M.P.; Vela, J.; Castaño, A.; Ramos, B.; del Río, J.C.; Vitorica, J.; Ruano, D. Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol. Aging 2006, 27, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.G.; Ramaiah, K.V. Reduced eIF2α phosphorylation and increased proapoptotic proteins in aging. Biochem. Biophys. Res. Commun. 2007, 355, 365–370. [Google Scholar] [CrossRef]
- Naidoo, N.; Ferber, M.; Master, M.; Zhu, Y.; Pack, A.I. Aging Impairs the Unfolded Protein Response to Sleep Deprivation and Leads to Proapoptotic Signaling. J. Neurosci. 2008, 28, 6539–6548. [Google Scholar] [CrossRef] [PubMed]
- Keck, S.; Nitsch, R.; Grune, T.; Ullrich, O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J. Neurochem. 2003, 85, 115–122. [Google Scholar] [CrossRef]
- Tseng, B.P.; Green, K.N.; Chan, J.L.; Blurton-Jones, M.; LaFerla, F.M. Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol. Aging 2008, 29, 1607–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickey, C.A.; Kamal, A.; Lundgren, K.; Klosak, N.; Bailey, R.M.; Dunmore, J.; Ash, P.; Shoraka, S.; Zlatkovic, J.; Eckman, C.B.; et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J. Clin. Investig. 2007, 117, 648–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, L.J.; Nordhues, B.A.; Hill, S.E.; Scaglione, K.M.; O’Leary, J.C., 3rd; Fontaine, S.N.; Breydo, L.; Zhang, B.; Li, P.; Wang, L.; et al. Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J. Clin. Investig. 2013, 123, 4158–4169. [Google Scholar] [CrossRef] [Green Version]
- Jaeger, P.A.; Pickford, F.; Sun, C.-H.; Lucin, K.M.; Masliah, E.; Wyss-Coray, T. Regulation of Amyloid Precursor Protein Processing by the Beclin 1 Complex. PLoS ONE 2010, 5, e11102. [Google Scholar] [CrossRef]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar]
- Costa-Mattioli, M.; Sossin, W.S.; Klann, E.; Sonenberg, N. Translational Control of Long-Lasting Synaptic Plasticity and Memory. Neuron 2009, 61, 10–26. [Google Scholar] [CrossRef] [Green Version]
- Costa-Mattioli, M.; Gobert, D.; Harding, H.; Herdy, B.; Azzi, M.; Bruno, M.; Bidinosti, M.; Ben Mamou, C.; Marcinkiewicz, E.; Yoshida, M.; et al. Translational control of hippocampal synaptic plasticity and memory by the eIF2α kinase GCN2. Nature 2005, 436, 1166–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buffington, S.A.; Huang, W.; Costa-Mattioli, M. Translational Control in Synaptic Plasticity and Cognitive Dysfunction. Annu. Rev. Neurosci. 2014, 37, 17–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinh, M.A.; Kaphzan, H.; Wek, R.C.; Pierre, P.; Cavener, D.R.; Klann, E. Brain-Specific Disruption of the eIF2α Kinase PERK Decreases ATF4 Expression and Impairs Behavioral Flexibility. Cell Rep. 2012, 1, 676–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.; Muzzio, I.A.; Malleret, G.; Bartsch, D.; Verbitsky, M.; Pavlidis, P.; Yonan, A.L.; Vronskaya, S.; Grody, M.B.; Cepeda, I.L.; et al. Inducible Enhancement of Memory Storage and Synaptic Plasticity in Transgenic Mice Expressing an Inhibitor of ATF4 (CREB-2) and C/EBP Proteins. Neuron 2003, 39, 655–669. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.; Trinh, M.A.; Wexler, A.J.; Bourbon, C.; Gatti, E.; Pierre, P.; Cavener, D.R.; Klann, E. Suppression of eIF2alpha kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci. 2013, 16, 1299–12305. [Google Scholar] [CrossRef] [Green Version]
- Segev, Y.; Barrera, I.; Ounallah-Saad, H.; Wibrand, K.; Sporild, I.; Livne, A.; Rosenberg, T.; David, O.; Mints, M.; Bramham, C.R.; et al. PKR Inhibition Rescues Memory Deficit and ATF4 Overexpression in ApoE epsilon4 Human Replacement Mice. J. Neurosci. 2015, 35, 12986–12993. [Google Scholar] [CrossRef] [Green Version]
- Lourenco, M.V.; Clarke, J.R.; Frozza, R.L.; Bomfim, T.R.; Forny-Germano, L.; Batista, A.F.; Sathler, L.B.; Brito-Moreira, J.; Amaral, O.B.; Silva, C.A.; et al. TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys. Cell Metab. 2013, 18, 831–843. [Google Scholar] [CrossRef] [Green Version]
- Devi, L.; Ohno, M. PERK mediates eIF2alpha phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2272–2281. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Zhou, X.; Zimmermann, H.R.; Cavener, D.R.; Klann, E.; Ma, T. Repression of the eIF2alpha kinase PERK alleviates mGluR-LTD impairments in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2016, 41, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, M.M.; Lourenco, M.V.; Longo, F.; Kasica, N.P.; Yang, W.; Ureta, G.; Ferreira, D.D.P.; Mendonça, P.H.J.; Bernales, S.; Ma, T.; et al. Correction of eIF2-dependent defects in brain protein synthesis, synaptic plasticity, and memory in mouse models of Alzheimer’s disease. Sci. Signal. 2021, 14, eabc5429. [Google Scholar] [CrossRef]
- Duran-Aniotz, C.; Cornejo, V.H.; Espinoza, S.; Ardiles, Á.O.; Medinas, D.B.; Salazar, C.; Foley, A.; Gajardo, I.; Thielen, P.; Iwawaki, T.; et al. IRE1 signaling exacerbates Alzheimer’s disease pathogenesis. Acta Neuropathol. 2017, 134, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Martínez, G.; Vidal, R.L.; Mardones, P.; Serrano, F.G.; Ardiles, A.O.; Wirth, C.; Valdés, P.; Thielen, P.; Schneider, B.L.; Kerr, B.; et al. Regulation of Memory Formation by the Transcription Factor XBP1. Cell Rep. 2016, 14, 1382–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cissé, M.; Duplan, E.; Lorivel, T.; Dunys, J.; Bauer, C.; Meckler, X.; Gerakis, Y.; Lauritzen, I.; Checler, F. The transcription factor XBP1s restores hippocampal synaptic plasticity and memory by control of the Kalirin-7 pathway in Alzheimer model. Mol. Psychiatry 2017, 22, 1562–1575. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, A.; Kasahara, T.; Kametani, M.; Kato, T. Attenuated BDNF-induced upregulation of GABAergic markers in neurons lacking Xbp1. Biochem. Biophys. Res. Commun. 2008, 376, 758–763. [Google Scholar] [CrossRef]
- Chung, C.Y.; Khurana, V.; Auluck, P.K.; Tardiff, D.F.; Mazzulli, J.R.; Soldner, F.; Baru, V.; Lou, Y.; Freyzon, Y.; Cho, S.; et al. Identification and Rescue of α-Synuclein Toxicity in Parkinson Patient–Derived Neurons. Science 2013, 342, 983–987. [Google Scholar] [CrossRef] [Green Version]
- Heman-Ackah, S.M.; Manzano, R.; Hoozemans, J.J.M.; Scheper, W.; Flynn, R.; Haerty, W.; Cowley, S.A.; Bassett, A.R.; Wood, M.J.A. Alpha-synuclein induces the unfolded protein response in Parkinson’s disease SNCA triplication iPSC-derived neurons. Hum. Mol. Genet. 2017, 26, 4441–4450. [Google Scholar] [CrossRef] [Green Version]
- Colla, E.; Coune, P.; Liu, Y.; Pletnikova, O.; Troncoso, J.C.; Iwatsubo, T.; Schneider, B.L.; Lee, M.K. Endoplasmic Reticulum Stress Is Important for the Manifestations of alpha-Synucleinopathy In Vivo. J. Neurosci. 2012, 32, 3306–3320. [Google Scholar] [CrossRef]
- Mercado, G.; Castillo, V.; Soto, P.; Sidhu, A. ER stress and Parkinson’s disease: Pathological inputs that converge into the secretory pathway. Brain Res. 2016, 1648, 626–632. [Google Scholar] [CrossRef]
- Conn, K.J.; Gao, W.; McKee, A.; Lan, M.S.; Ullman, M.D.; Eisenhauer, P.B.; Fine, R.E.; Wells, J.M. Identification of the protein disulfide isomerase family member PDIp in experimental Parkinson’s disease and Lewy body pathology. Brain Res. 2004, 1022, 164–172. [Google Scholar] [CrossRef]
- Slodzinski, H.; Moran, L.B.; Michael, G.J.; Wang, B.; Novoselov, S.; Cheetham, M.E.; Pearce, R.K.B.; Graeber, M.B. Homocysteine-induced endoplasmic reticulum protein (herp) is up-regulated in parkinsonian substantia nigra and present in the core of Lewy bodies. Clin. Neuropathol. 2009, 28, 333–343. [Google Scholar]
- Ryu, E.J.; Harding, H.P.; Angelastro, J.M.; Vitolo, O.V.; Ron, D.; Greene, L.A. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson’s disease. J. Neurosci. 2002, 22, 10690–10698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtz, W.A.; O’Malley, K.L. Parkinsonian Mimetics Induce Aspects of Unfolded Protein Response in Death of Dopaminergic Neurons. J. Biol. Chem. 2003, 278, 19367–19377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoozemans, J.J.; van Haastert, E.S.; Eikelenboom, P.; de Vos, R.A.; Rozemuller, J.M.; Scheper, W. Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun. 2007, 354, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Matsuda, N. Proteostasis and neurodegeneration: The roles of proteasomal degradation and autophagy. Biochim. Biophys. Acta 2014, 1843, 197–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbatyuk, M.S.; Shabashvili, A.; Chen, W.; Meyers, C.; Sullivan, L.F.; Salganik, M.; Lin, J.H.; Lewin, A.S.; Muzyczka, N.; Gorbatyuk, O.S. Glucose Regulated Protein 78 Diminishes α-Synuclein Neurotoxicity in a Rat Model of Parkinson Disease. Mol. Ther. 2012, 20, 1327–1337. [Google Scholar] [CrossRef]
- Silva, R.M.; Ries, V.; Oo, T.F.; Yarygina, O.; Jackson-Lewis, V.; Ryu, E.J.; Lu, P.D.; Marciniak, S.; Ron, D.; Przedborski, S.; et al. CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J. Neurochem. 2005, 95, 974–986. [Google Scholar] [CrossRef] [Green Version]
- Sado, M.; Yamasaki, Y.; Iwanaga, T.; Onaka, Y.; Ibuki, T.; Nishihara, S.; Mizuguchi, H.; Momota, H.; Kishibuchi, R.; Hashimoto, T.; et al. Protective effect against Parkinson’s disease-related insults through the activation of XBP1. Brain Res. 2009, 1257, 16–24. [Google Scholar] [CrossRef]
- Valdés, P.; Mercado, G.; Vidal, R.L.; Molina, C.; Parsons, G.; Court, F.A.; Martinez, A.; Galleguillos, D.; Armentano, D.; Schneider, B.L.; et al. Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc. Natl. Acad. Sci. USA 2014, 111, 6804–6809. [Google Scholar] [CrossRef] [Green Version]
- Borghi, R.; Marchese, R.; Negro, A.; Marinelli, L.; Forloni, G.; Zaccheo, D.; Abbruzzese, G.; Tabaton, M. Full length alpha-synuclein is present in cerebrospinal fluid from Parkinson’s disease and normal subjects. Neurosci. Lett. 2000, 287, 65–67. [Google Scholar] [CrossRef]
- Lee, H.-J.; Patel, S.; Lee, S.-J. Intravesicular Localization and Exocytosis of α-Synuclein and its Aggregates. J. Neurosci. 2005, 25, 6016–6024. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.; Salem, S.A.; Paleologou, K.E.; Cooper, L.J.; Fullwood, N.J.; Gibson, M.J.; Curran, M.D.; Court, J.A.; Mann, D.M.; Ikeda, S.; et al. Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003, 17, 1945–1947. [Google Scholar] [CrossRef] [PubMed]
- El-Agnaf, O.M.; Salem, S.A.; Paleologou, K.E.; Curran, M.D.; Gibson, M.J.; Court, J.A.; Schlossmacher, M.G.; Allsop, D. Detection of oligomeric forms of alpha-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J. 2006, 20, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Devic, I.; Hwang, H.; Edgar, J.S.; Izutsu, K.; Presland, R.; Pan, C.; Goodlett, D.R.; Wang, Y.; Armaly, J.; Tumas, V.; et al. Salivary alpha-synuclein and DJ-1: Potential biomarkers for Parkinson’s disease. Brain 2011, 134, e178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foulds, P.G.; Mitchell, J.D.; Parker, A.; Turner, R.; Green, G.; Diggle, P.; Hasegawa, M.; Taylor, M.; Mann, D.; Allsop, D. Phosphorylated alpha-synuclein can be detected in blood plasma and is potentially a useful biomarker for Parkinson’s disease. FASEB J. 2011, 25, 4127–4137. [Google Scholar] [CrossRef] [PubMed]
- Nam, D.; Lee, J.-Y.; Lee, M.; Kim, J.; Seol, W.; Son, I.; Ho, D.H. Detection and Assessment of α-Synuclein Oligomers in the Urine of Parkinson’s Disease Patients. J. Park. Dis. 2020, 10, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Maass, F.; Rikker, S.; Dambeck, V.; Warth, C.; Tatenhorst, L.; Csoti, I.; Schmitz, M.; Zerr, I.; Leha, A.; Bähr, M.; et al. Increased alpha-synuclein tear fluid levels in patients with Parkinson’s disease. Sci. Rep. 2020, 10, 8507. [Google Scholar] [CrossRef] [PubMed]
- Desplats, P.; Lee, H.-J.; Bae, E.-J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.-J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.R.; Park, S.J.; Park, S.M. Molecular events underlying the cell-to-cell transmission of alpha-synuclein. FEBS J. 2021, 288, 6593–6602. [Google Scholar] [CrossRef]
- Jang, A.; Lee, H.-J.; Suk, J.-E.; Jung, J.-W.; Kim, K.P.; Lee, S.-J. Non-classical exocytosis of α-synuclein is sensitive to folding states and promoted under stress conditions. J. Neurochem. 2010, 113, 1263–1274. [Google Scholar] [CrossRef]
- Da Fonseca, T.L.; Villar-Piqué, A.; Outeiro, T.F. The Interplay between Alpha-Synuclein Clearance and Spreading. Biomolecules 2015, 5, 435–471. [Google Scholar] [CrossRef] [Green Version]
- Decressac, M.; Bjorklund, A. TFEB: Pathogenic role and therapeutic target in Parkinson disease. Autophagy 2013, 9, 1244–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winslow, A.R.; Chen, C.-W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. α-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.W.; Kordower, J.H. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to alpha-synuclein inclusions. Neurobiol. Dis. 2009, 35, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Decressac, M.; Mattsson, B.; Weikop, P.; Lundblad, M.; Jakobsson, J.; Bjorklund, A. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc. Natl. Acad. Sci. USA 2013, 110, E1817–E1826. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.-C.; Follenzi, A.; et al. Dopamine-modified α-synuclein blocks chaperone-mediated autophagy. J. Clin. Investig. 2008, 118, 777–788. [Google Scholar] [CrossRef] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Imai, Y.; Soda, M.; Takahashi, R. Parkin Suppresses Unfolded Protein Stress-induced Cell Death through Its E3 Ubiquitin-protein Ligase Activity. J. Biol. Chem. 2000, 275, 35661–35664. [Google Scholar] [CrossRef] [Green Version]
- Bouman, L.; Schlierf, A.; Lutz, A.K.; Shan, J.; Deinlein, A.; Kast, J.; Galehdar, Z.; Palmisano, V.; Patenge, N.; Berg, D.; et al. Parkin is transcriptionally regulated by ATF4: Evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ. 2011, 18, 769–782. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Soda, M.; Inoue, H.; Hattori, N.; Mizuno, Y.; Takahashi, R. An Unfolded Putative Transmembrane Polypeptide, which Can Lead to Endoplasmic Reticulum Stress, Is a Substrate of Parkin. Cell 2001, 105, 891–902. [Google Scholar] [CrossRef] [Green Version]
- Duplan, E.; Giaime, E.; Viotti, J.; Sévalle, J.; Corti, O.; Brice, A.; Ariga, H.; Qi, L.; Checler, F.; da Costa, C.A. ER-stress-associated functional link between Parkin and DJ-1 via a transcriptional cascade involving the tumor suppressor p53 and the spliced X-box binding protein XBP-1. J. Cell Sci. 2013, 126, 2124–2133. [Google Scholar] [CrossRef] [Green Version]
- Pizzo, P.; Pozzan, T. Mitochondria–endoplasmic reticulum choreography: Structure and signaling dynamics. Trends Cell Biol. 2007, 17, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Ghribi, O.; Herman, M.M.; Pramoonjago, P.; Savory, J. MPP+ induces the endoplasmic reticulum stress response in rabbit brain involving activation of the ATF-6 and NF-kappaB signaling pathways. J. Neuropathol. Exp. Neurol. 2003, 62, 1144–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cali, T.; Ottolini, D.; Brini, M. Mitochondria, calcium, and endoplasmic reticulum stress in Parkinson’s disease. Biofactors 2011, 37, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Salthun-Lassalle, B.; Hirsch, E.C.; Wolfart, J.; Ruberg, M.; Michel, P.P. Rescue of Mesencephalic Dopaminergic Neurons in Culture by Low-Level Stimulation of Voltage-Gated Sodium Channels. J. Neurosci. 2004, 24, 5922–5930. [Google Scholar] [CrossRef] [Green Version]
- Arduino, D.M.; Esteves, A.R.; Cardoso, S.M.; Oliveira, C.R. Endoplasmic reticulum and mitochondria interplay mediates apoptotic cell death: Relevance to Parkinson’s disease. Neurochem. Int. 2009, 55, 341–348. [Google Scholar] [CrossRef]
- Calì, T.; Ottolini, D.; Negro, A.; Brini, M. α-Synuclein Controls Mitochondrial Calcium Homeostasis by Enhancing Endoplasmic Reticulum-Mitochondria Interactions. J. Biol. Chem. 2012, 287, 17914–17929. [Google Scholar] [CrossRef] [Green Version]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rüb, C.; Liu, Y.; Magrané, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. alpha-Synuclein Is Localized to Mitochondria-Associated ER Membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef]
- Wellington, C.L.; Singaraja, R.; Ellerby, L.; Savill, J.; Roy, S.; Leavitt, B.; Cattaneo, E.; Hackam, A.; Sharp, A.; Thornberry, N.; et al. Inhibiting Caspase Cleavage of Huntingtin Reduces Toxicity and Aggregate Formation in Neuronal and Nonneuronal Cells. J. Biol. Chem. 2000, 275, 19831–19838. [Google Scholar] [CrossRef] [Green Version]
- Martindale, D.; Hackam, A.; Wieczorek, A.; Ellerby, L.; Wellington, C.; McCutcheon, K.; Singaraja, R.; Kazemi-Esfarjani, P.; Devon, R.; Kim, S.U.; et al. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat. Genet. 1998, 18, 150–154. [Google Scholar] [CrossRef]
- Xia, J.; Lee, D.H.; Taylor, J.; van Delft, M.; Truant, R. Huntingtin contains a highly conserved nuclear export signal. Hum. Mol. Genet. 2003, 12, 1393–1403. [Google Scholar] [CrossRef]
- Kegel, K.B.; Meloni, A.R.; Yi, Y.; Kim, Y.J.; Doyle, E.; Cuiffo, B.G.; Sapp, E.; Wang, Y.; Qin, Z.-H.; Chen, J.D.; et al. Huntingtin Is Present in the Nucleus, Interacts with the Transcriptional Corepressor C-terminal Binding Protein, and Represses Transcription. J. Biol. Chem. 2002, 277, 7466–7476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutekunst, C.-A.; Li, S.-H.; Yi, H.; Mulroy, J.S.; Kuemmerle, S.; Jones, R.; Rye, D.; Ferrante, R.J.; Hersch, S.M.; Li, X.-J. Nuclear and Neuropil Aggregates in Huntington’s Disease: Relationship to Neuropathology. J. Neurosci. 1999, 19, 2522–2534. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.W.; Turmaine, M.; Cozens, B.A.; DiFiglia, M.; Sharp, A.H.; Ross, C.A.; Scherzinger, E.; Wanker, E.E.; Mangiarini, L.; Bates, G.P. Formation of Neuronal Intranuclear Inclusions Underlies the Neurological Dysfunction in Mice Transgenic for the HD Mutation. Cell 1997, 90, 537–548. [Google Scholar] [CrossRef] [Green Version]
- Landles, C.; Sathasivam, K.; Weiss, A.; Woodman, B.; Moffitt, H.; Finkbeiner, S.; Sun, B.; Gafni, J.; Ellerby, L.M.; Trottier, Y.; et al. Proteolysis of Mutant Huntingtin Produces an Exon 1 Fragment That Accumulates as an Aggregated Protein in Neuronal Nuclei in Huntington Disease. J. Biol. Chem. 2010, 285, 8808–8823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornett, J.; Cao, F.; Wang, C.-E.; A Ross, C.A.; Bates, G.P.; Li, S.-H.; Li, X.-J. Polyglutamine expansion of huntingtin impairs its nuclear export. Nat. Genet. 2005, 37, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Saudou, F.; Finkbeiner, S.; Devys, D.; Greenberg, M.E. Huntingtin Acts in the Nucleus to Induce Apoptosis but Death Does Not Correlate with the Formation of Intranuclear Inclusions. Cell 1998, 95, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Peters, M.F.; Nucifora, C.F., Jr.; Kushia, J.; Seaman, H.C.; Cooper, J.K.; Herring, W.J.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Nuclear Targeting of Mutant Huntingtin Increases Toxicity. Mol. Cell. Neurosci. 1999, 14, 121–128. [Google Scholar] [CrossRef] [PubMed]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of Huntingtin in Neuronal Intranuclear Inclusions and Dystrophic Neurites in Brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef]
- Li, S.-H.; Cheng, A.L.; Li, H.; Li, X.-J. Cellular Defects and Altered Gene Expression in PC12 Cells Stably Expressing Mutant Huntingtin. J. Neurosci. 1999, 19, 5159–5172. [Google Scholar] [CrossRef]
- Kotliarova, S.; Jana, N.; Sakamoto, N.; Kurosawa, M.; Miyazaki, H.; Nekooki, M.; Doi, H.; Machida, Y.; Wong, H.-K.; Suzuki, T.; et al. Decreased expression of hypothalamic neuropeptides in Huntington disease transgenic mice with expanded polyglutamine-EGFP fluorescent aggregates. J. Neurochem. 2005, 93, 641–653. [Google Scholar] [CrossRef]
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.Z.; Gohler, H.; Wanker, E.E.; Bates, G.P.; Housman, D.E.; Thompson, L.M. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.C.; Faber, P.W.; Persichetti, F.; Mittal, V.; Vonsattel, J.-P.; Macdonald, M.E.; Gusella, J.F. Amyloid formation by mutant huntingtin: Threshold, progressivity and recruitment of normal polyglutamine proteins. Somat. Cell Mol. Genet. 1998, 24, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Kazantsev, A.; Preisinger, E.; Dranovsky, A.; Goldgaber, D.; Housman, D. Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc. Natl. Acad. Sci. USA 1999, 96, 11404–11409. [Google Scholar] [CrossRef] [Green Version]
- Cha, J.-H.J.; Kosinski, C.M.; Kerner, J.A.; Alsdorf, S.A.; Mangiarini, L.; Davies, S.W.; Penney, J.B.; Bates, G.P.; Young, A.B. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human Huntington disease gene. Proc. Natl. Acad. Sci. USA 1998, 95, 6480–6485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luthi-Carter, R.; Strand, A.; Peters, N.L.; Solano, S.M.; Hollingsworth, Z.R.; Menon, A.S.; Frey, A.S.; Spektor, B.S.; Penney, E.B. Decreased expression of striatal signaling genes in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2000, 9, 1259–12571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gidalevitz, T.; Ben-Zvi, A.; Ho, K.H.; Brignull, H.R.; Morimoto, R.I. Progressive Disruption of Cellular Protein Folding in Models of Polyglutamine Diseases. Science 2006, 311, 1471–1474. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Kasturi, P.; Bracher, A.; Loew, C.; Zheng, M.; Villella, A.; Garza, D.; Hartl, F.U.; Raychaudhuri, S. Firefly luciferase mutants as sensors of proteome stress. Nat. Methods 2011, 8, 879–884. [Google Scholar] [CrossRef]
- Muchowski, P.J.; Schaffar, G.; Sittler, A.; Wanker, E.E.; Hayer-Hartl, M.K.; Hartl, F.U. Hsp70 and Hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2000, 97, 7841–7846. [Google Scholar] [CrossRef] [Green Version]
- Wacker, J.L.; Zareie, M.H.; Fong, H.; Sarikaya, M.; Muchowski, P.J. Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat. Struct. Mol. Biol. 2004, 11, 1215–1222. [Google Scholar] [CrossRef]
- Jana, N.R.; Tanaka, M.; Wang, G.; Nukina, N. Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: Their role in suppression of aggregation and cellular toxicity. Hum. Mol. Genet. 2000, 9, 2009–2018. [Google Scholar] [CrossRef] [Green Version]
- Chan, H.Y.; Warrick, J.M.; Gray-Board, G.L.; Paulson, H.L.; Bonini, N.M. Mechanisms of chaperone suppression of polyglutamine disease: Selectivity, synergy and modulation of protein solubility in Drosophila. Hum. Mol. Genet. 2000, 9, 2811–2820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazemi-Esfarjani, P.; Benzer, S. Genetic Suppression of Polyglutamine Toxicity in Drosophila. Science 2000, 287, 1837–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, M.; Takaki, E.; Hayashi, T.; Kitaura, Y.; Tanaka, Y.; Inouye, S.; Nakai, A. Active HSF1 Significantly Suppresses Polyglutamine Aggregate Formation in Cellular and Mouse Models. J. Biol. Chem. 2005, 280, 34908–34916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, D.G.; Sathasivam, K.; Tobaben, S.; Stahl, B.; Marber, M.; Mestril, R.; Mahal, A.; Smith, D.L.; Woodman, B.; Bates, G.P. Progressive decrease in chaperone protein levels in a mouse model of Huntington’s disease and induction of stress proteins as a therapeutic approach. Hum. Mol. Genet. 2004, 13, 1389–1405. [Google Scholar] [CrossRef] [Green Version]
- Leitman, J.; Ulrich Hartl, F.; Lederkremer, G.Z. Soluble forms of polyQ-expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nat. Commun. 2013, 4, 2753. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Zhong, X.; Ballar, P.; Luo, S.; Shen, Y.; Rubinsztein, D.C.; Monteiro, M.J.; Fang, S. Ubiquitin ligase Hrd1 enhances the degradation and suppresses the toxicity of polyglutamine-expanded huntingtin. Exp. Cell Res. 2007, 313, 538–550. [Google Scholar] [CrossRef]
- Ghosh, D.K.; Roy, A.; Ranjan, A. The ATPase VCP/p97 functions as a disaggregase against toxic Huntingtin-exon1 aggregates. FEBS Lett. 2018, 592, 2680–2692. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Liu, C.; Zhong, Y.; Luo, S.; Monteiro, M.J.; Fang, S. Huntingtin interacts with the cue domain of gp78 and inhibits gp78 binding to ubiquitin and p97/VCP. PLoS ONE 2010, 5, e8905. [Google Scholar] [CrossRef]
- Duennwald, M.L.; Lindquist, S. Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev. 2008, 22, 3308–3319. [Google Scholar] [CrossRef] [Green Version]
- Hirabayashi, M.; Inoue, K.; Tanaka-Yamamoto, K.; Nakadate, K.; Ohsawa, Y.; Kamei, Y.; Popiel, A.H.; Sinohara, A.; Iwamatsu, A.; Kimura, Y.; et al. VCP/p97 in abnormal protein aggregates, cytoplasmic vacuoles, and cell death, phenotypes relevant to neurodegeneration. Cell Death Differ. 2001, 8, 977–984. [Google Scholar] [CrossRef] [Green Version]
- Trettel, F.; Rigamonti, D.; Hilditch-Maguire, P.; Wheeler, V.C.; Sharp, A.H.; Persichetti, F.; Cattaneo, E.; Macdonald, M.E. Dominant phenotypes produced by the HD mutation in STHdhQ111 striatal cells. Hum. Mol. Genet. 2000, 9, 2799–2809. [Google Scholar] [CrossRef]
- Carnemolla, A.; Fossale, E.; Agostoni, E.; Michelazzi, S.; Calligaris, R.; De Maso, L.; Del Sal, G.; MacDonald, M.E.; Persichetti, F. Rrs1 Is Involved in Endoplasmic Reticulum Stress Response in Huntington Disease. J. Biol. Chem. 2009, 284, 18167–18173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leitman, J.; Barak, B.; Benyair, R.; Shenkman, M.; Ashery, U.; Ulrich Hartl, F.; Lederkremer, G.Z. ER Stress-Induced eIF2-alpha Phosphorylation Underlies Sensitivity of Striatal Neurons to Pathogenic Huntingtin. PLoS ONE 2014, 9, e90803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reijonen, S.; Putkonen, N.; Nørremølle, A.; Lindholm, D.; Korhonen, L. Inhibition of endoplasmic reticulum stress counteracts neuronal cell death and protein aggregation caused by N-terminal mutant huntingtin proteins. Exp. Cell Res. 2008, 314, 950–960. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Fernandez, M.R.; Ferrer, I.; Lucas, J.J. Impaired ATF6alpha processing, decreased Rheb and neuronal cell cycle re-entry in Huntington’s disease. Neurobiol. Dis. 2011, 41, 23–32. [Google Scholar] [CrossRef]
- Ganz, J.; Shacham, T.; Kramer, M.; Shenkman, M.; Eiger, H.; Weinberg, N.; Iancovici, O.; Roy, S.; Simhaev, L.; Da’adoosh, B.; et al. A novel specific PERK activator reduces toxicity and extends survival in Huntington’s disease models. Sci. Rep. 2020, 10, 6875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, R.L.; Figueroa, A.; Court, F.A.; Thielen, P.; Molina, C.; Wirth, C.; Caballero, B.; Kiffin, R.; Segura-Aguilar, J.; Cuervo, A.M.; et al. Targeting the UPR transcription factor XBP1 protects against Huntington’s disease through the regulation of FoxO1 and autophagy. Hum. Mol. Genet. 2012, 21, 2245–2262. [Google Scholar] [CrossRef] [PubMed]
- Ince, P.G.; Lowe, J.; Shaw, P.J. Amyotrophic lateral sclerosis: Current issues in classification, pathogenesis and molecular pathology. Neuropathol. Appl. Neurobiol. 1998, 24, 104–117. [Google Scholar] [CrossRef]
- Tu, P.H.; Raju, P.; Robinson, K.A.; Gurney, M.E.; Trojanowski, J.Q.; Lee, V.M. Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc. Natl. Acad. Sci. USA 1996, 93, 3155–3160. [Google Scholar] [CrossRef] [Green Version]
- Marino, M.; Papa, S.; Crippa, V.; Nardo, G.; Peviani, M.; Cheroni, C.; Trolese, M.C.; Lauranzano, E.; Bonetto, V.; Poletti, A.; et al. Differences in protein quality control correlate with phenotype variability in 2 mouse models of familial amyotrophic lateral sclerosis. Neurobiol. Aging 2015, 36, 492–504. [Google Scholar] [CrossRef]
- Rakhit, R.; Crow, J.P.; Lepock, J.R.; Kondejewski, L.H.; Cashman, N.R.; Chakrabartty, A. Monomeric Cu,Zn-superoxide Dismutase Is a Common Misfolding Intermediate in the Oxidation Models of Sporadic and Familial Amyotrophic Lateral Sclerosis. J. Biol. Chem. 2004, 279, 15499–15504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiskinis, E.; Sandoe, J.; Williams, L.A.; Boulting, G.L.; Moccia, R.; Wainger, B.J.; Han, S.; Peng, T.; Thams, S.; Mikkilineni, S.; et al. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 2014, 14, 781–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyanagi, K.; Yamazaki, M.; Takahashi, H.; Watabe, K.; Wada, M.; Komori, T.; Morita, T.; Mizutani, T. Spinal anterior horn cells in sporadic amyotrophic lateral sclerosis show ribosomal detachment from, and cisternal distention of the rough endoplasmic reticulum. Neuropathol. Appl. Neurobiol. 2008, 34, 650–658. [Google Scholar] [CrossRef]
- Saxena, S.; Caroni, P. Selective Neuronal Vulnerability in Neurodegenerative Diseases: From Stressor Thresholds to Degeneration. Neuron 2011, 71, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Saxena, S.; Cabuy, E.; Caroni, P. A role for motoneuron subtype–selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 2009, 12, 627–636. [Google Scholar] [CrossRef]
- Nagata, T.; Ilieva, H.; Murakami, T.; Shiote, M.; Narai, H.; Ohta, Y.; Hayashi, T.; Shoji, M.; Abe, K. Increased ER stress during motor neuron degeneration in a transgenic mouse model of amyotrophic lateral sclerosis. Neurol. Res. 2007, 29, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, E.V.; Ayala, V.; Jové, M.; Dalfó, E.; Cacabelos, D.; Povedano, M.; Bellmunt, M.J.; Ferrer, I.; Pamplona, R.; Portero-Otín, M. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain 2007, 130, 3111–3123. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, H.; Almer, G.; Yamashita, S.; Guegan, C.; Nagai, M.; Xu, Z.; Sosunov, A.A.; McKhann, G.M., 2nd; Przedborski, S. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc. Natl. Acad. Sci. USA 2006, 103, 6025–6030. [Google Scholar] [CrossRef] [Green Version]
- Parakh, S.; Atkin, J.D. Protein folding alterations in amyotrophic lateral sclerosis. Brain Res. 2016, 1648, 633–649. [Google Scholar] [CrossRef]
- Hetz, C.; Thielen, P.; Matus, S.; Nassif, M.; Court, F.; Kiffin, R.; Martinez, G.; Cuervo, A.M.; Brown, R.H.; Glimcher, L.H. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009, 23, 2294–2306. [Google Scholar] [CrossRef] [Green Version]
- Wate, R.; Ito, H.; Zhang, J.H.; Ohnishi, S.; Nakano, S.; Kusaka, H. Expression of an endoplasmic reticulum-resident chaperone, glucose-regulated stress protein 78, in the spinal cord of a mouse model of amyotrophic lateral sclerosis. Acta Neuropathol. 2005, 110, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Jeon, G.S.; Nakamura, T.; Lee, J.-S.; Choi, W.-J.; Ahn, S.-W.; Lee, K.-W.; Sung, J.-J.; Lipton, S.A. Potential Effect of S-Nitrosylated Protein Disulfide Isomerase on Mutant SOD1 Aggregation and Neuronal Cell Death in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2014, 49, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.K.; Shin, K.S.; Yuan, J.; Kang, S.J. Superoxide dismutase 1 mutants related to amyotrophic lateral sclerosis induce endoplasmic stress in neuro2a cells. J. Neurochem. 2008, 104, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, J.; Crary, J.F.; Malagelada, C.; Sulzer, D.; Greene, L.A.; Levy, O.A. ATF4 protects against neuronal death in cellular Parkinson’s disease models by maintaining levels of parkin. J. Neurosci. 2013, 33, 2398–2407. [Google Scholar] [CrossRef] [PubMed]
- Farg, M.A.; Soo, K.Y.; Walker, A.K.; Pham, H.; Orian, J.; Horne, M.K.; Warraich, S.T.; Williams, K.L.; Blair, I.P.; Atkin, J.D. Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulfide-isomerase. Neurobiol. Aging 2012, 33, 2855–2868. [Google Scholar] [CrossRef]
- Walker, A.K.; Soo, K.Y.; Sundaramoorthy, V.; Parakh, S.; Ma, Y.; Farg, M.A.; Wallace, R.H.; Crouch, P.; Turner, B.J.; Horne, M.K.; et al. ALS-Associated TDP-43 Induces Endoplasmic Reticulum Stress, Which Drives Cytoplasmic TDP-43 Accumulation and Stress Granule Formation. PLoS ONE 2013, 8, e81170. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-J.; Raphael, A.R.; LaDow, E.S.; McGurk, L.; Weber, R.A.; Trojanowski, J.Q.; Lee, V.M.-Y.; Finkbeiner, S.; Gitler, A.D.; Bonini, N.M. Therapeutic modulation of eIF2α phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat. Genet. 2014, 46, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y.; Yamada, M.; Tanaka, H.; Aida, K.; Tsuruma, K.; Shimazawa, M.; Hozumi, I.; Inuzuka, T.; Takahashi, H.; Hara, H. Involvement of CHOP, an ER-stress apoptotic mediator, in both human sporadic ALS and ALS model mice. Neurobiol. Dis. 2009, 36, 470–476. [Google Scholar] [CrossRef]
- Nishitoh, H.; Kadowaki, H.; Nagai, A.; Maruyama, T.; Yokota, T.; Fukutomi, H.; Noguchi, T.; Matsuzawa, A.; Takeda, K.; Ichijo, H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008, 22, 1451–1464. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Russelakis-Carneiro, M.; Maundrell, K.; Castilla, J.; Soto, C. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J 2003, 22, 5435–5445. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Xiang, W.; Terry, L.; Kretzschmar, H.A.; Windl, O. Transcriptional analysis implicates endoplasmic reticulum stress in bovine spongiform encephalopathy. PLoS ONE 2010, 5, e14207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, B.C.; Krapfenbauer, K.; Cairns, N.; Belay, G.; Bajo, M.; Lubec, G. Overexpressed protein disulfide isomerase in brains of patients with sporadic Creutzfeldt-Jakob disease. Neurosci. Lett. 2002, 334, 196–200. [Google Scholar] [CrossRef]
- Hetz, C.; Russelakis-Carneiro, M.; Walchli, S.; Carboni, S.; Vial-Knecht, E.; Maundrell, K.; Castilla, J.; Soto, C. The disulfide isomerase Grp58 is a protective factor against prion neurotoxicity. J. Neurosci. 2005, 25, 2793–2802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, M.; Medinas, D.B.; Matamala, J.M.; Woehlbier, U.; Cornejo, V.H.; Solda, T.; Andreu, C.; Rozas, P.; Matus, S.; Munoz, N.; et al. The Protein-disulfide Isomerase ERp57 Regulates the Steady-state Levels of the Prion Protein. J. Biol. Chem. 2015, 290, 23631–23645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunziante, M.; Ackermann, K.; Dietrich, K.; Wolf, H.; Gadtke, L.; Gilch, S.; Vorberg, I.; Groschup, M.; Schatzl, H.M. Proteasomal dysfunction and endoplasmic reticulum stress enhance trafficking of prion protein aggregates through the secretory pathway and increase accumulation of pathologic prion protein. J. Biol. Chem. 2011, 286, 33942–33953. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Castilla, J.; Soto, C. Perturbation of endoplasmic reticulum homeostasis facilitates prion replication. J. Biol. Chem. 2007, 282, 12725–12733. [Google Scholar] [CrossRef] [Green Version]
- Moreno, J.A.; Radford, H.; Peretti, D.; Steinert, J.R.; Verity, N.; Martin, M.G.; Halliday, M.; Morgan, J.; Dinsdale, D.; Ortori, C.A.; et al. Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature 2012, 485, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Moreno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A.; Willis, A.E.; Fischer, P.M.; Barrett, D.A.; et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 2013, 268, 206ra138. [Google Scholar] [CrossRef]
- Kristiansen, M.; Deriziotis, P.; Dimcheff, D.E.; Jackson, G.S.; Ovaa, H.; Naumann, H.; Clarke, A.R.; van Leeuwen, F.W.; Menendez-Benito, V.; Dantuma, N.P.; et al. Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol. Cell 2007, 26, 175–188. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, X.; Li, L.; Zheng, J. Inhibition of amyloid-beta aggregation in Alzheimer’s disease. Curr. Pharm. Des. 2014, 20, 1223–1243. [Google Scholar] [CrossRef]
- Wang, L.; Bharti; Kumar, R.; Pavlov, P.F.; Winblad, B. Small molecule therapeutics for tauopathy in Alzheimer’s disease: Walking on the path of most resistance. Eur. J. Med. Chem. 2021, 209, 112915. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, K.-T. Therapeutic Approaches for Inhibition of Protein Aggregation in Huntington’s Disease. Exp. Neurobiol. 2014, 23, 36–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, B.; Kim, T.; Huh, Y.-J.; Lee, J.; Lee, Y.-I. Amelioration of Mitochondrial Quality Control and Proteostasis by Natural Compounds in Parkinson’s Disease Models. Int. J. Mol. Sci. 2019, 20, 5208. [Google Scholar] [CrossRef] [Green Version]
- Uliassi, E.; Nikolic, L.; Bolognesi, M.L.; Legname, G. Therapeutic strategies for identifying small molecules against prion diseases. Cell Tissue Res. 2022, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, A.; Tauffenberger, A.; Aggad, D.; Rouleau, G.; Drapeau, P.; Parker, J.A. Mutant TDP-43 and FUS Cause Age-Dependent Paralysis and Neurodegeneration in C. elegans. PLoS ONE 2012, 7, e31321. [Google Scholar] [CrossRef]
- Vaccaro, A.; Patten, S.A.; Ciura, S.; Maios, C.; Therrien, M.; Drapeau, P.; Kabashi, E.; Parker, J.A. Methylene Blue Protects against TDP-43 and FUS Neuronal Toxicity in C. elegans and D. rerio. PLoS ONE 2012, 7, e42117. [Google Scholar] [CrossRef] [Green Version]
- Paganoni, S.; Macklin, E.A.; Hendrix, S.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; Quick, A.; et al. Trial of Sodium Phenylbutyrate–Taurursodiol for Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2020, 383, 919–930. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Chambers, J.E.; Ron, D. Pharmacological targeting of endoplasmic reticulum stress in disease. Nat. Rev. Drug Discov. 2021, 21, 115–140. [Google Scholar] [CrossRef]
- Radford, H.; Moreno, J.A.; Verity, N.; Halliday, M.; Mallucci, G.R. PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 2015, 130, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Mercado, G.; Castillo, V.; Soto, P.; Lopez, N.; Axten, J.M.; Sardi, S.P.; Hoozemans, J.J.M.; Hetz, C. Targeting PERK signaling with the small molecule GSK2606414 prevents neurodegeneration in a model of Parkinson’s disease. Neurobiol. Dis. 2018, 112, 136–148. [Google Scholar] [CrossRef]
- Grande, V.; Ornaghi, F.; Comerio, L.; Restelli, E.; Masone, A.; Corbelli, A.; Tolomeo, D.; Capone, V.; Axten, J.M.; Laping, N.J.; et al. PERK inhibition delays neurodegeneration and improves motor function in a mouse model of Marinesco-Sjögren syndrome. Hum. Mol. Genet. 2018, 27, 2477–2489. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.W.; Jiang, H.; Pei, Z.; Tanaka, Y.; Morita, H.; Sawa, A.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet. 2005, 14, 3801–3811. [Google Scholar] [CrossRef] [PubMed]
- Costa-Mattioli, M.; Gobert, D.; Stern, E.; Gamache, K.; Colina, R.; Cuello, C.; Sossin, W.; Kaufman, R.; Pelletier, J.; Rosenblum, K.; et al. eIF2α Phosphorylation Bidirectionally Regulates the Switch from Short- to Long-Term Synaptic Plasticity and Memory. Cell 2007, 129, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Bruch, J.; Xu, H.; Rosler, T.W.; De Andrade, A.; Kuhn, P.H.; Lichtenthaler, S.F.; Arzberger, T.; Winklhofer, K.F.; Muller, U.; Hoglinger, G.U. PERK activation mitigates tau pathology in vitro and in vivo. EMBO Mol. Med. 2017, 9, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Zyryanova, A.; Weis, F.; Faille, A.; Alard, A.A.; Crespillo-Casado, A.; Sekine, Y.; Harding, H.; Allen, F.; Parts, L.; Fromont, C.; et al. Binding of ISRIB reveals a regulatory site in the nucleotide exchange factor eIF2B. Science 2018, 359, 1533–1536. [Google Scholar] [CrossRef] [Green Version]
- Bugallo, R.; Marlin, E.; Baltanás, A.; Toledo, E.; Ferrero, R.; Vinueza-Gavilanes, R.; Larrea, L.; Arrasate, M.; Aragón, T. Fine tuning of the unfolded protein response by ISRIB improves neuronal survival in a model of amyotrophic lateral sclerosis. Cell Death Dis. 2020, 11, 397. [Google Scholar] [CrossRef]
- Hughes, D.T.; Halliday, M.; Smith, H.L.; Verity, N.C.; Molloy, C.; Radford, H.; Butcher, A.J.; Mallucci, G.R. Targeting the kinase insert loop of PERK selectively modulates PERK signaling without systemic toxicity in mice. Sci Signal 2020, 13, eabb4749. [Google Scholar] [CrossRef]
- Das, I.; Krzyzosiak, A.; Schneider, K.; Wrabetz, L.; D’Antonio, M.; Barry, N.; Sigurdardottir, A.; Bertolotti, A. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 2015, 348, 239–242. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.-Q.; Ren, M.; Wang, J.; Zhang, J.; Yin, X.; Wang, S.-Y.; Qi, Y.; Wang, X.-D.; Feng, H.-L. Guanabenz delays the onset of disease symptoms, extends lifespan, improves motor performance and attenuates motor neuron loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neuroscience 2014, 277, 132–138. [Google Scholar] [CrossRef]
- Wang, L.; Popko, B.; Tixier, E.; Roos, R.P. Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1-induced amyotrophic lateral sclerosis. Neurobiol. Dis. 2014, 71, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Celardo, I.; Costa, A.C.; Lehmann, S.; Jones, C.; Wood, N.; Mencacci, N.E.; Mallucci, G.R.; Loh, S.H.; Martins, L.M. Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease. Cell Death Dis. 2016, 7, e2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliday, M.J.; Radford, H.; Sekine, Y.; Moreno, J.J.; Verity, N.; Le Quesne, J.P.C.; Ortori, C.A.; Barrett, D.A.; Fromont, C.; Fischer, P.M.; et al. Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 2015, 6, e1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliday, M.; Radford, H.; Zents, K.A.M.; Molloy, C.; Moreno, J.A.; Verity, N.C.; Smith, E.; Ortori, C.A.; Barrett, D.A.; Bushell, M.; et al. Repurposed drugs targeting eIF2α-P-mediated translational repression prevent neurodegeneration in mice. Brain J. Neurol. 2017, 140, 1768–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandjean, J.M.D.; Madhavan, A.; Cech, L.; Seguinot, B.O.; Paxman, R.J.; Smith, E.; Scampavia, L.; Powers, E.T.; Cooley, C.B.; Plate, L.; et al. Pharmacologic IRE1/XBP1s activation confers targeted ER proteostasis reprogramming. Nat. Chem. Biol. 2020, 16, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Plate, L.; Cooley, C.B.; Chen, J.J.; Paxman, R.; Gallagher, C.M.; Madoux, F.; Genereux, J.C.; Dobbs, W.; Garza, D.; Spicer, T.P.; et al. Small molecule proteostasis regulators that reprogram the ER to reduce extracellular protein aggregation. eLife 2016, 5, e15550. [Google Scholar] [CrossRef] [PubMed]
- Hoffstrom, B.G.; Kaplan, A.; Letso, R.; Schmid, R.S.; Turmel, G.J.; Lo, D.C.; Stockwell, B.R. Inhibitors of protein disulfide isomerase suppress apoptosis induced by misfolded proteins. Nat. Chem. Biol. 2010, 6, 900–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Lee, J.; Hernandez, M.A.S.; Mazitschek, R.; Ozcan, U. Treatment of Obesity with Celastrol. Cell 2015, 161, 999–1011. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Zhang, Q.; Luo, P.; Gu, L.; Shen, S.; Tang, H.; Zhang, Y.; Lyu, M.; Shi, Q.; Yang, C.; et al. Neuroprotective Effects of Celastrol in Neurodegenerative Diseases-Unscramble Its Major Mechanisms of Action and Targets. Aging Dis. 2022, 13, 815. [Google Scholar] [CrossRef]
- Hyun, S.; Shin, D. Chemical-Mediated Targeted Protein Degradation in Neurodegenerative Diseases. Life 2021, 11, 607. [Google Scholar] [CrossRef]




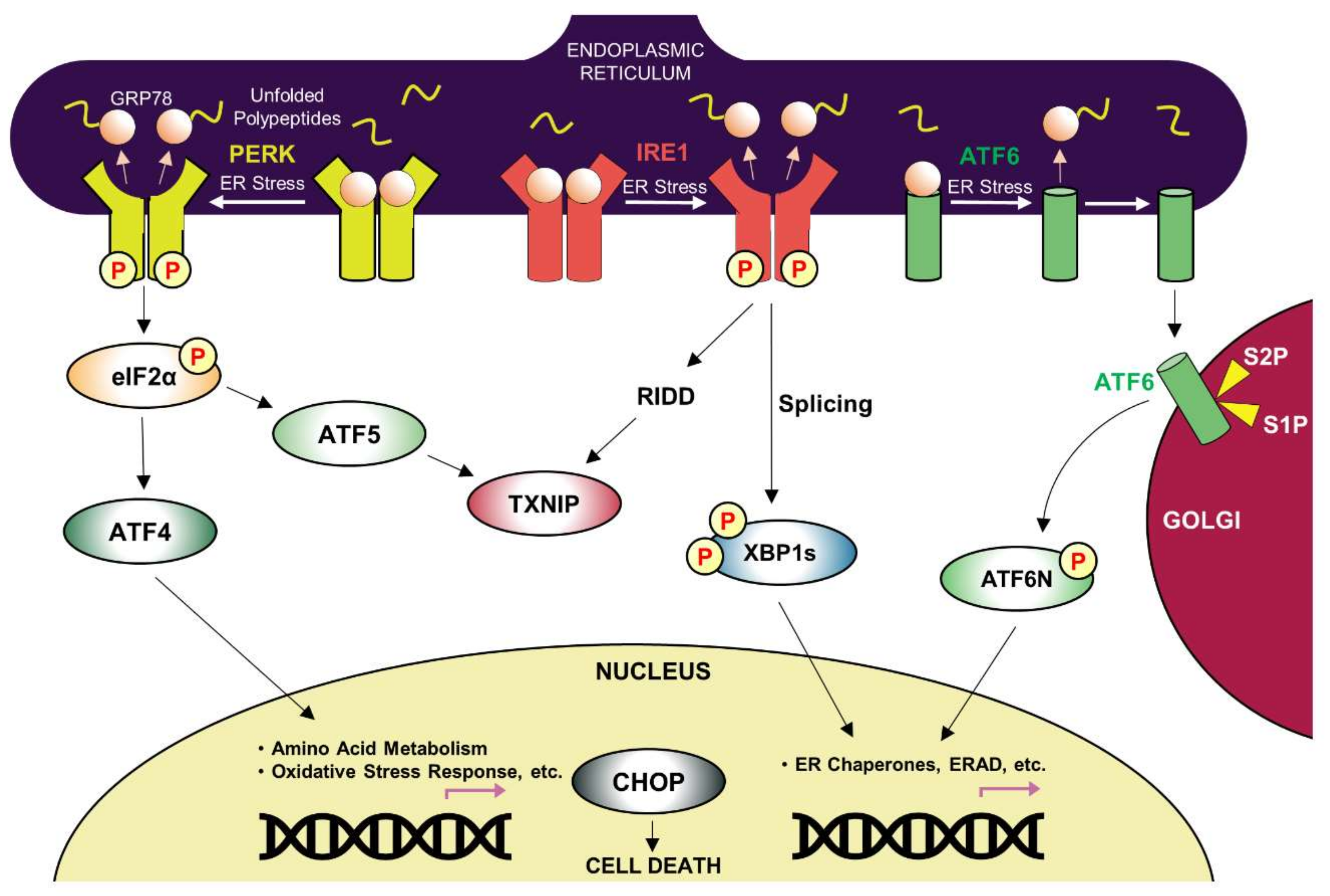{kind=link}
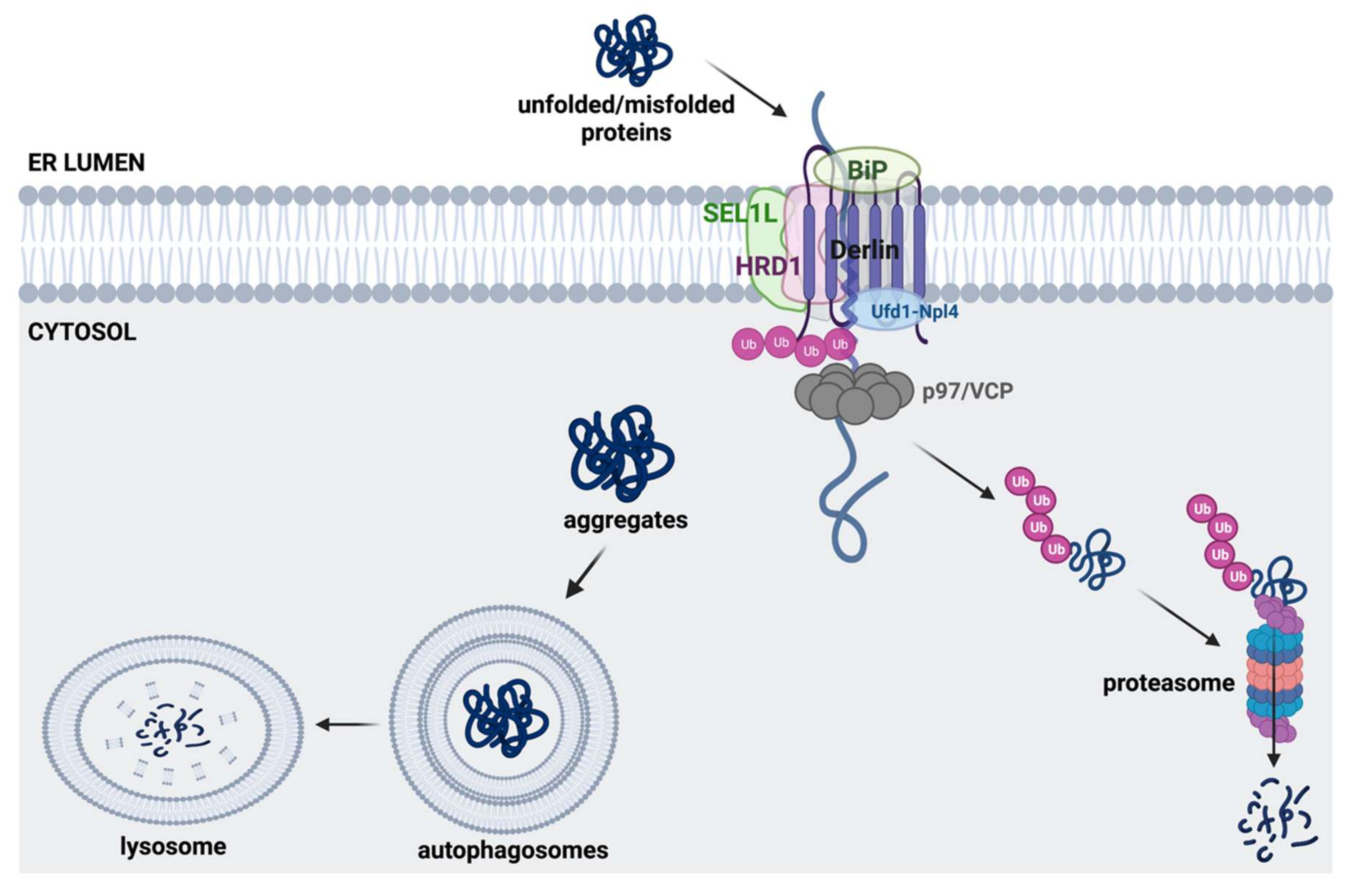{kind=link}
{kind=link}
| Disease | Affected Brain Regions | Disease-Causing Protein Deposited/Mutant | Effective Chemical Chaperones and Other Small Molecules |
|---|---|---|---|
| Alzheimer disease | Cortex, hippocampus, basal forebrain, brain stem | Amyloid β peptide derived from APP/ mutation in APP, presenilin1 or presenilin2, APOE4 allele | Congo red, polyphenol-based compounds, curcumin, thioflavin-T |
| Hyperphosphorylated tau | Curcumin derivatives (e.g.,Dibenzoylmethane), methylene blue, N744, rhodanines, aminothienopyridazines (ATPZs) | ||
| Parkinson disease | Substantia nigra, cortex, locus coeruleus, raphe, etc. | α-Synuclein | Polyphenol-based compounds, curcumin, myricetin, tanshinones, ginsenoside Rb1 |
| Huntington disease | Cortex, striatum, other basal ganglia, etc. | Huntington with polyglutamine expansion (exon1) | Congo red, trehalose, C2-8 |
| Amyotrophic lateral sclerosis | Spinal motor neurons and motor cortex | Mutations in C9orf72 (40~50%), SOD1 (20~25%), TDP-43 (4~5%), FUS (4~5%), etc. | 4-PBA, TUDCA, methylene blue |
| Prion disease | Cortex, thalamus, brain stem, cerebellum, etc. | Prion protein (PrPSc) | Diphenylmethane derivative (GN8), carbazole derivative (5y), small aromatic molecules (NPRs) |
| UPR Target | Molecule | Target Pathology | Reference | |
|---|---|---|---|---|
| PERK signaling activators | CCT020312 |  | HD, tauopathy | [236,284] |
| MK-28 |  | |||
| eIF2α phosphatase inhibitors | Salubrinal |  | HD, α-synucleinopathies | [157,234,282] |
| Sephin1 |  | ALS | [288] | |
| Guanabenz |  | ALS | [289,290] | |
| PERK kinase inhibitor | GSK2606414 |  | tau-related pathology (AD, frontotemporal dementia), Prion, PD, Marinesco-Sjögren syndrome, ALS | [257,268,279,280,281,291] |
| Downstream inhibitors of PERK signaling | ISRIB |  | AD, Prion | [150,292] |
| Trazodone |  | Prion, tauopathy-frontotemporal dementia | [293] | |
| Dibenzoylmethane |  | |||
| IRE1/XBP1s activation | IXA1 |  | AD | [294] |
| IXA4 |  | |||
| IXA6 |  | |||
| Activation of ATF6 transcriptional activity | AA147 (N-(2-Hydroxy-5-methylphenyl)-3-phenylpropanamide) |  | Amyloid aggregates-related pathology | [295] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Kim, D.K.; Jeong, S.; Lee, J. The Common Cellular Events in the Neurodegenerative Diseases and the Associated Role of Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2022, 23, 5894. https://doi.org/10.3390/ijms23115894
Kim S, Kim DK, Jeong S, Lee J. The Common Cellular Events in the Neurodegenerative Diseases and the Associated Role of Endoplasmic Reticulum Stress. International Journal of Molecular Sciences. 2022; 23(11):5894. https://doi.org/10.3390/ijms23115894
Chicago/Turabian StyleKim, Soojeong, Doo Kyung Kim, Seho Jeong, and Jaemin Lee. 2022. "The Common Cellular Events in the Neurodegenerative Diseases and the Associated Role of Endoplasmic Reticulum Stress" International Journal of Molecular Sciences 23, no. 11: 5894. https://doi.org/10.3390/ijms23115894