1. Introduction
The importance of a finely balanced T cell response—strong enough to prevent cancer and to protect from infectious diseases while not damaging its own tissue—is reflected in the increasing amount of therapeutic approaches targeting T cells. These strategies aim to either suppress T cell activity, as in the case of autoimmunity and organ transplantation, or boost it, which is preferable for anti-tumor approaches. The latter applies, for example, for an adoptive transfer of T cells (ACT) expressing a chimeric antigen receptor as a therapeutic tool. In this regard, it is of great interest to understand the molecular switches of T cell activation, to modulate their responsiveness and the T cell-mediated immune response outcome. Thus, there is a strong need for physiological model systems to test potential modulators of T cell responses.
However, since T cells demonstrate an enormous range (estimated about 10
6–10
8, [
1]) of distinct T cell receptors (TCR), finding appropriate tools for investigations is still challenging. In vitro, the issues of unknown antigen-specificity and low precursor frequency can be circumvented by polyclonal stimulation, using, for example, antibodies targeting the TCR-associated CD3ε-chain and the CD28 co-receptor, phorbol esters and ionomycin, which act intracellularly to activate the signaling cascade downstream of the TCR, or mitogens, such as concanavalin A. Nonetheless, these strong stimuli are quite artificial and therefore not truly reflecting the physiological stimulation through the TCR via peptide-loaded MHCs (major histocompatibility complex) on cells that additionally provide signals by co-stimulatory receptors or secreted cytokines. One step towards a more physiological in vitro T cell activation approach can be reached by a so-called allogenic stimulation. Allogenicity was first noted in the 1950s, when George Snell bred two mouse strains, which were nearly identical but differing in tissue compatibility upon transplantation [
2]. Jean Dausset observed an immune response against donor leukocytes during human blood transfusion [
3], which later on led to the discovery of the MHC locus. Briefly, in an allogenic setting, T cells confound a non-self MHC donor peptide complex with a self-MHC, presenting a foreign peptide. This leads then to an erroneous T cell activation, even in the absence of any signs of danger. Besides the significance of this process for transplantation medicine, the system can also be hijacked for T cell studies using in vitro mixed lymphocyte reactions (MLR). In this setting, T cells of interest are mixed with at least 3-fold more murine splenocytes or peripheral blood mononuclear cells (PBMCs) of a different MHC haplotype [
4,
5]. To avoid proliferation of the stimulator splenocytes, these are either irradiated or mitomycin C-treated. However, these treatments lead to many dead cells within the co-culture that might influence the responder T cells’ behavior. Alternatively, a “one-way MLR” can be applied that uses for stimulation much less allogenic dendritic cells, which are matured by LPS treatment but are not treated in any other way, leading to cell cycle arrest and thus cell death.
Although providing stimulation through the TCR, all these in vitro models face the problem of not taking into account migration and localization within tissues, representing an environment lacking the stimulatory or inhibitory factors provided by other cell types. This obstacle can only be overcome using in vivo models.
Thus, for example, mice can be studied upon immunization with aluminum hydroxide-emulsified ovalbumin (OVA-alum). However, the tracking of T cell activation using this in vivo approach is challenging due to the low frequency of (naïve) antigen-specific precursors (approx. 0.8 to 10 cells per million naïve CD4
+ T cells; [
6]). Thus, with this approach, only a small number of cells can be analyzed at the endpoint, which makes statements about activation/exhaustion difficult, even though antigen-specific T cells can be detected via MHC-tetramer technology. Moreover, using tetramers covers only the response to one specific peptide, which does not reflect the whole T cell response to the protein.
Other in vivo models are based on an ACT of TCR-transgenic (tgTCR) T cells, using a monoclonal TCR, followed by immunization with the cognate antigen. For instances, OT-I and OT-II mice are used, whose CD8
+ and CD4
+ T cells, respectively, harbor a TCR specific for a peptide from chicken ovalbumin [
7,
8]. In this way, the frequency of antigen-specific T cells is much higher and therefore easier to investigate, especially when antibodies specific for the tgTCR-T cells are available. However, when using this approach, one has to consider the drawbacks of this system. For example, tgTCR-T cells experience an artificially strong activation and demonstrate an altered thymic development; e.g., OT-I mice develop almost just CD8
+ T cells due to MHC class I-mediated positive selection. Furthermore, no polyclonal response is analyzed using this approach.
Another possibility to address T cell responses in vivo is based on the above-described MLR approach, which increases the frequency of potential responders to approximately 7% [
9]. Thereby, recipient mice, which are lymphodepleted or totally body irradiated to prevent rejection of the transferred cells, receive an allogenic T cell-depleted bone marrow transplantation (TCD-BMT) along with allogenic T cells. This setup is used as a MHC-mismatched mouse model of T cell-mediated acute graft-versus-host disease (aGVHD). Direct cytotoxic effects of donor T cells on recipient tissues lead to damage, and the release of pro-inflammatory cytokines contributes to severe immunopathology, which is usually lethal, at the latest, four weeks after transfer (reviewed in [
10]).
Unlike the aGVHD model described above, the so-called parent-into-F1 (P → F1) model avoids rejection of the transferred T cells of a certain haplotype by using semi-allogenic F1-hybrids of this haplotype and of an allogenic haplotype as a host. For instance, C57BL/6 splenocytes or sorted T cells (MHC haplotype b) are transferred to offspring of a C57BL/6 × DBA2 breeding (b/d haplotype). Using this approach, the irradiation of the recipients is not necessary. The transferred parental strain T cells will respond vigorously to the mismatched MHC d haplotype of the recipient.
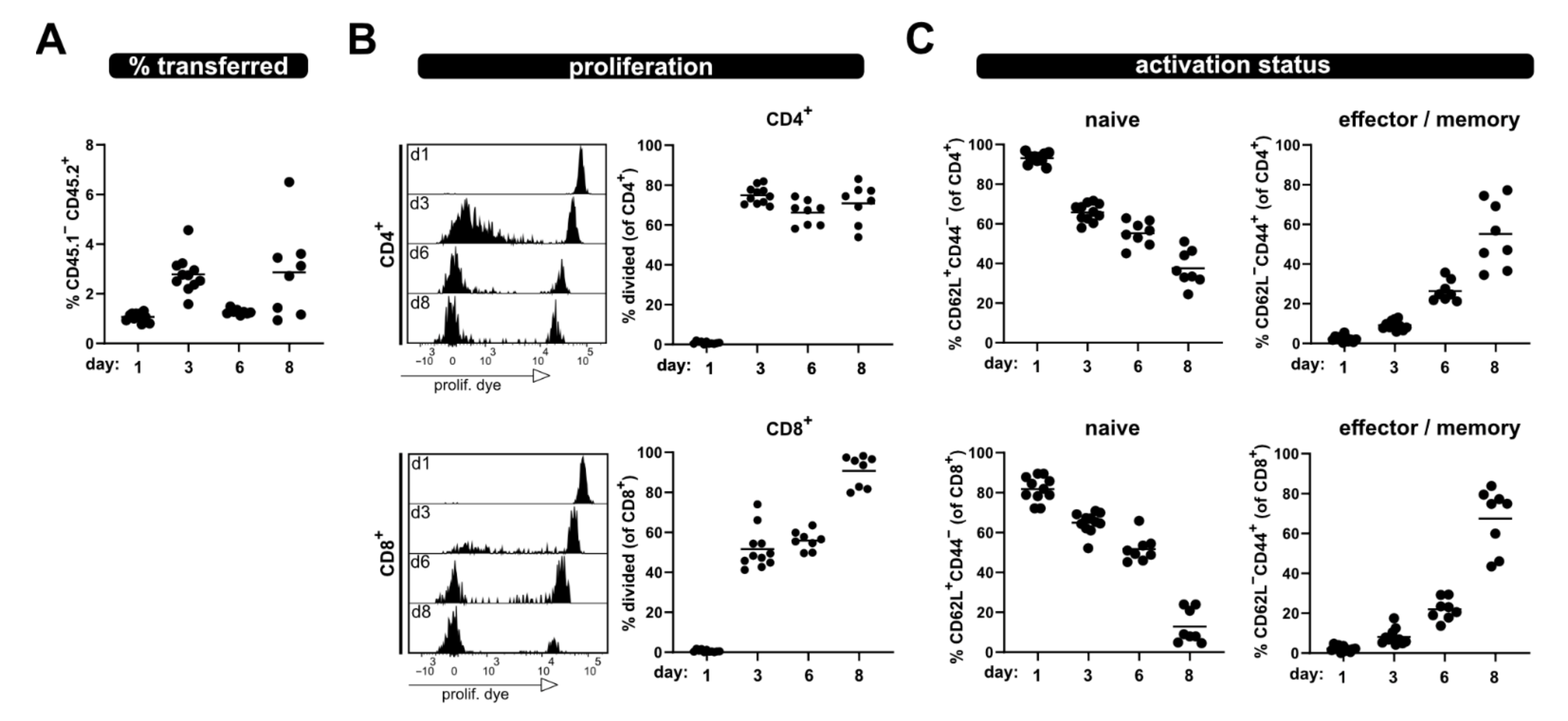
Here, we combined the P → F1 model with the congenic marker of CD45, which allows us to track the activation of transferred T cells on single cell level. Our analyses were performed within eight days after ACT before the recipient mice develop any obvious signs of aGVHD. Using this approach, we have additionally analyzed T cells derived from
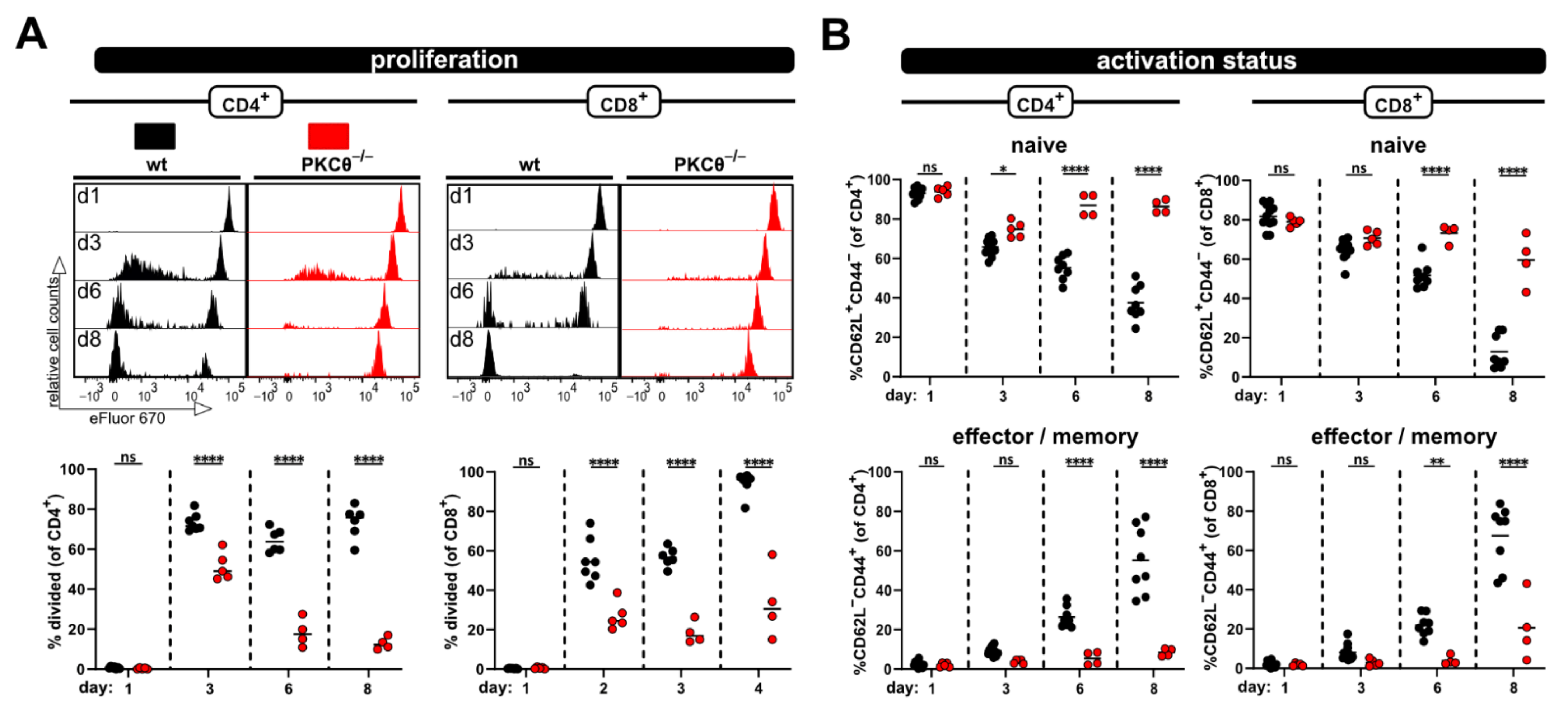
PKCθ-knockout mice. PKCθ is a central signaling molecule downstream of the TCR, activated by DAG-binding, whose deficiency is known to impair T cell activation resulting in reduced interleukin (IL)-2 expression and proliferation ([
11,
12]). Thus, we have validated the model for positive regulators of T cell activation.
3. Discussion
The P → F1 model, established more than 40 years ago [
16], has been used in several studies as a disease model to understand T cell regulating factors important for the development of aGVHD or lupus-like cGVHD. Initially, activated donor CD4
+ T cells expand and provide help for host B cells, which proliferate and produce autoantibodies. In addition, donor CD8
+ T cells will be activated and differentiate into cytotoxic T lymphocytes (CTL) that eliminate host lymphocytes, primarily B cells. The latter response leads to aGVHD with a profound immunodeficiency by day 14, counteracting donor-CD4
+/host-B cell-mediated autoimmune symptoms (reviewed in [
13]). Of note, the choice of the parental donor determines the disease phenotype. For example, while a transfer of T cells from a C57BL/6 donor to a B6D2F1 recipient (B6 → F1) results in an aGVHD phenotype, the transfer of DBA-donor T cells (DBA → F1) induces a lupus-like cGVHD. This might be due to the differences in the strain-specific quality of CD8
+ CTL responses. For instance, for the C57BL/6 mouse strain, in contrast to DBA mice, high proportions of cytotoxic CD8
+ T cells have been described [
17]. Furthermore, CD4
+ T cells of DBA mice have been demonstrated to be poor IL-2 producers and thus provide less help for CD8
+ T cells [
18]. Altogether, these studies highlight the importance of fine-tuning effector T cell responses of the distinct T cell subpopulations to impact disease outcome. In this regard, if CD4
+ T cells are transferred to F1-hosts without donor CD8
+ T cells, a lupus-like cGVHD develops instead of aGVHD [
19]. Hence, under these circumstances, donor CD8
+ CTL responses are beneficial, since they counteract autoantibody production by killing host B cells.
Of note, using the P → F1 model, different approaches have been conducted to modulate T cell responses and thereby affect the disease outcome to induce either acute or chronic GVHD. In brief, while the blocking of CTLA-4 simultaneous with ACT prevented the development of acute and chronic GVHD, other treatments promoted either one or the other disease phenotype [
20]. Treatment of mice with an agonistic anti-CD40 antibody, an antibody blocking CD80, recombinant IL-12 or IL-21 promotes CTL-development and leads to aGVHD in the DBA → F1 model, which usually develop cGVHD [
21,
22,
23,
24]. In contrast, the transfer of IL-21R-deficient donor T cells, early neutralization of TNF-α, or administration of IL-233, a hybrid cytokine composed of IL-2 and IL-33, impaired the donor CD8
+ T cell expansion and maturation, thereby attenuating aGVHD symptoms in the B6 → F1 model [
22,
25,
26].
The mentioned findings focus primarily on the outcome of the two major P → F1 disease phenotypes, which can be mainly distinguished by the extent of donor T cell engraftment, elimination or expansion of host B cells and serum anti-DNA antibody levels, at later time points (10 days to months). However, we are interested in rather early events of T cell activation on a cellular and molecular level. Thus, we modified the P → F1 model to assess how genetic alterations influence T cell activation by analyzing the phenotype of T cells from knockout mice within 8 days after transfer. Using this time frame for our analyses, no massive killing of endogenous cells and also no severe health issues are expected from what is known from earlier studies [
13]. This model provides an in vivo T cell stimulation without the need of irradiation or lymphodepletion of the recipients. Of note, in contrast to many other studies applying the P → F1 model, we only transfer donor T cells instead of huge numbers of unfractionated parental strain splenocytes (typically 50 × 10
6 C67BL/6 donor T cells are used [
27]). Furthermore, since we use CD45 isoforms to distinguish donor and host T cells, similar to what has been performed by Suchin et al., we are able to track transferred T cells and determine their proliferation even when the proliferation dye labeling has already been diluted to unstained levels [
9,
28]. Besides endpoint analyses addressing the splenic T cell subsets, it is also possible to follow the donor T cell phenotypes over time by taking blood samples in a minimal invasive way. Of note, the frequency of transferred T cells was lower at day 6 in blood than at day 3 or day 8. We can only speculate why this is the case: the trapping of the allogenic T cells in organs, where they recognize their cognate antigen and also re-distribution to peripheral tissues, which is a characteristic of effector T cells, might contribute.
We believe that this model based on transferring congenic allo-reactive T cells is a valuable tool for the investigation of genes of interest regarding a physiological T cell activation and effector function in vivo. In addition to the here described readouts, it is also possible to investigate tissue localization of the transferred T cells, for example by immunohistochemistry, and tissue infiltrating cells, e.g., from the gut, could be analyzed by flow cytometry.
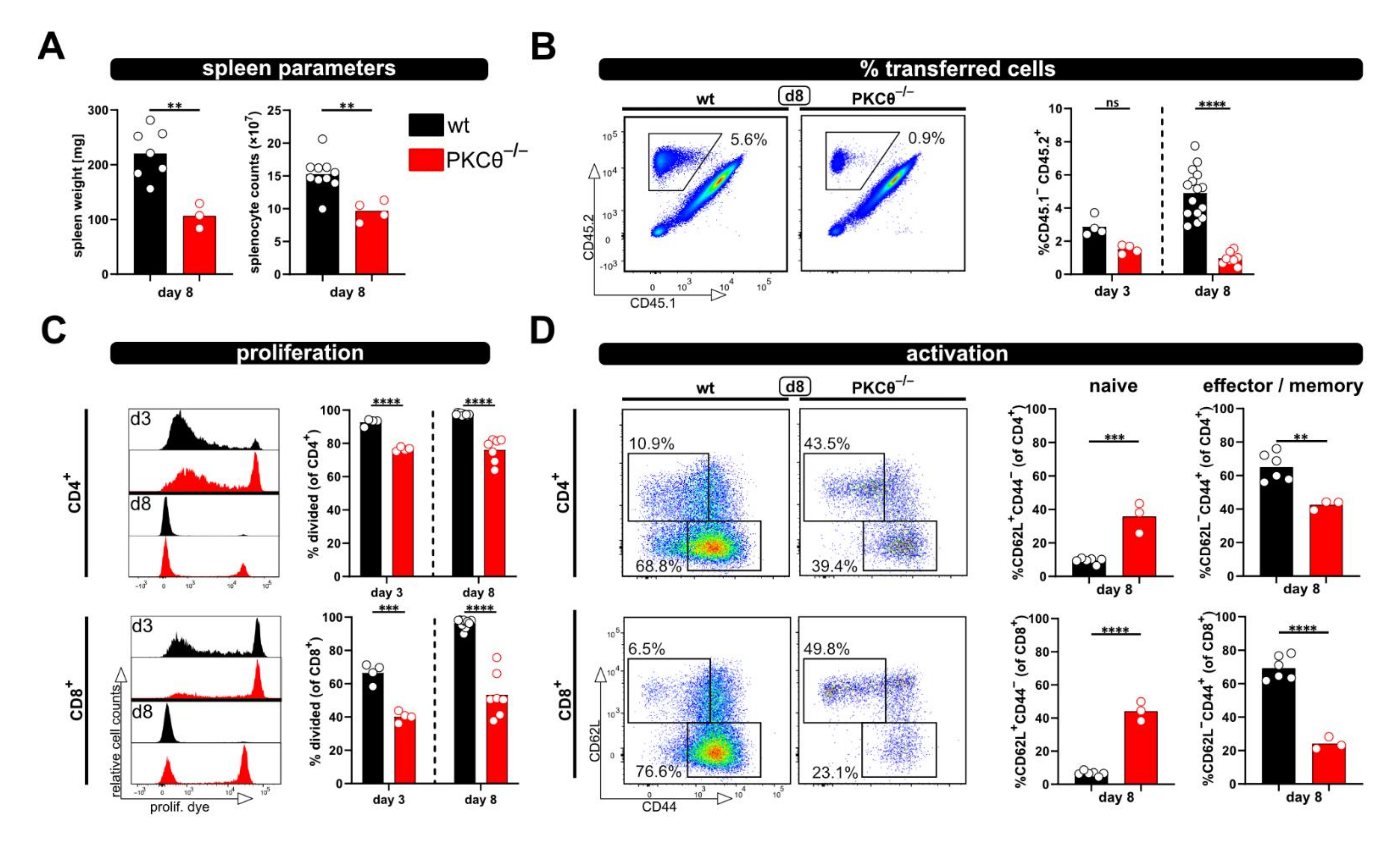
Using this approach, we observed enlarged spleens as well as increased splenocyte counts on day 3 and 8 after ACT, accompanied by the massive proliferation of CD4
+ as well as CD8
+ T cells in allogenic but not syngenic recipients. In this regard, the flow cytometric analysis revealed that the donor T cell phenotype changes from a dominantly naive to an exclusively effector/memory state and acquires exhausted profiles on day 8 after ACT. This can also be observed in a blood kinetic over time (d1, d3, d6, d8) with the accumulation of proliferated cells and a switch from a naive to an effector phenotype. However, this was all impaired in the case of PKCθ-deficient donor T cells, supporting our attempt to use this model in the future to determine genes that might be positive regulators of T cell functions. Our results, demonstrating reduced allogenic proliferation of T cells lacking PKCθ, are in line with what is known from the literature implicating a role of PKCθ in alloreactivity and GVHD. Thus, PKCθ-deficient donor T cells demonstrated reduced ability to induce disease in MHC-mismatched hosts (using TCD-BMT GVHD models [
29,
30], fitting to impaired in vivo proliferation in allogenic settings at day 3 or 4 after ACT [
29,
31]. Additionally, inhibitors for PKCθ, but also targeting PKCα, could prevent GVHD [
30].
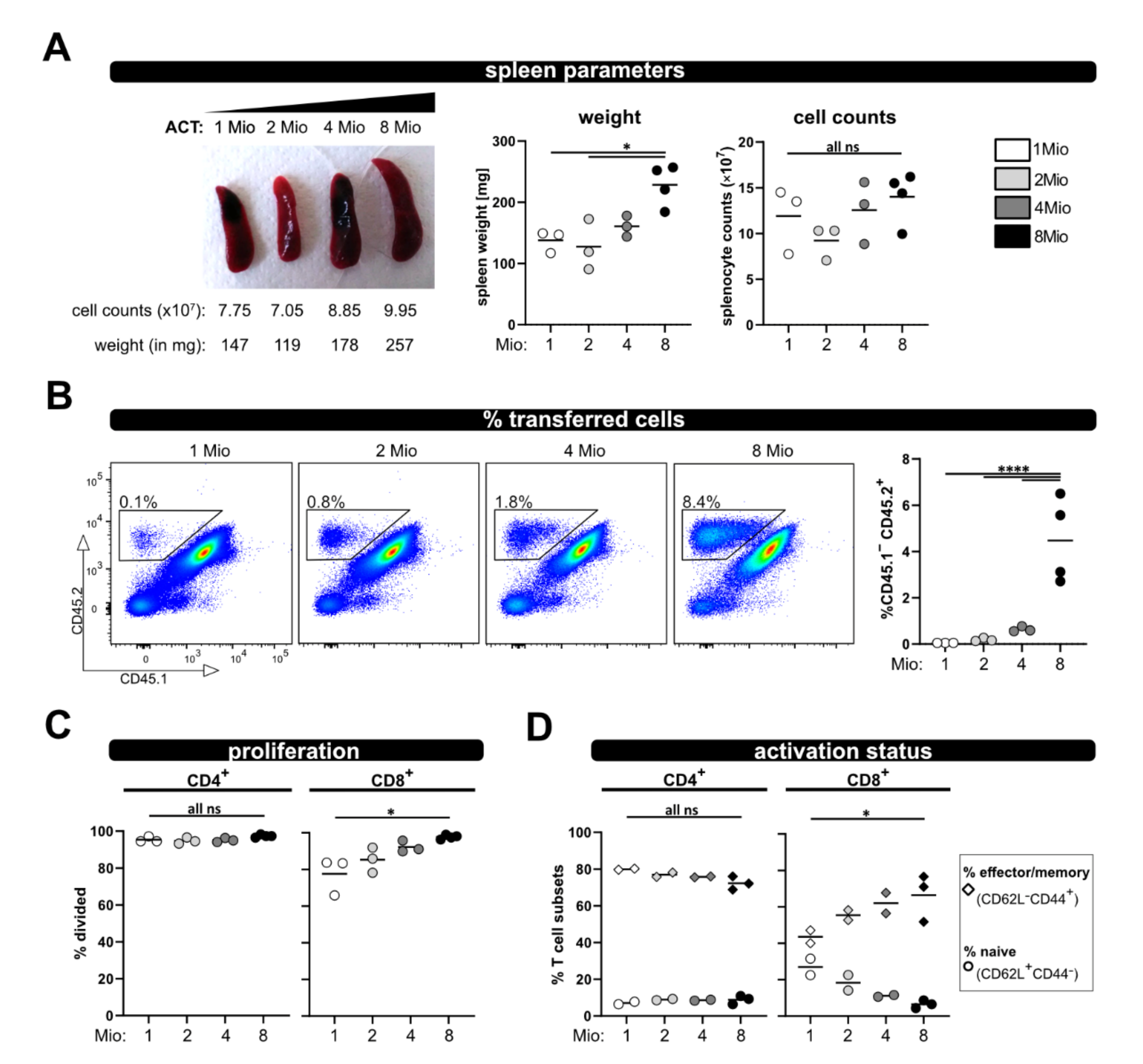
Furthermore, we could demonstrate that downscaling the number of transferred T cells is possible. In this case, it is feasible to use the model if less cells are available; we tested as few as one million transferred T cells. However, one has to be aware that decreasing numbers of transferred T cells might impede the analysis. In particular, the tracking of parameters over time in blood samples will be difficult, since there is only limited availability of blood, which can be harvested per time point from a living animal. Moreover, we observed that CD8
+ T cell activation was slightly stronger the more T cells have been transferred. Therefore, it is obviously important that equal T cell numbers are transferred; especially when comparing wild type and knockout T cells. In this regard, it has been demonstrated that the transfer of suboptimal donor T cells numbers (in their study ≤ 30 × 10
6 unfractionated splenocytes) does not induce aGVHD [
27]. Nevertheless, we could demonstrate that it is possible to analyze the allogenic response on a single cell level even if reduced T cell numbers are used for ACT. Thus, the model could be also extended to investigations of T cell subpopulations, e.g., transferring CD4
+ or CD8
+ T cells separately or performing a transfer of genetically modified CD4
+ T cells together with wild type CD8
+ T cells, or the other way around, depending on the research question.
Taken together, our findings prove that the here described model is well suited to investigate regulators of T cell responses at a single cell level in vivo. Here, we focused on a basic setting, where genetically modified T cells are transferred to allogenic recipients serving as a starting point for a highly flexible, physiological model system investigating T cell activation in vivo, which can be adapted to one’s specific research questions and technical possibilities.
4. Materials and Methods
4.1. Mice
Male DBA/2J mice were purchased from The Jackson Laboratory (strain #000671) and crossed with B6.SJL-Ptprca Pepcb/BoyJ (strain #002014; “CD45.1
+/1
+”) females to receive a hybrid (F1 generation; B6D2F1). Syngenic controls were obtained by the breeding of C57BL/6J (strain #000664) to B6.SJL-Ptprca Pepcb/BoyJ mice. PKCθ-deficient mice ([
11]) were used to investigate hypo-responses. Mice were maintained under specific pathogen-free (SPF) conditions. All animal experiments were performed in accordance with national and European guidelines and authorized by the committee on animal experiments (2020-0.345.526 and BMWFW-66.011/0076-WF/V/3b/2018). Experimental mice were chosen randomly from litters.
4.2. Splenocyte Isolation, T Cell Sorting and Proliferation Dye Labeling
Single cell suspensions of spleens were prepared by mechanical disintegration using metal sieves or 100 µM cell strainers (Falcon). Thereafter, erythrocytes were removed by lysis (0.15 M NH4Cl, 10 mM KHCO3 and 0.1 mM EDTA) for 2-6 min at room temperature (RT), followed by a wash/ filter step using 1× PBS, 0.5% BSA, 0.5 M EDTA and 40 µM cell strainers (Falcon). Viable cell counts were determined after staining with acridine orange and propidium iodide (AO/PI) Cell Viability Kit (F23001-LG, Biocat, Heidelberg, Germany) on a LUNA Automated Cell Counter (Logos Biosystems, Villeneuve d’Ascq, France).
CD3+ T cells were sorted untouched by negative selection using MACS technology-based isolation kits (130-095-130, pan-T cells) along with pre-separation filters, LS columns and a QuadroMACS separator (all Miltenyi Biotec, Bergisch Gladbach, Germany), according to the manufacturer’s instructions. Sort purity was checked by flow cytometry.
Isolated CD3+ T cells were labeled with cell proliferation dye eFluor670 (ebiosciences 65-0840). Therefore, a twofold labeling solution (10 µM) was prepared in HBSS with Mg2+ and Ca2+ (HBSS++). T cells were washed twice with HBSS++ at RT and adjusted to 1 × 107 cells/mL. An equal volume of the twofold labeling solution was added and mixed immediately with the cells by inverting the tube. Cells were incubated for 5 min at 37 °C in the dark. The reaction was stopped with cold FCS and RPMI followed by a wash step with RPMI and subsequently with HBSS++. Finally, cells were resuspended in HBSS++ (8 × 106/100 µL) and injected intravenously.
4.3. P → F1 Model
CD3+ T cells from C57BL/6 mice (CD45.2, MHC haplotype b) were isolated as described above, labeled with proliferation dye and transferred to allogenic B6D2F1 (MHC haplotype b/d) or syngeneic control (MHC haplotype b) recipients. A total of 1, 2, 4 or 8 × 106 T cells resuspended in 100 µL HBSS++ were injected into the lateral tail vein. For endpoint analyses, spleens from recipient mice were harvested and weighted on day 3 or day 8 after ACT. Total splenocyte counts were determined from single cell suspension after the lysis of erythrocytes. For T cell analyses from blood, the mice were bled from the facial vein using a lancet into tubes containing heparin. Subsequently, splenocytes or blood samples (after lysis of erythrocytes) were analyzed by flow cytometry.
4.4. Flow Cytometry
Flow cytometric analyses were performed on FACS Canto II (4-2-2 configuration, BD Biosciences, Franklin Lakes, NJ, USA) and the subsequent data processing was conducted with the FlowJo software 10.8.0 (BD, Ashland, OR, USA). For surface stainings, the cells were incubated for 5 min with FcR-block (anti-CD16/32; BD Biosciences) prior to the addition of the antibody mix in PBS, 0.5% BSA, 0.5 M EDTA. Subsequently, the cells were incubated for 20 min with the antibody solution. Lastly, the cells were washed with PBS, 0.5% BSA, 0.5 M EDTA and transferred into FACS tubes (all steps at 4 °C). The following antibodies were used for surface staining: CD4 V500 (clone RM4-5; BioLegend, San Diego, CA, USA), CD8 PerCPCy5.5, CD8 BV510, CD8 APC-Cy7 (all clone 53-6.7; BioLegend), CD44 PE-Cy7, CD44 FITC (both clone IM7; BioLegend), CD45.1 PB, CD45.1 APC-Cy7 (both clone A20; BioLegend), CD45.2 FITC, CD45.2 PE-Cy7 (both clone 104; BioLegend), CD45.2 V500 (clone 104; BD Biosciences) and CD62L APC-Cy7 (clone MEL-14; BioLegend). To exclude dead cells, the cells were stained with the Fixable Viability Stain 780 (BD Biosciences, diluted 1:2000 in HBSS++) for 10 min at RT in the dark according to the manufacturer’s instructions. The following gating strategy was applied: (1) exclusion of dead cells via viability staining, (2) gating on lymphocytes in the FSC/SSC plot, (3) double cell exclusion, (4) gating on transferred cells via CD45.1/2 and (5) CD4 or CD8 blotted against proliferation dye and (6) gating on the parameter of interest (proliferation dye, CD44 vs. CD62L).
4.5. Statistical Analysis
Data were analyzed using the GraphPad Prism 9.0.1 software (GraphPad Software, San Diego, CA, USA). The data were analyzed for statistical significance by one-way ANOVA following Bonferroni’s post hoc test (for comparisons on more time points or more groups) or by the Student’s t-test (for simple comparisons between two groups), as indicated in the figure legends. A p-value of <0.05 was considered statistically significant. Symbols used in the figures are: * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




