
Specialized Proresolving Lipid Mediators: A Potential Therapeutic Target for Atherosclerosis

 , , , , , , , , and
, , , , , , , , and
Abstract
:1. Introduction
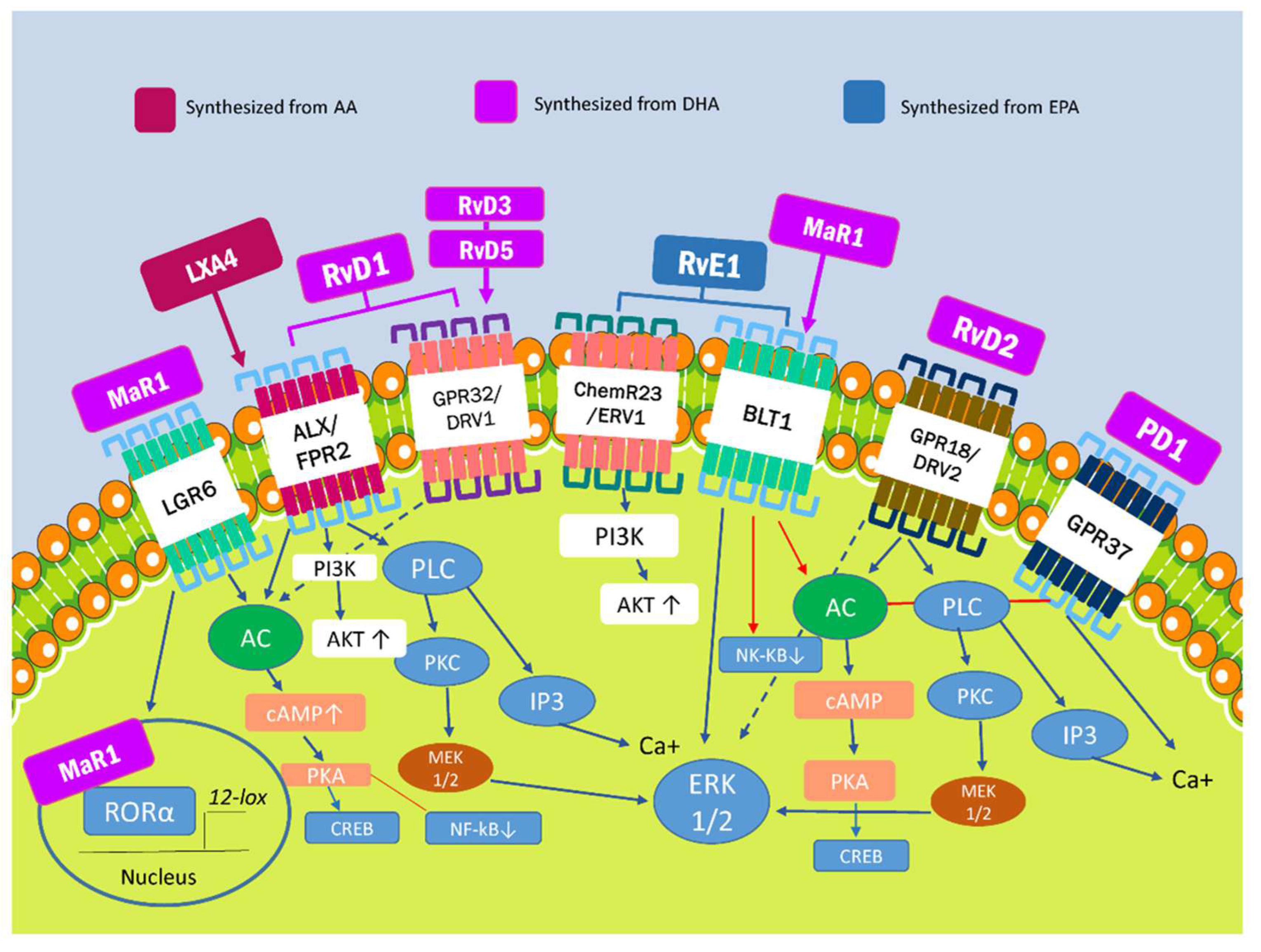
2. Specialized Proresolving Lipid Mediators and Modulation of Proinflammatory Pathways
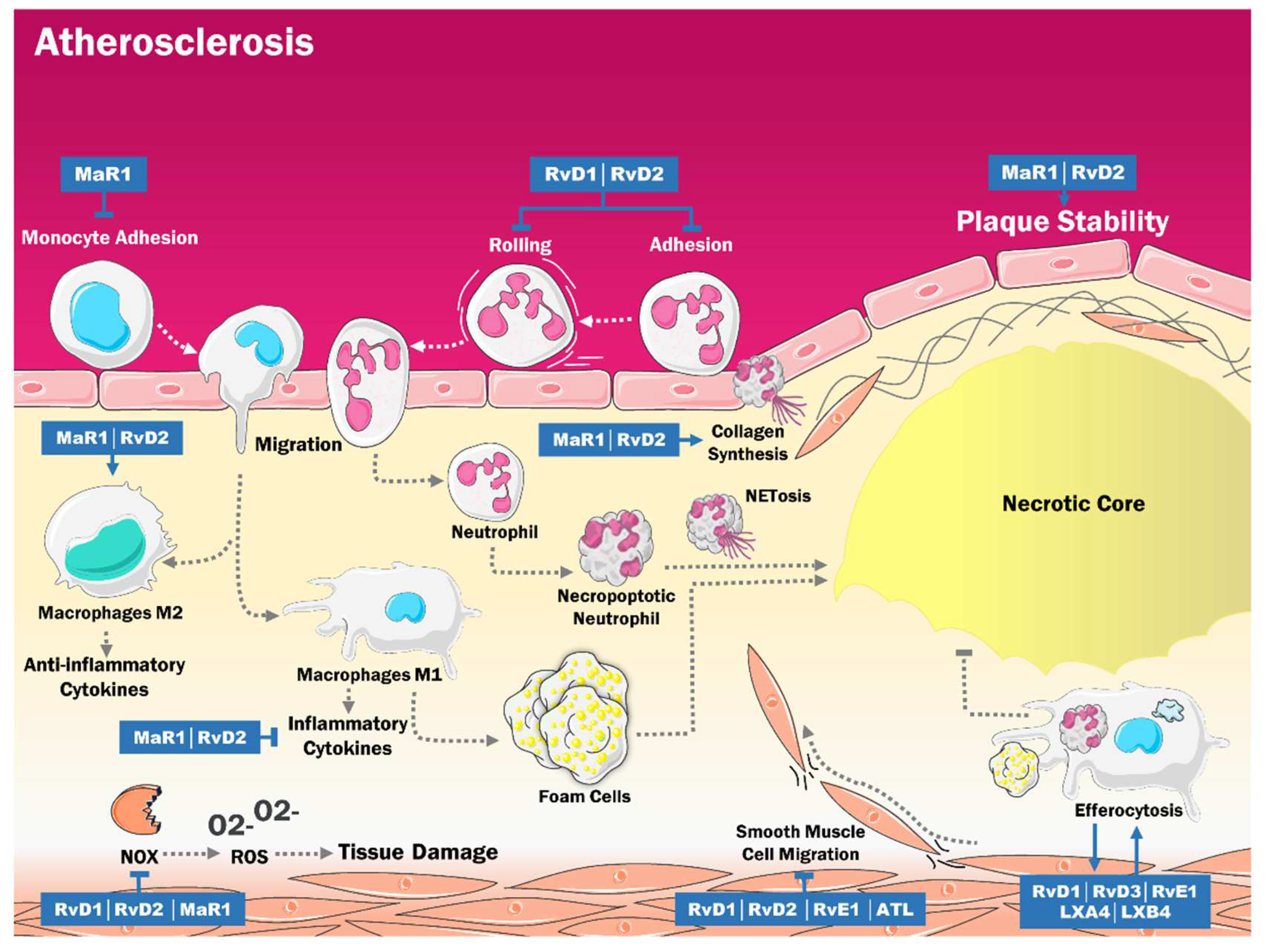
3. Role of SPMs in the Pathogenesis of Atherosclerosis
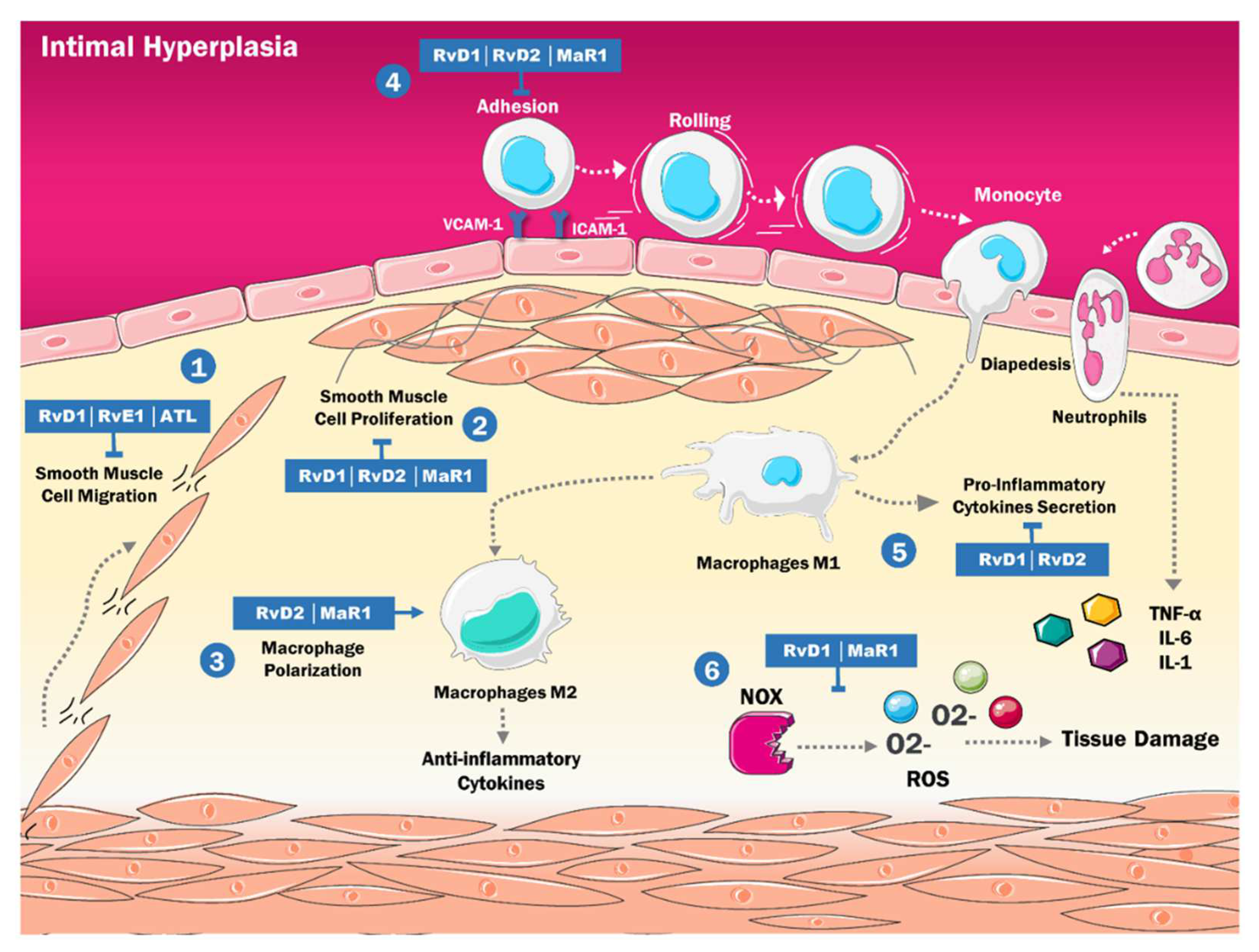
4. SPMs and the Physiopathology of Intimal Hyperplasia
5. Protective Effects of SPMs during Reperfusion Injury
6. SPMs as Therapeutic Target in Atherosclerosis
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Capó, X.; Martorell, M.; Busquets-Cortés, C.; Tejada, S.; Tur, J.A.; Pons, A.; Sureda, A. Resolvins as proresolving inflammatory mediators in cardiovascular disease. Eur. J. Med. Chem. 2018, 153, 123–130. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Cardiovascular Diseases (CVDs). Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 18 August 2021).
- Morales Aguilar, R.; Lastre-Amell, G.; Pardo Vásquez, A. Estilos de vida relacionados con factores de riesgo cardiovascular. AVFT Arch. Venez. Farm. Ter. 2018, 37, 50–62. [Google Scholar]
- Recchiuti, A.; Serhan, C.N. Pro-Resolving Lipid Mediators (SPMs) and Their Actions in Regulating miRNA in Novel Resolution Circuits in Inflammation. Front. Immunol. 2012, 3, 298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, M.A.; Sousa, L.P.; Pinho, V.; Perretti, M.; Teixeira, M.M. Resolution of Inflammation: What Controls Its Onset? Front. Immunol. 2016, 7, 160. [Google Scholar] [CrossRef] [Green Version]
- Nathan, C.; Ding, A. Nonresolving Inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Headland, S.E.; Norling, L.V. The resolution of inflammation: Principles and challenges. Semin. Immunol. 2015, 27, 149–160. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Buckley, C.D.; Gilroy, D.W.; Serhan, C.N. Pro-Resolving lipid mediators and Mechanisms in the resolution of acute inflammation. Immunity 2014, 40, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Basil, M.C.; Levy, B.D. Specialized pro-resolving mediators: Endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 2016, 16, 51–67. [Google Scholar] [CrossRef]
- Park, J.; Langmead, C.J.; Riddy, D.M. New Advances in Targeting the Resolution of Inflammation: Implications for Specialized Pro-Resolving Mediator GPCR Drug Discovery. ACS Pharmacol. Transl. Sci. 2020, 3, 88–106. [Google Scholar] [CrossRef]
- Doran, A.C. Inflammation Resolution: Implications for Atherosclerosis. Circ. Res. 2022, 130, 130–148. [Google Scholar] [CrossRef]
- de Gaetano, M.; McEvoy, C.; Andrews, D.; Cacace, A.; Hunter, J.; Brennan, E.; Godson, C. Specialized Pro-resolving Lipid Mediators: Modulation of Diabetes-Associated Cardio-, Reno-, and Retino-Vascular Complications. Front. Pharmacol. 2018, 9, 1488. [Google Scholar] [CrossRef] [PubMed]
- Doyle, R.; Sadlier, D.M.; Godson, C. Pro-resolving lipid mediators: Agents of anti-ageing? Semin. Immunol. 2018, 40, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Viola, J.R.; Lemnitzer, P.; Jansen, Y.; Csaba, G.; Winter, C.; Neideck, C.; Silvestre-Roig, C.; Dittmar, G.; Döring, Y.; Drechsler, M.; et al. Resolving Lipid Mediators Maresin 1 and Resolvin D2 Prevent Atheroprogression in Mice. Circ. Res. 2016, 119, 1030–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredman, G.; Spite, M. Specialized pro-resolving mediators in cardiovascular diseases. Mol. Asp. Med. 2017, 58, 65–71. [Google Scholar] [CrossRef]
- Kasikara, C.; Doran, A.C.; Cai, B.; Tabas, I. The role of non-resolving inflammation in atherosclerosis. J. Clin. Investig. 2018, 128, 2713–2723. [Google Scholar] [CrossRef] [Green Version]
- Satish, M.; Agrawal, D.K. Pro-resolving lipid mediators in the resolution of neointimal hyperplasia pathogenesis in atherosclerotic diseases. Expert Rev. Cardiovasc. Ther. 2019, 17, 177–184. [Google Scholar] [CrossRef]
- Halade, G.V.; Black, L.M.; Verma, M. Paradigm shift—Metabolic transformation of docosahexaenoic and eicosapentaenoic acids to bioactives exemplify the promise of fatty acid drug discovery. Biotechnol. Adv. 2018, 36, 935–953. [Google Scholar] [CrossRef]
- Halade, G.V.; Kain, V.; Ingle, K.A.; Prabhu, S.D. Interaction of 12/15-lipoxygenase with fatty acids alters the leukocyte kinetics leading to improved postmyocardial infarction healing. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H89–H102. [Google Scholar] [CrossRef]
- Halade, G.V.; Kain, V.; Tourki, B.; Jadapalli, J.K. Lipoxygenase drives lipidomic and metabolic reprogramming in ischemic heart failure. Metabolism 2019, 96, 22–32. [Google Scholar] [CrossRef]
- Chiang, N.; Serhan, C.N. Structural elucidation and physiologic functions of specialized pro-resolving mediators and their receptors. Mol. Asp. Med. 2017, 58, 114–129. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.; Xie, Y.-K.; Zhang, Z.-J.; Wang, Z.; Xu, Z.-Z.; Ji, R.-R. GPR37 regulates macrophage phagocytosis and resolution of inflammatory pain. J. Clin. Investig. 2018, 128, 3568–3582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalli, J.; Serhan, C.N. Identification and structure elucidation of the pro-resolving mediators provides novel leads for resolution pharmacology. Br. J. Pharmacol. 2019, 176, 1024–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.-H.; Lee, K.; Saha, A.; Han, J.; Choi, H.; Noh, M.; Lee, Y.-H.; Lee, M.-O. Specialized Proresolving Mediators for Therapeutic Interventions Targeting Metabolic and Inflammatory Disorders. Biomol. Ther. 2021, 29, 455–464. [Google Scholar] [CrossRef]
- Kang, G.J.; Kim, E.J.; Lee, C.H. Therapeutic Effects of Specialized Pro-Resolving Lipids Mediators on Cardiac Fibrosis via NRF2 Activation. Antioxidants 2020, 9, 1259. [Google Scholar] [CrossRef]
- Recchiuti, A. Resolvin D1 and its GPCRs in resolution circuits of inflammation. Prostaglandins Other Lipid Mediat. 2013, 107, 64–76. [Google Scholar] [CrossRef]
- McCrary, M.R.; Jiang, M.Q.; Giddens, M.M.; Zhang, J.Y.; Owino, S.; Wei, Z.Z.; Zhong, W.; Gu, X.; Xin, H.; Hall, R.A.; et al. Protective effects of GPR37 via regulation of inflammation and multiple cell death pathways after ischemic stroke in mice. FASEB J. 2019, 33, 10680–10691. [Google Scholar] [CrossRef]
- Dalli, J.; Serhan, C.N. Pro-Resolving Mediators in Regulating and Conferring Macrophage Function. Front. Immunol. 2017, 8, 1400. [Google Scholar] [CrossRef] [Green Version]
- Yokomizo, T. Two distinct leukotriene B4 receptors, BLT1 and BLT2. J. Biochem. 2015, 157, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Colas, R.A.; Dalli, J.; Chiang, N.; Vlasakov, I.; Sanger, J.M.; Riley, I.R.; Serhan, C.N. Identification and Actions of the Maresin 1 Metabolome in Infectious Inflammation. J. Immunol. 2016, 197, 4444–4452. [Google Scholar] [CrossRef] [Green Version]
- Chiang, N.; Libreros, S.; Norris, P.C.; De La Rosa, X.; Serhan, C.N. Maresin 1 activates LGR6 receptor promoting phagocyte immunoresolvent functions. J. Clin. Investig. 2019, 129, 5294–5311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flak, M.B.; Koenis, D.S.; Sobrino, A.; Smith, J.; Pistorius, K.; Palmas, F.; Dalli, J. GPR101 mediates the pro-resolving actions of RvD5n-3 DPA in arthritis and infections. J. Clin. Investig. 2020, 130, 359–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chattopadhyay, R.; Mani, A.M.; Singh, N.K.; Rao, G.N. Resolvin D1 blocks H2O2-mediated inhibitory crosstalk between SHP2 and PP2A and suppresses endothelial-monocyte interactions. Free Radic. Biol. Med. 2018, 117, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, R.; Raghavan, S.; Rao, G.N. Resolvin D1 via prevention of ROS-mediated SHP2 inactivation protects endothelial adherens junction integrity and barrier function. Redox Biol. 2017, 12, 438–455. [Google Scholar] [CrossRef]
- Carracedo, M.; Artiach, G.; Arnardottir, H.; Bäck, M. The resolution of inflammation through omega-3 fatty acids in atherosclerosis, intimal hyperplasia, and vascular calcification. Semin. Immunopathol. 2019, 41, 757–766. [Google Scholar] [CrossRef] [Green Version]
- Krishnamoorthy, S.; Recchiuti, A.; Chiang, N.; Yacoubian, S.; Lee, C.-H.; Yang, R.; Petasis, N.A.; Serhan, C.N. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc. Natl. Acad. Sci. USA 2010, 107, 1660–1665. [Google Scholar] [CrossRef] [Green Version]
- So, J.; Wu, D.; Lichtenstein, A.H.; Tai, A.K.; Matthan, N.R.; Maddipati, K.R.; Lamon-Fava, S. EPA and DHA differentially modulate monocyte inflammatory response in subjects with chronic inflammation in part via plasma specialized pro-resolving lipid mediators: A randomized, double-blind, crossover study. Atherosclerosis 2020, 316, 90–98. [Google Scholar] [CrossRef]
- Lopez, E.F.; Kabarowski, J.H.; Ingle, K.A.; Kain, V.; Barnes, S.; Crossman, D.; Lindsey, M.; Halade, G.V. Obesity superimposed on aging magnifies inflammation and delays the resolving response after myocardial infarction. Am. J. Physiol. Circ. Physiol. 2015, 308, H269–H280. [Google Scholar] [CrossRef] [Green Version]
- Halade, G.V.; Kain, V.; Black, L.M.; Prabhu, S.D.; Ingle, K.A. Aging dysregulates D- and E-series resolvins to modulate cardiosplenic and cardiorenal network following myocardial infarction. Aging 2016, 8, 2611–2634. [Google Scholar] [CrossRef] [Green Version]
- Jadapalli, J.K.; Wright, G.W.; Kain, V.; Sherwani, M.A.; Sonkar, R.; Yusuf, N.; Halade, G.V. Doxorubicin triggers splenic contraction and irreversible dysregulation of COX and LOX that alters the inflammation-resolution program in the myocardium. Am. J. Physiol. Circ. Physiol. 2018, 315, H1091–H1100. [Google Scholar] [CrossRef]
- Halade, G.V.; Kain, V.; Wright, G.M.; Jadapalli, J.K. Subacute treatment of carprofen facilitate splenocardiac resolution deficit in cardiac injury. J. Leukoc. Biol. 2018, 104, 1173–1186. [Google Scholar] [CrossRef] [PubMed]
- Kain, V.; Jadapalli, J.K.; Tourki, B.; Halade, G.V. Inhibition of FPR2 impaired leukocytes recruitment and elicited non-resolving inflammation in acute heart failure. Pharmacol. Res. 2019, 146, 104295. [Google Scholar] [CrossRef] [PubMed]
- Brennan, E.; Kantharidis, P.; Cooper, M.E.; Godson, C. Pro-resolving lipid mediators: Regulators of inflammation, metabolism and kidney function. Nat. Rev. Nephrol. 2021, 17, 725–739. [Google Scholar] [CrossRef] [PubMed]
- Jadapalli, J.K.; Halade, G.V. Unified nexus of macrophages and maresins in cardiac reparative mechanisms. FASEB J. 2018, 32, 5227–5237. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharan, J.A.; Sharma-Walia, N. Lipoxins: Nature’s way to resolve inflammation. J. Inflamm. Res. 2015, 8, 181–192. [Google Scholar]
- Ho, K.J.; Spite, M.; Owens, C.D.; Lancero, H.; Kroemer, A.H.; Pande, R.; Creager, M.A.; Serhan, C.N.; Conte, M.S. Aspirin-Triggered Lipoxin and Resolvin E1 Modulate Vascular Smooth Muscle Phenotype and Correlate with Peripheral Atherosclerosis. Am. J. Pathol. 2010, 177, 2116–2123. [Google Scholar] [CrossRef]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, Indicators, Risk Factors and New Hopes. Int. J. Prev. Med. 2014, 5, 927–946. [Google Scholar]
- Verónica Mora, D.E.; Morr, C.; Paciotti, S.; Prospert, O.P.; Quiroz, J. Células inflamatorias en lesiones ateroescleróticas de las arterias coronarias humanas. Gac. Médica Caracas 2020, 124, 298–307. [Google Scholar]
- Fredman, G.; Tabas, I. Boosting Inflammation Resolution in Atherosclerosis: The Next Frontier for Therapy. Am. J. Pathol. 2017, 187, 1211–1221. [Google Scholar] [CrossRef] [Green Version]
- Rojas, J.; Salazar, J.; Martínez, M.S.; Palmar, J.; Bautista, J.; Chávez-Castillo, M.; Gómez, A.; Bermudez, V. Macrophage Heterogeneity and Plasticity: Impact of Macrophage Biomarkers on Atherosclerosis. Scientifica 2015, 2015, 1–17. [Google Scholar] [CrossRef]
- Viola, J.; Soehnlein, O. Atherosclerosis—A matter of unresolved inflammation. Semin. Immunol. 2015, 27, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Sansbury, B.E.; Spite, M. Resolution of Acute Inflammation and the Role of Resolvins in Immunity, Thrombosis, and Vascular Biology. Circ. Res. 2016, 119, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Kain, V.; Van Der Pol, W.; Mariappan, N.; Ahmad, A.; Eipers, P.; Morrow, C.; Gibson, D.L.; Gladine, C.; Vigor, C.; Durand, T.; et al. Obesogenic diet in aging mice disrupts gut microbe composition and alters neutrophil:lymphocyte ratio leading to inflamed milieu in acute heart failure. FASEB J. 2019, 33, 6456–6469. [Google Scholar] [CrossRef]
- Chatterjee, A.; Sharma, A.; Chen, M.; Toy, R.; Mottola, G.; Conte, M.S. The Pro-Resolving Lipid Mediator Maresin 1 (MaR1) Attenuates Inflammatory Signaling Pathways in Vascular Smooth Muscle and Endothelial Cells. PLoS ONE 2014, 9, e113480. [Google Scholar] [CrossRef] [Green Version]
- Cherpokova, D.; Jouvene, C.C.; Libreros, S.; DeRoo, E.P.; Chu, L.; De La Rosa, X.; Norris, P.C.; Wagner, D.D.; Serhan, C.N. Resolvin D4 attenuates the severity of pathological thrombosis in mice. Blood 2019, 134, 1458–1468. [Google Scholar] [CrossRef] [PubMed]
- Miyahara, T.; Runge, S.; Chatterjee, A.; Chen, M.; Mottola, G.; Fitzgerald, J.M.; Serhan, C.N.; Conte, M.S. D-series resolvin attenuates vascular smooth muscle cell activation and neointimal hyperplasia following vascular injury. FASEB J. 2013, 27, 2220–2232. [Google Scholar] [CrossRef] [Green Version]
- Dalli, J.; Serhan, C.N. Specific lipid mediator signatures of human phagocytes: Microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood 2012, 120, e60–e72. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, S.; Thomas, G.; Harvey, K.; Cottell, D.; Reville, K.; Berlasconi, G.; Petasis, N.; Erwig, L.; Rees, A.J.; Savill, J.; et al. Lipoxins, Aspirin-Triggered Epi-Lipoxins, Lipoxin Stable Analogues, and the Resolution of Inflammation: Stimulation of Macrophage Phagocytosis of Apoptotic Neutrophils In Vivo. J. Am. Soc. Nephrol. 2002, 13, 2497–2507. [Google Scholar] [CrossRef] [Green Version]
- Petri, M.H.; Laguna-Fernández, A.; Gonzalez-Diez, M.; Paulsson-Berne, G.; Hansson, G.K.; Bäck, M. The role of the FPR2/ALX receptor in atherosclerosis development and plaque stability. Cardiovasc. Res. 2015, 105, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Norling, L.V.; Dalli, J.; Flower, R.J.; Serhan, C.N.; Perretti, M. Resolvin D1 limits polymorphonuclear leukocyte recruitment to inflammatory loci: Receptor-dependent actions. Arter. Thromb. Vasc. Biol. 2012, 32, 1970–1978. [Google Scholar] [CrossRef] [Green Version]
- Spite, M.; Norling, L.V.; Summers, L.; Yang, R.; Cooper, D.; Petasis, N.A.; Flower, R.J.; Perretti, M.; Serhan, C.N. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 2009, 461, 1287–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roig, C.S.; Daemen, M.; Lutgens, E.; Soehnlein, O.; Hartwig, H. Neutrophils in atherosclerosis. A brief overview. Hamostaseologie 2015, 35, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Yin, Y.; Wang, T.; Li, W.; Li, C.; Zeng, X.; Yang, W.; Zhang, R.; Tang, Y.; Shi, L.; et al. Maresin 1 mitigates concanavalin A-induced acute liver injury in mice by inhibiting ROS-mediated activation of NF-κB signaling. Free Radic. Biol. Med. 2020, 147, 23–36. [Google Scholar] [CrossRef]
- Zhuang, Y.; Liu, H.; Zhou, X.E.; Verma, R.K.; De Waal, P.W.; Jang, W.; Xu, T.-H.; Wang, L.; Meng, X.; Zhao, G.; et al. Structure of formylpeptide receptor 2-Gi complex reveals insights into ligand recognition and signaling. Nat. Commun. 2020, 11, 885. [Google Scholar] [CrossRef] [Green Version]
- Romano, M.; Cianci, E.; Simiele, F.; Recchiuti, A. Lipoxins and aspirin-triggered lipoxins in resolution of inflammation. Eur. J. Pharmacol. 2015, 760, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Elajami, T.K.; Colas, R.A.; Dalli, J.; Chiang, N.; Serhan, C.N.; Welty, F.K. Specialized proresolving lipid mediators in patients with coronary artery disease and their potential for clot remodeling. FASEB J. 2016, 30, 2792–2801. [Google Scholar] [CrossRef] [Green Version]
- Mottola, G.; Chatterjee, A.; Wu, B.; Chen, M.; Conte, M.S. Aspirin-triggered resolvin D1 attenuates PDGF-induced vascular smooth muscle cell migration via the cyclic adenosine monophosphate/protein kinase A (cAMP/PKA) pathway. PLoS ONE 2017, 12, e0174936. [Google Scholar] [CrossRef]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef]
- Nakayama, A.; Albarrán-Juárez, J.; Liang, G.; Roquid, K.A.; Iring, A.; Tonack, S.; Chen, M.; Müller, O.J.; Weinstein, L.S.; Offermanns, S. Disturbed flow–induced Gs-mediated signaling protects against endothelial inflammation and atherosclerosis. JCI Insight 2020, 5, 140485. [Google Scholar] [CrossRef]
- Fredman, G.; Hellmann, J.; Proto, J.D.; Kuriakose, G.; Colas, R.A.; Dorweiler, B.; Connolly, E.S.; Solomon, R.; Jones, D.M.; Heyer, E.J.; et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat. Commun. 2016, 7, 12859. [Google Scholar] [CrossRef]
- Lee, H.-N.; Surh, Y.-J. Resolvin D1-mediated NOX2 inactivation rescues macrophages undertaking efferocytosis from oxidative stress-induced apoptosis. Biochem. Pharmacol. 2013, 86, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G.; Ozcan, L.; Spolitu, S.; Hellmann, J.; Spite, M.; Backs, J.; Tabas, I. Resolvin D1 limits 5-lipoxygenase nuclear localization and leukotriene B4 synthesis by inhibiting a calcium-activated kinase pathway. Proc. Natl. Acad. Sci. USA 2014, 111, 14530–14535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doran, A.C.; Yurdagul, A.; Tabas, I. Efferocytosis in health and disease. Nat. Rev. Immunol. 2019, 20, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Thorp, E.B.; Doran, A.C.; Subramanian, M.; Sansbury, B.E.; Lin, C.-S.; Spite, M.; Fredman, G.; Tabas, I. MerTK cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc. Natl. Acad. Sci. USA 2016, 113, 6526–6531. [Google Scholar] [CrossRef] [Green Version]
- Rymut, N.; Heinz, J.; Sadhu, S.; Hosseini, Z.; Riley, C.O.; Marinello, M.; Maloney, J.; MacNamara, K.C.; Spite, M.; Fredman, G. Resolvin D1 promotes efferocytosis in aging by limiting senescent cell-induced MerTK cleavage. FASEB J. 2019, 34, 597–609. [Google Scholar] [CrossRef] [Green Version]
- Hosseini, Z.; Marinello, M.; Decker, C.; Sansbury, B.E.; Sadhu, S.; Gerlach, B.D.; Ramos, R.B.; Adam, A.P.; Spite, M.; Fredman, G. Resolvin D1 Enhances Necroptotic Cell Clearance Through Promoting Macrophage Fatty Acid Oxidation and Oxidative Phosphorylation. Arter. Thromb. Vasc. Biol. 2021, 41, 1062–1075. [Google Scholar] [CrossRef]
- Schif-Zuck, S.; Gross, N.; Assi, S.; Rostoker, R.; Serhan, C.N.; Ariel, A. Saturated-efferocytosis generates pro-resolving CD11b low macrophages: Modulation by resolvins and glucocorticoids. Eur. J. Immunol. 2011, 41, 366–379. [Google Scholar] [CrossRef]
- Dalli, J.; Winkler, J.W.; Colas, R.A.; Arnardottir, H.; Cheng, C.-Y.C.; Chiang, N.; Petasis, N.A.; Serhan, C.N. Resolvin D3 and Aspirin-Triggered Resolvin D3 Are Potent Immunoresolvents. Chem. Biol. 2013, 20, 188–201. [Google Scholar] [CrossRef] [Green Version]
- Welty, F.K.; Schulte, F.; Alfaddagh, A.; Elajami, T.K.; Bistrian, B.R.; Hardt, M. Regression of human coronary artery plaque is associated with a high ratio of (18-hydroxy-eicosapentaenoic acid + resolvin E1) to leukotriene B 4. FASEB J. 2021, 35, e21448. [Google Scholar] [CrossRef]
- Wu, B.; Mottola, G.; Schaller, M.; Upchurch, G.R.; Conte, M.S. Resolution of vascular injury: Specialized lipid mediators and their evolving therapeutic implications. Mol. Asp. Med. 2017, 58, 72–82. [Google Scholar] [CrossRef] [Green Version]
- Efremova, O.A.; Starodubov, O.D.; Kamyshnikova, L.A.; Bolkhovitina, O.A.; Obolonkova, N.I. Clinical-functional changes of myocardium after percutaneous coronary interventions in patients with chronic heart failure. Lat. Hipertens. 2019, 14, 256–261. [Google Scholar]
- Espinoza, R.; Arab, G.; Wiliam, Z.; Carrasquero, L.; Subero, L.; Colina, R.; Ana, T.; Jorge, P.; José, T.; Antonio, V.; et al. Evaluación de la incidencia de trombosis coronaria post Stent, posterior a la comercialización de segundas marcas de Clopidogrel. Hospital Miguel Pérez Carreño. Caracas. Venezuela. AVFT Arch. Venez. Farm. Ter. 2015, 33, 7–12. [Google Scholar]
- Shah, P.K. Inflammation, Neointimal Hyperplasia, and Restenosis. Circulation 2003, 107, 2175–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newby, A.C.; Zaltsman, A.B. Molecular mechanisms in intimal hyperplasia. J. Pathol. 2000, 190, 300–309. [Google Scholar] [CrossRef]
- Conte, M.S.; Desai, T.A.; Wu, B.; Schaller, M.; Werlin, E. Pro-resolving lipid mediators in vascular disease. J. Clin. Investig. 2018, 128, 3727–3735. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Gong, Y.; Zhang, R.; Piao, L.; Li, X.; Liu, Q.; Yan, S.; Shen, Y.; Guo, S.; Zhu, M.; et al. Resolvin E1 attenuates injury-induced vascular neointimal formation by inhibition of inflammatory responses and vascular smooth muscle cell migration. FASEB J. 2018, 32, 5413–5425. [Google Scholar] [CrossRef]
- Petri, M.H.; Laguna-Fernandez, A.; Tseng, C.-N.; Hedin, U.; Perretti, M.; Bäck, M. Aspirin-triggered 15-epi-lipoxin A4 signals through FPR2/ALX in vascular smooth muscle cells and protects against intimal hyperplasia after carotid ligation. Int. J. Cardiol. 2015, 179, 370–372. [Google Scholar] [CrossRef] [Green Version]
- Akagi, D.; Chen, M.; Toy, R.; Chatterjee, A.; Conte, M.S. Systemic delivery of proresolving lipid mediators resolvin D 2 and maresin 1 attenuates intimal hyperplasia in mice. FASEB J. 2015, 29, 2504–2513. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Mottola, G.; Chatterjee, A.; Lance, K.D.; Chen, M.; Siguenza, I.O.; Desai, T.A.; Conte, M.S. Perivascular delivery of resolvin D1 inhibits neointimal hyperplasia in a rat model of arterial injury. J. Vasc. Surg. 2016, 65, 207–217.e3. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Chen, Q.; Mei, L.; Wen, G.; An, W.; Zhou, X.; Niu, K.; Liu, C.; Ren, M.; Sun, K.; et al. Neutrophil elastase promotes neointimal hyperplasia by targeting toll-like receptor 4 (TLR4)-NF-κB signalling. Br. J. Pharmacol. 2021, 178, 4048–4068. [Google Scholar] [CrossRef]
- Li, Y.; Wang, N.; Ma, Z.; Wang, Y.; Yuan, Y.; Zhong, Z.; Hong, Y.; Zhao, M. Lipoxin A4 protects against paraquat-induced acute lung injury by inhibiting the TLR4/MyD88-mediated activation of the NF-κB and PI3K/AKT pathways. Int. J. Mol. Med. 2021, 47, 86. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Qu, M.; Yang, Q.; Chang, Y. Lipoxin A4 ameliorates renal ischaemia–reperfusion-induced acute lung injury in rats. Clin. Exp. Pharmacol. Physiol. 2019, 46, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, Z.; Wang, Q. Resolvin D1 Attenuates Myocardial Infarction in a Rodent Model with the Participation of the HMGB1 Pathway. Cardiovasc. Drugs Ther. 2019, 33, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Xue, C.; Auerbach, B.J.; Fan, J.; Bashore, A.C.; Cui, J.; Yang, D.Y.; Trignano, S.B.; Liu, W.; Shi, J.; et al. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation 2020, 142, 2060–2075. [Google Scholar] [CrossRef]
- Feil, S.; Fehrenbacher, B.; Lukowski, R.; Essmann, F.; Schulze-Osthoff, K.; Schaller, M.; Feil, R. Transdifferentiation of Vascular Smooth Muscle Cells to Macrophage-Like Cells During Atherogenesis. Circ. Res. 2014, 115, 662–667. [Google Scholar] [CrossRef]
- Yang, J.; Li, M.; Hu, X.; Lu, J.; Wang, Q.; Lu, S.; Gao, F.; Jin, S.; Zheng, S. Protectin DX promotes epithelial injury repair and inhibits fibroproliferation partly via ALX/PI3K signalling pathway. J. Cell. Mol. Med. 2020, 24, 14001–14012. [Google Scholar] [CrossRef]
- Zheng, S.; Wang, Q.; D’Souza, V.; Bartis, D.; Dancer, R.; Parekh, D.; Gao, F.; Lian, Q.; Jin, S.; Thickett, D.R. ResolvinD1 stimulates epithelial wound repair and inhibits TGF-β-induced EMT whilst reducing fibroproliferation and collagen production. Lab. Investig. 2018, 98, 130–140. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; D’Souza, V.K.; Bartis, D.; Dancer, R.C.; Parekh, D.; Naidu, B.; Gao-Smith, F.; Wang, Q.; Jin, S.; Lian, Q.; et al. Lipoxin A4promotes lung epithelial repair whilst inhibiting fibroblast proliferation. ERJ Open Res. 2016, 2, 00079–02015. [Google Scholar] [CrossRef]
- Frank, A.; Bonney, M.; Bonney, S.; Weitzel, L.; Koeppen, M.; Eckle, T. Myocardial ischemia reperfusion injury: From basic science to clinical bedside. Semin. Cardiothorac. Vasc. Anesth. 2012, 16, 123–132. [Google Scholar] [CrossRef]
- Bermúdez Arias, D.F. La recuperación del miocardio hibernado mejora el pronóstico de la cardiopatía isquémica metabólica. Gac. Médica Caracas 2020, 113, 19–41. [Google Scholar]
- Wu, M.-Y.; Yiang, G.-T.; Liao, W.-T.; Tsai, A.P.Y.; Cheng, Y.-L.; Cheng, P.-W.; Li, C.-Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell. Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wu, Z.; Huang, C.; Zhao, Y.; Zhou, Y.; Zhou, X.; Lu, X.; Mao, L.; Li, S. Effect of Lipoxin A4 on Myocardial Ischemia Reperfusion Injury Following Cardiac Arrest in a Rabbit Model. Inflammation 2013, 36, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-Q.; Wu, S.-H.; Zhou, Y.; Tang, Y.-R. Involvement of K+ channel-dependant pathways in lipoxin A4-induced protective effects on hypoxia/reoxygenation injury of cardiomyocytes. Prostaglandins Leukot. Essent. Fat. Acids 2013, 88, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Chen, P.; Zhong, J.; Cheng, Y.; Chen, H.; He, Y.; Chen, C. HIF-1α in myocardial ischemia-reperfusion injury. Mol. Med. Rep. 2021, 23, 352. [Google Scholar] [CrossRef]
- Zhao, Q.; Shao, L.; Hu, X.; Wu, G.; Du, J.; Xia, J.; Qiu, H. Lipoxin A4Preconditioning and Postconditioning Protect Myocardial Ischemia/Reperfusion Injury in Rats. Mediat. Inflamm. 2013, 2013, e231351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Rong, J. Pro-resolving lipid mediators as therapeutic leads for cardiovascular diseases. Expert Opin. Ther. Targets 2019, 23, 423–436. [Google Scholar] [CrossRef]
- Halade, G.V.; Norris, P.C.; Kain, V.; Serhan, C.N.; Ingle, K.A. Splenic leukocytes define the resolution of inflammation in heart failure. Sci. Signal. 2018, 11, eaao1818. [Google Scholar] [CrossRef] [Green Version]
- Halade, G.V.; Kain, V.; Dillion, C.; Beasley, M.; Dudenbostel, T.; Oparil, S.; Limdi, N.A. Race-based and sex-based differences in bioactive lipid mediators after myocardial infarction. ESC Heart Fail. 2020, 7, 1700–1710. [Google Scholar] [CrossRef]
- Tourki, B.; Kain, V.; Pullen, A.B.; Norris, P.C.; Patel, N.; Arora, P.; Leroy, X.; Serhan, C.N.; Halade, G.V. Lack of resolution sensor drives age-related cardiometabolic and cardiorenal defects and impedes inflammation-resolution in heart failure. Mol. Metab. 2020, 31, 138–149. [Google Scholar] [CrossRef]
- Keyes, K.T.; Ye, Y.; Lin, Y.; Zhang, C.; Perez-Polo, J.R.; Gjorstrup, P.; Birnbaum, Y. Resolvin E1 protects the rat heart against reperfusion injury. Am. J. Physiol. Circ. Physiol. 2010, 299, H153–H164. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, K.; Bernier, J.; Godbout, R.; Rousseau, G. Resolvin D1, a Metabolite of Omega-3 Polyunsaturated Fatty Acid, Decreases Post-Myocardial Infarct Depression. Mar. Drugs 2014, 12, 5396–5407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, K.; Bernier, J.; Bourque-Riel, V.; Malick, M.; Rousseau, G. Resolvin D1 Reduces Infarct Size Through a Phosphoinositide 3-Kinase/Protein Kinase B Mechanism. J. Cardiovasc. Pharmacol. 2015, 66, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Kain, V.; Ingle, K.A.; Colas, R.A.; Dalli, J.; Prabhu, S.D.; Serhan, C.N.; Joshi, M.D.; Halade, G.V. Resolvin D1 activates the inflammation resolving response at splenic and ventricular site following myocardial infarction leading to improved ventricular function. J. Mol. Cell. Cardiol. 2015, 84, 24–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, S.K.; Colas, R.A.; Dalli, J.; Chiang, N.; Serhan, C.N. Proresolving actions of a new resolvin D1 analog mimetic qualifies as an immunoresolvent. Am. J. Physiol. Cell. Mol. Physiol. 2015, 308, L904–L911. [Google Scholar] [CrossRef] [Green Version]
- Pirault, J.; Bäck, M. Lipoxin and Resolvin Receptors Transducing the Resolution of Inflammation in Cardiovascular Disease. Front. Pharmacol. 2018, 9, 1273. [Google Scholar] [CrossRef]
- Sánchez-Hernández, C.D.; Torres-Alarcón, L.A.; González-Cortés, A.; Peón, A.N. Ischemia/Reperfusion Injury: Pathophysiology, Current Clinical Management, and Potential Preventive Approaches. Mediat. Inflamm. 2020, 2020, e8405370. [Google Scholar] [CrossRef]
- Heinz, J.; Marinello, M.; Fredman, G. Pro-resolution therapeutics for cardiovascular diseases. Prostaglandins Other Lipid Mediat. 2017, 132, 12–16. [Google Scholar] [CrossRef]
- Ayala, J.; López, C.; Hong, A.; Oberto, C.; Paiva, A.; Lares, M. Efecto de los ácidos grasos poliinsaturados (omega 3) sobre la agregación plaquetaria. Lat. Hipertens. 2009, 4, 71–78. [Google Scholar]
- Hasturk, H.; Abdallah, R.; Kantarci, A.; Nguyen, D.; Giordano, N.; Hamilton, J.; Van Dyke, T.E. Resolvin E1 (RvE1) Attenuates Atherosclerotic Plaque Formation in Diet and Inflammation-Induced Atherogenesis. Arter. Thromb. Vasc. Biol. 2015, 35, 1123–1133. [Google Scholar] [CrossRef] [Green Version]
- Salic, K.; Morrison, M.C.; Verschuren, L.; Wielinga, P.Y.; Wu, L.; Kleemann, R.; Gjorstrup, P.; Kooistra, T. Resolvin E1 attenuates atherosclerosis in absence of cholesterol-lowering effects and on top of atorvastatin. Atherosclerosis 2016, 250, 158–165. [Google Scholar] [CrossRef] [Green Version]
- Petri, M.H.; Laguna-Fernandez, A.; Arnardottir, H.; Wheelock, C.E.; Perretti, M.; Hansson, G.K.; Bäck, M. Aspirin-triggered lipoxin A4 inhibits atherosclerosis progression in apolipoprotein E-/- mice. Br. J. Pharmacol. 2017, 174, 4043–4054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makino, Y.; Miyahara, T.; Nitta, J.; Miyahara, K.; Seo, A.; Kimura, M.; Suhara, M.; Akai, A.; Akagi, D.; Yamamoto, K.; et al. Proresolving Lipid Mediators Resolvin D1 and Protectin D1 Isomer Attenuate Neointimal Hyperplasia in the Rat Carotid Artery Balloon Injury Model. J. Surg. Res. 2019, 233, 104–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thul, S.; Labat, C.; Temmar, M.; Benetos, A.; Bäck, M. Low salivary resolvin D1 to leukotriene B4 ratio predicts carotid intima media thickness: A novel biomarker of non-resolving vascular inflammation. Eur. J. Prev. Cardiol. 2017, 24, 903–906. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.L.; Steg, P.G.; Miller, M.; Brinton, E.A.; Jacobson, T.A.; Ketchum, S.B.; Doyle, R.T., Jr.; Juliano, R.A.; Jiao, L.; Granowitz, C.; et al. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N. Engl. J. Med. 2019, 380, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Gromovsky, A.D.; Schugar, R.C.; Brown, A.L.; Helsley, R.N.; Burrows, A.C.; Ferguson, D.; Zhang, R.; Sansbury, B.E.; Lee, R.G.; Morton, R.E.; et al. Δ-5 Fatty Acid Desaturase FADS1 Impacts Metabolic Disease by Balancing Proinflammatory and Proresolving Lipid Mediators. Arter. Thromb. Vasc. Biol. 2018, 38, 218–231. [Google Scholar] [CrossRef] [Green Version]
- Skarke, C.; Alamuddin, N.; Lawson, J.A.; Li, X.; Ferguson, J.F.; Reilly, M.P.; FitzGerald, G.A. Bioactive products formed in humans from fish oils. J. Lipid Res. 2015, 56, 1808–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bäck, M.; Yurdagul, A., Jr.; Tabas, I.; Öörni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef]
- Zhou, X.; Cai, J.; Liu, W.; Wu, X.; Gao, C. Cysteinyl leukotriene receptor type 1 (CysLT1R) antagonist zafirlukast protects against TNF-α-induced endothelial inflammation. Biomed. Pharmacother. 2019, 111, 452–459. [Google Scholar] [CrossRef]
- Medina-Leyte, D.; Zepeda-García, O.; Domínguez-Pérez, M.; González-Garrido, A.; Villarreal-Molina, T.; Jacobo-Albavera, L. Endothelial Dysfunction, Inflammation and Coronary Artery Disease: Potential Biomarkers and Promising Therapeutical Approaches. Int. J. Mol. Sci. 2021, 22, 3850. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N. Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Castillo, M.; Ortega, A.; Cudris-Torres, L.; Duran, P.; Rojas, M.; Manzano, A.; Garrido, B.; Salazar, J.; Silva, A.; Rojas-Gomez, D.M.; et al. Specialized Pro-Resolving Lipid Mediators: The Future of Chronic Pain Therapy? Int. J. Mol. Sci. 2021, 22, 10370. [Google Scholar] [CrossRef] [PubMed]
- Gila-Diaz, A.; Carrillo, G.; Singh, P.; Ramiro-Cortijo, D. Specialized Pro-Resolving Lipid Mediators in Neonatal Cardiovascular Physiology and Diseases. Antioxidants 2021, 10, 933. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| SPM | Receptors | Characteristic | Actions | References |
|---|---|---|---|---|
| LXA4 | ALX/FPR2 | Synthesized from ω-6-derived from AA | -Anti-inflammatory cytokine production and macrophage polarization. -Transcription of antioxidant genes, such as HO-1, NQO-1, SOD and TXN. | [24,26] |
| LXB4 | Not identified | |||
| RvE1 | ChemR23/ERV1, BLT1 | Synthesized from EPA derived from ω-3 | -Inhibition of transformation suppressor programmed cell death protein 4 (PDCD4) transcription that results in the increase of IL-10. -Inhibition of NF-kb inflammatory effects. | [25,27,28,30,31] |
| RvE2-6 | Not identified | |||
| RvD1 | ALX/FPR2, GPR32/DRV1 | Synthesized from DHA derived from ω-3 | ||
| RvD2 | GPR18/DRV2 | |||
| RvD3-6 | GPR32/DRV1 | |||
| PD1 | GPR37 | Synthesized from DHA derived from ω-3 | -Stimulation of phagocytosis and regulation of cytokine production. -Mitigation of neutrophil chemotaxis and oxidative metabolism. | [23,28] |
| MaR1 | BLT1, LGR6 | Synthesized from DHA derived from ω-3 | -Regulation of M2 macrophage polarization and anti-inflammatory properties. -Aid phagocytosis and efferocytosis. | [14,32] |
| MaR2 | Not identified |
| SPMs | AUTHOR | MODELS | EFFECTS |
|---|---|---|---|
| RvD2 y MaR1 | Viola et al. [15] | ApoE-deficient mice. | Stabilization of the atherosclerotic plaque. |
| MaR1 | Chatterjee et al. [55] | Primary cultures of EC and VSMC from human saphen veins. | Anti-inflammatory effects in human EC and VSMC. |
| RvD4 | Cherpokova et al. [56] | Mouse models of deep vein thrombosis. | Decrease severity of thromboinflammatory disease in vivo and improved resolution of thrombus. |
| RvD1 y RvD2 | Miyahara et al. [57] | Rabbit models of angioplasty. | Modulation of superoxide production in VSMC. |
| RvD1, RvD2 y RvE2 | Dalli et al. [58] | Macrophage cultures. | Efferocytosis increased SPM biosynthesis. |
| LXA4 y LXB4 | Mitchell et al. [59] | Rodent models of peritonitits. | Significative improvement of efferocytosis. |
| SPMs | Author | Methodology | Results |
|---|---|---|---|
| RvE1 | Hasturk et al. [120] | 39 rabbits with a dietary regimen for 13 weeks, were treated with topical RvE1 3 times per week. | RvE1 decreased atherogenesis and C reactive protein levels (p < 0.05). |
| Salic et al. [121] | 80 ApoE*3Leiden mice who were fed with a high-fat diet were administered different doses of RvE1. | High-dose and low-dose RvE1 reduce the size of the atherosclerotic lesion to the same degree (35%. p < 0.05). | |
| Liu et al. [94] | Male mice were fed with rodent food and treated with RvE1. | RvE1 was generated from EPA with the aid of ASA. | |
| RvD2 and MaR1 | Viola et al. [15] | ApoE -/- mice who were fed with a high-fat diet, were administered doses of RvD2 and MaR1. | RvD2 and MaR1 were signs of atheromatous plaque stability. |
| Akagi et al. [89] | Smooth muscle cells from the aorta of adult male mice received an intraperitoneal injection of RvD2 and MaR1. | ASMC chemotaxis was reduced in 74% after treatment with RvD2 and MaR1. | |
| ATL | Petri et al. [122] | Four mice who presented Fpr2 deficiency were fed with a high-fat diet for 4 weeks and later treated with ATL. | ATL blocked the progression at the root of atherosclerosis. |
| RvD1 and PD1 | Makino et al. [123] | Male injured mice who were subjected to carotid artery angioplasty received 1 μg of RvD1 or PD1 intravenously. | RvD1 and PD1 mitigated muscle cell proliferation, as well as leukocyte infiltration. |
| RvD1 and RvD2 | Miyahara et al. [57] | Cultures of human greater saphenous veins VSMC were isolated and treated with RvD1 and RvD2. | RvD1 and RvD2 inhibited VSMC and monocyte proliferation, migration, and adhesion. |
| RvD1 | Kain et al. [114] | Male mice with coronary artery ligation were administered liposomes with RvD1 or Lipo-RvD1. | RvD1 decreased macrophage density. |
| Orr et al. [115] | 0.5 g of EOR and BDA-RvD1 were given to mice. | BDA-RvD1 was resistant to EOR and reduced neutrophil infiltration in the lungs. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salazar, J.; Pirela, D.; Nava, M.; Castro, A.; Angarita, L.; Parra, H.; Durán-Agüero, S.; Rojas-Gómez, D.M.; Galbán, N.; Añez, R.; et al. Specialized Proresolving Lipid Mediators: A Potential Therapeutic Target for Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3133. https://doi.org/10.3390/ijms23063133
Salazar J, Pirela D, Nava M, Castro A, Angarita L, Parra H, Durán-Agüero S, Rojas-Gómez DM, Galbán N, Añez R, et al. Specialized Proresolving Lipid Mediators: A Potential Therapeutic Target for Atherosclerosis. International Journal of Molecular Sciences. 2022; 23(6):3133. https://doi.org/10.3390/ijms23063133
Chicago/Turabian StyleSalazar, Juan, Daniela Pirela, Manuel Nava, Ana Castro, Lissé Angarita, Heliana Parra, Samuel Durán-Agüero, Diana Marcela Rojas-Gómez, Néstor Galbán, Roberto Añez, and et al. 2022. "Specialized Proresolving Lipid Mediators: A Potential Therapeutic Target for Atherosclerosis" International Journal of Molecular Sciences 23, no. 6: 3133. https://doi.org/10.3390/ijms23063133