
VPA and TSA Interrupt the Interplay between mutp53 and HSP70, Leading to CHK1 and RAD51 Down-Regulation and Sensitizing Pancreatic Cancer Cells to AZD2461 PARP Inhibitor

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
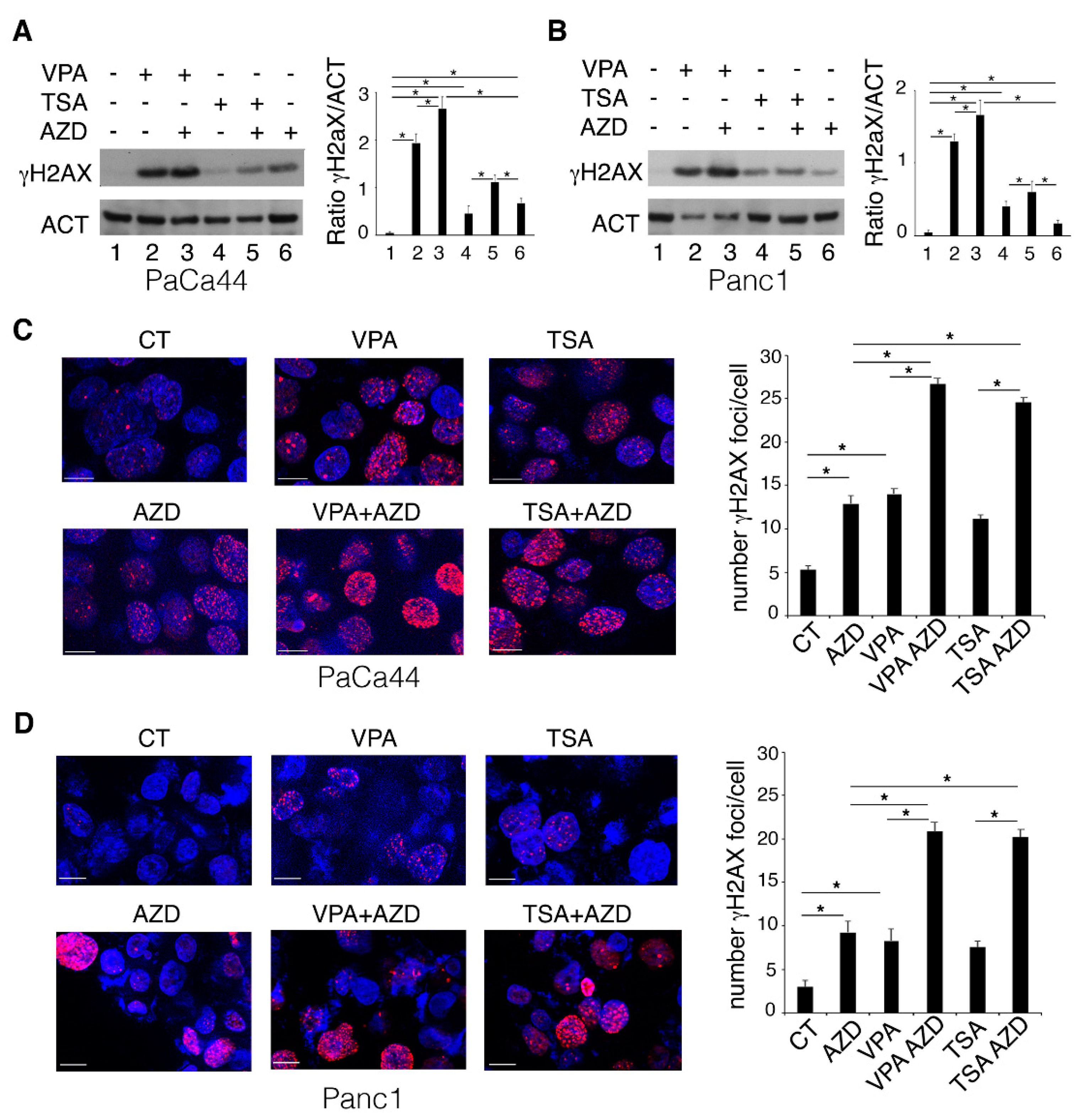
2.1. VPA and TSA Increase the Cytotoxic Effect and DNA Damage Induced by AZD2461 PARP Inhibitor
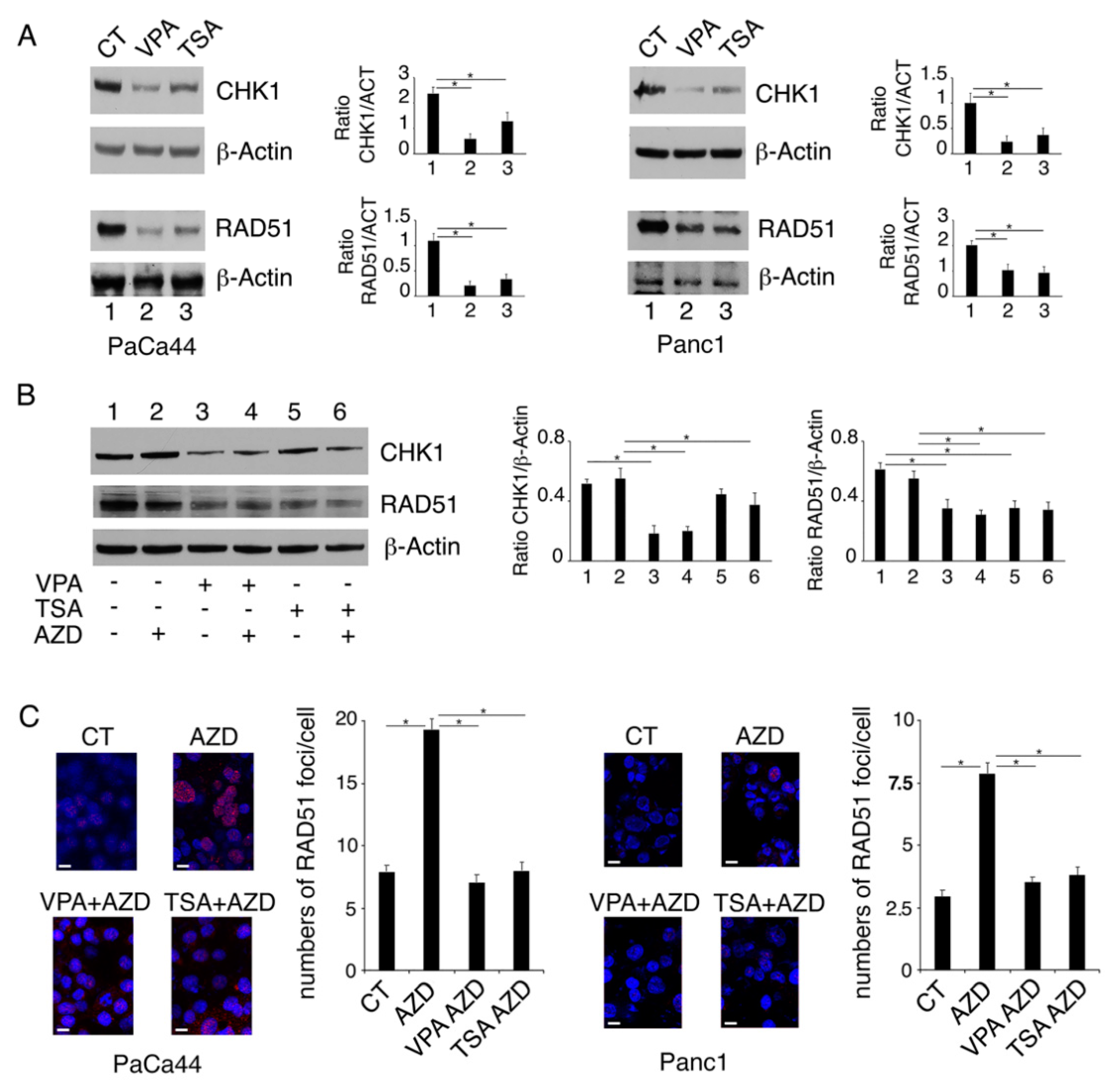
2.2. VPA and TSA Down-Regulate RAD51 and CHK1 and Reduce the Formation of RAD51-Positive Foci in Pancreatic Cancer Cells
2.3. Proteasomal Degradation Is Involved in the Downregulation of CHK1 and RAD51
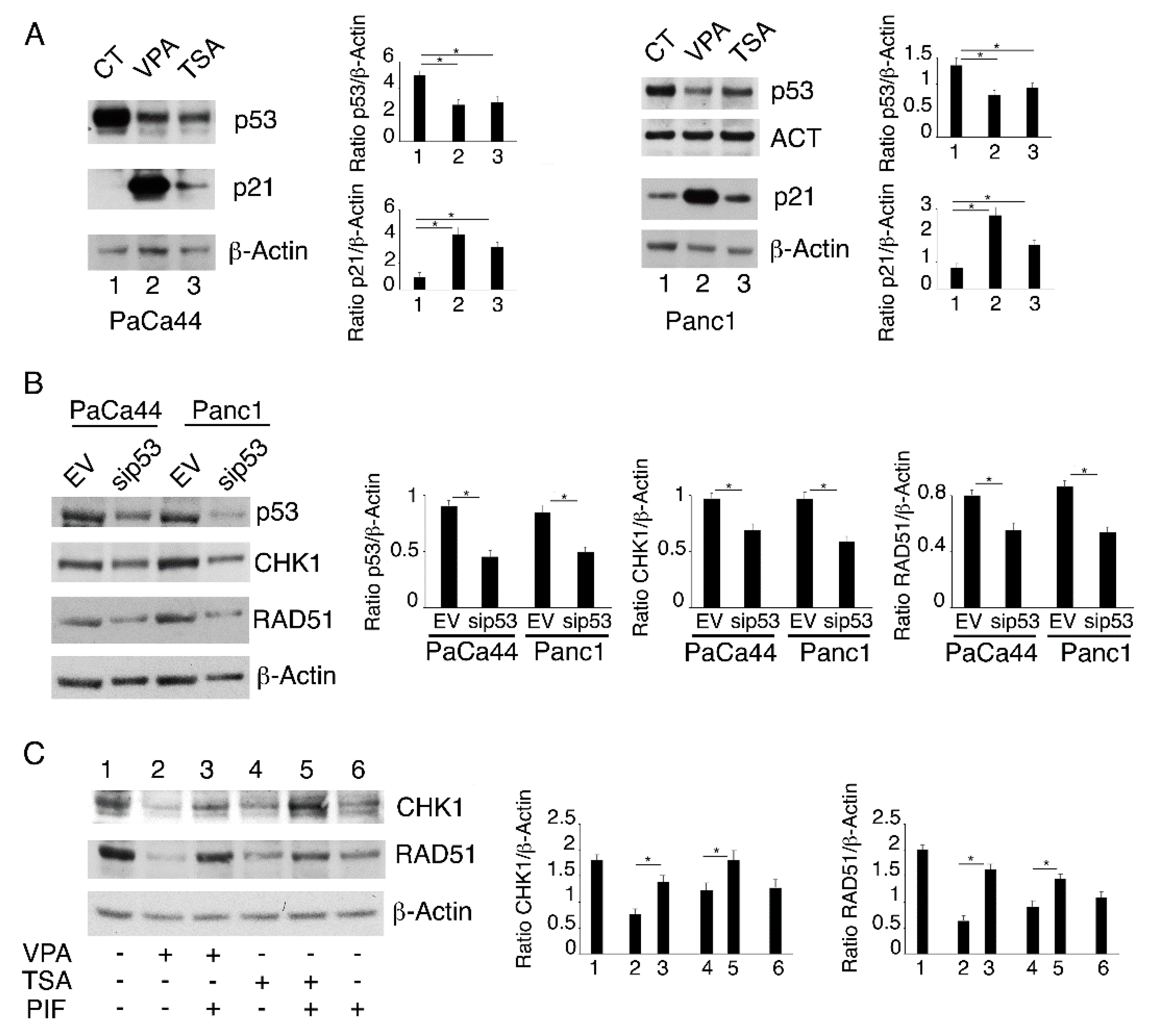
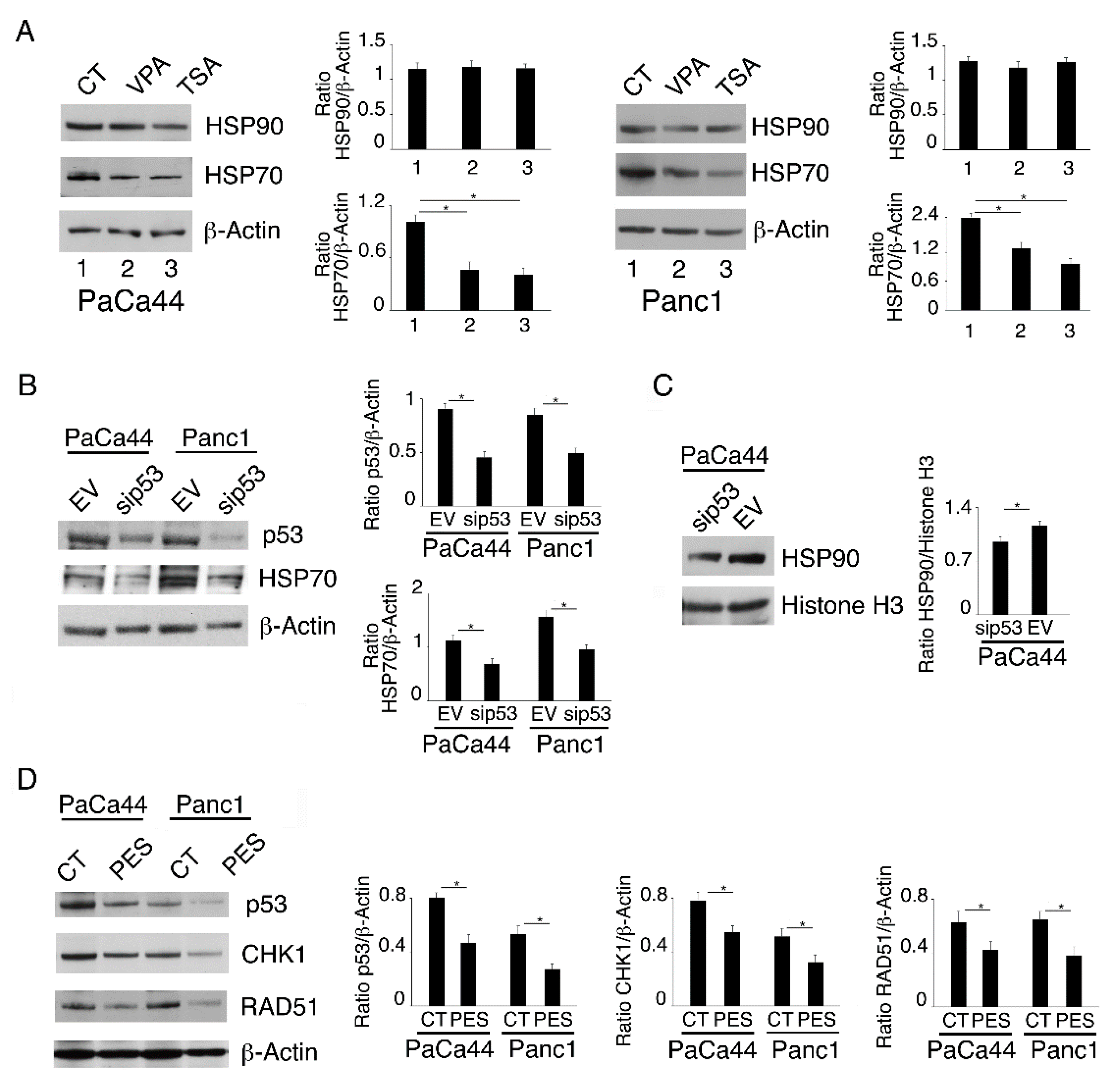
2.4. Both mutp53 Reduction and wtp53 Reactivation by VPA and TSA Contribute to the Down-Regulation of RAD51 and CHK1
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Treatments
4.2. Trypan Blue Assay
4.3. MTT Assay
4.4. p53 Silencing
4.5. Cell Cycle Analysis
4.6. Indirect Immunofluorescence Assay (IFA)
4.7. Western Blot Analysis
4.8. Antibodies
4.9. RNA Isolation and Quantitative Real Time Polymerase Chain Reaction (qRT-PCR)
4.10. Densitometric Analysis
4.11. Statistical Analysis
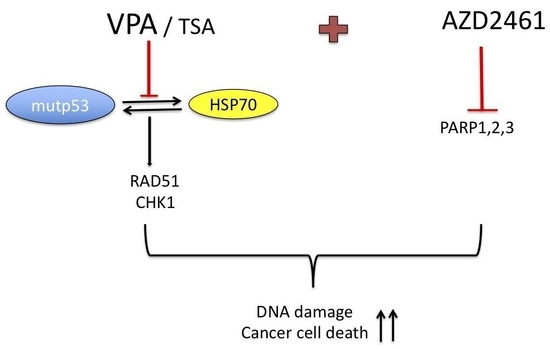
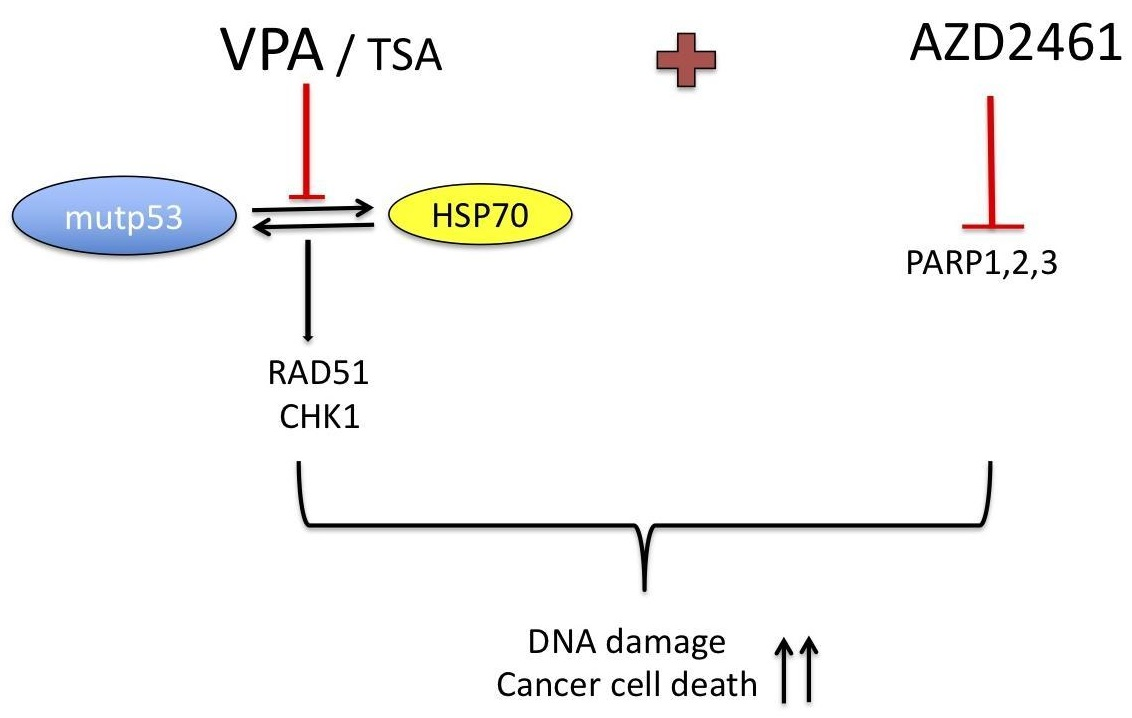
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schneider, G.; Schmid, R.M. Genetic alterations in pancreatic carcinoma. Mol. Cancer 2003, 2, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Orazi, G.; Cordani, M.; Cirone, M. Oncogenic pathways activated by pro-inflammatory cytokines promote mutant p53 stability: Clue for novel anticancer therapies. Cell Mol. Life Sci. 2021, 78, 1853–1860. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Gheorghiu, L.; Drumm, M.; Clayman, R.; Eidelman, A.; Wszolek, M.F.; Olumi, A.; Feldman, A.; Wang, M.; Marcar, L.; et al. PARP-1 inhibition with or without ionizing radiation confers reactive oxygen species-mediated cytotoxicity preferentially to cancer cells with mutant TP53. Oncogene 2018, 37, 2793–2805. [Google Scholar] [CrossRef] [PubMed]
- Romeo, M.A.; Montani, M.S.G.; Benedetti, R.; Arena, A.; Maretto, M.; Bassetti, E.; Caiazzo, R.; D’Orazi, G.; Cirone, M. Anticancer effect of AZD2461 PARP inhibitor against colon cancer cells carrying wt or dysfunctional p53. Exp. Cell Res. 2021, 408, 112879. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Zajac-Kaye, M.; Rubinstein, L.; Voeller, D.; Tomaszewski, J.E.; Kummar, S.; Chen, A.P.; Pommier, Y.; Doroshow, J.H.; Yang, S.X. Poly(ADP-ribose) polymerase inhibition enhances p53-dependent and -independent DNA damage responses induced by DNA damaging agent. Cell Cycle 2011, 10, 4074–4082. [Google Scholar] [CrossRef] [Green Version]
- Pazzaglia, S.; Pioli, C. Multifaceted Role of PARP-1 in DNA Repair and Inflammation: Pathological and Therapeutic Implications in Cancer and Non-Cancer Diseases. Cells 2019, 9, 41. [Google Scholar] [CrossRef] [Green Version]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Wiegmans, A.P.; Yap, P.Y.; Ward, A.; Lim, Y.C.; Khanna, K.K. Differences in Expression of Key DNA Damage Repair Genes after Epigenetic-Induced BRCAness Dictate Synthetic Lethality with PARP1 Inhibition. Mol. Cancer Ther. 2015, 14, 2321–2331. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Shi, B.; Liu, X.; An, H.X. Acetylation and Deacetylation of DNA Repair Proteins in Cancers. Front. Oncol. 2020, 10, 573502. [Google Scholar] [CrossRef]
- Zhao, J.; Xie, C.; Edwards, H.; Wang, G.; Taub, J.W.; Ge, Y. Histone deacetylases 1 and 2 cooperate in regulating BRCA1, CHK1, and RAD51 expression in acute myeloid leukemia cells. Oncotarget 2017, 8, 6319–6329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, K.; Fiskus, W.; Choi, D.S.; Bhaskara, S.; Cerchietti, L.; Devaraj, S.G.; Shah, B.; Sharma, S.; Chang, J.C.; Melnick, A.M.; et al. Histone deacetylase inhibitor treatment induces ‘BRCAness’ and synergistic lethality with PARP inhibitor and cisplatin against human triple negative breast cancer cells. Oncotarget 2014, 5, 5637–5650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blagosklonny, M.V.; Trostel, S.; Kayastha, G.; Demidenko, Z.N.; Vassilev, L.T.; Romanova, L.Y.; Bates, S.; Fojo, T. Depletion of mutant p53 and cytotoxicity of histone deacetylase inhibitors. Cancer Res. 2005, 65, 7386–7392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernet, L.; Faure, V.; Gilquin, B.; Dufour-Guérin, S.; Khochbin, S.; Vourc’h, C. HDAC6-ubiquitin interaction controls the duration of HSF1 activation after heat shock. Mol. Biol. Cell 2014, 25, 4187–4194. [Google Scholar] [CrossRef] [Green Version]
- Wiech, M.; Olszewski, M.B.; Tracz-Gaszewska, Z.; Wawrzynow, B.; Zylicz, M.; Zylicz, A. Molecular mechanism of mutant p53 stabilization: The role of HSP70 and MDM2. PLoS ONE 2012, 7, e51426. [Google Scholar] [CrossRef] [Green Version]
- Genest, O.; Wickner, S.; Doyle, S.M. Hsp90 and Hsp70 chaperones: Collaborators in protein remodeling. J. Biol. Chem. 2019, 294, 2109–2120. [Google Scholar] [CrossRef] [Green Version]
- Montani, M.S.G.; Granato, M.; Santoni, C.; del Porto, P.; Merendino, N.; D’Orazi, G.; Faggioni, A.; Cirone, M. Histone deacetylase inhibitors VPA and TSA induce apoptosis and autophagy in pancreatic cancer cells. Cell Oncol. 2017, 40, 167–180. [Google Scholar] [CrossRef]
- Montani, M.S.G.; Benedetti, R.; Piconese, S.; Pulcinelli, F.M.; Timperio, A.M.; Romeo, M.A.; Masuelli, L.; Mattei, M.; Bei, R.; D’Orazi, G.; et al. PGE2 Released by Pancreatic Cancer Cells Undergoing ER Stress Transfers the Stress to DCs Impairing Their Immune Function. Mol. Cancer Ther. 2021, 20, 934–945. [Google Scholar] [CrossRef]
- Lee, H.S.; Park, S.B.; Kim, S.A.; Kwon, S.K.; Cha, H.; Lee, D.Y.; Ro, S.; Cho, J.M.; Song, S.Y. A novel HDAC inhibitor, CG200745, inhibits pancreatic cancer cell growth and overcomes gemcitabine resistance. Sci. Rep. 2017, 7, 41615. [Google Scholar] [CrossRef] [Green Version]
- Arias-Lopez, C.; Lazaro-Trueba, I.; Kerr, P.; Lord, C.J.; Dexter, T.; Iravani, M.; Ashworth, A.; Silva, A. p53 modulates homologous recombination by transcriptional regulation of the RAD51 gene. EMBO Rep. 2006, 7, 219–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arizti, P.; Fang, L.; Park, I.; Yin, Y.; Solomon, E.; Ouchi, T.; Aaronson, S.A.; Lee, S.W. Tumor suppressor p53 is required to modulate BRCA1 expression. Mol. Cell Biol. 2000, 20, 7450–7459. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Choy, M.L.; Ngo, L.; Foster, S.S.; Marks, P.A. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc. Natl. Acad. Sci. USA 2010, 107, 14639–14644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montani, M.S.G.; Cecere, N.; Granato, M.; Romeo, M.A.; Falcinelli, L.; Ciciarelli, U.; D’Orazi, G.; Faggioni, A.; Cirone, M. Mutant p53, Stabilized by Its Interplay with HSP90, Activates a Positive Feed-Back Loop Between NRF2 and p62 that Induces Chemo-Resistance to Apigenin in Pancreatic Cancer Cells. Cancers 2019, 11, 703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Krumm, A. The multifaceted influence of histone deacetylases on DNA damage signalling and DNA repair. Nucleic Acids Res. 2016, 44, 10017–10030. [Google Scholar] [CrossRef] [Green Version]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Chen, I.C.; Sethy, B.; Liou, J.P. Recent Update of HDAC Inhibitors in Lymphoma. Front. Cell Dev. Biol. 2020, 8, 576391. [Google Scholar] [CrossRef]
- Hii, L.W.; Chung, F.F.; Soo, J.S.; Tan, B.S.; Mai, C.W.; Leong, C.O. Histone deacetylase (HDAC) inhibitors and doxorubicin combinations target both breast cancer stem cells and non-stem breast cancer cells simultaneously. Breast Cancer Res. Treat. 2020, 179, 615–629. [Google Scholar] [CrossRef]
- Tharkar-Promod, S.; Johnson, D.P.; Bennett, S.E.; Dennis, E.M.; Banowsky, B.G.; Jones, S.S.; Shearstone, J.R.; Quayle, S.N.; Min, C.; Jarpe, M.; et al. HDAC1,2 inhibition and doxorubicin impair Mre11-dependent DNA repair and DISC to override BCR-ABL1-driven DSB repair in Philadelphia chromosome-positive B-cell precursor acute lymphoblastic leukemia. Leukemia 2018, 32, 49–60. [Google Scholar] [CrossRef]
- Camphausen, K.; Cerna, D.; Scott, T.; Sproull, M.; Burgan, W.E.; Cerra, M.A.; Fine, H.; Tofilon, P.J. Enhancement of in vitro and in vivo tumor cell radiosensitivity by valproic acid. Int. J. Cancer 2005, 114, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Chao, O.S.; Goodman, O.B. Synergistic loss of prostate cancer cell viability by coinhibition of HDAC and PARP. Mol. Cancer Res. 2014, 12, 1755–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, L.; Liu, Y.; Peng, Y.; Yu, X.; Gao, Y.; Yuan, B.; Zhu, Q.; Cao, T.; He, L.; Gong, Z.; et al. PARP inhibitor veliparib and HDAC inhibitor SAHA synergistically co-target the UHRF1/BRCA1 DNA damage repair complex in prostate cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Mawatari, T.; Ninomiya, I.; Inokuchi, M.; Harada, S.; Hayashi, H.; Oyama, K.; Makino, I.; Nakagawara, H.; Miyashita, T.; Tajima, H.; et al. Valproic acid inhibits proliferation of HER2-expressing breast cancer cells by inducing cell cycle arrest and apoptosis through Hsp70 acetylation. Int. J. Oncol. 2015, 47, 2073–2081. [Google Scholar] [CrossRef] [Green Version]
- Cecchinelli, B.; Lavra, L.; Rinaldo, C.; Iacovelli, S.; Gurtner, A.; Gasbarri, A.; Ulivieri, A.; del Prete, F.; Trovato, M.; Piaggio, G.; et al. Repression of the antiapoptotic molecule galectin-3 by homeodomain-interacting protein kinase 2-activated p53 is required for p53-induced apoptosis. Mol. Cell Biol. 2006, 26, 4746–4757. [Google Scholar] [CrossRef] [Green Version]
- Montani, M.S.G.; Granato, M.; Cuomo, L.; Valia, S.; di Renzo, L.; D’Orazi, G.; Faggioni, A.; Cirone, M. High glucose and hyperglycemic sera from type 2 diabetic patients impair DC differentiation by inducing ROS and activating Wnt/β-catenin and p38 MAPK. Biochim. Biophys. Acta 2016, 1862, 805–813. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romeo, M.A.; Gilardini Montani, M.S.; Benedetti, R.; Arena, A.; D’Orazi, G.; Cirone, M. VPA and TSA Interrupt the Interplay between mutp53 and HSP70, Leading to CHK1 and RAD51 Down-Regulation and Sensitizing Pancreatic Cancer Cells to AZD2461 PARP Inhibitor. Int. J. Mol. Sci. 2022, 23, 2268. https://doi.org/10.3390/ijms23042268
Romeo MA, Gilardini Montani MS, Benedetti R, Arena A, D’Orazi G, Cirone M. VPA and TSA Interrupt the Interplay between mutp53 and HSP70, Leading to CHK1 and RAD51 Down-Regulation and Sensitizing Pancreatic Cancer Cells to AZD2461 PARP Inhibitor. International Journal of Molecular Sciences. 2022; 23(4):2268. https://doi.org/10.3390/ijms23042268
Chicago/Turabian StyleRomeo, Maria Anele, Maria Saveria Gilardini Montani, Rossella Benedetti, Andrea Arena, Gabriella D’Orazi, and Mara Cirone. 2022. "VPA and TSA Interrupt the Interplay between mutp53 and HSP70, Leading to CHK1 and RAD51 Down-Regulation and Sensitizing Pancreatic Cancer Cells to AZD2461 PARP Inhibitor" International Journal of Molecular Sciences 23, no. 4: 2268. https://doi.org/10.3390/ijms23042268