
Effect of Oxidative Stress-Induced Apoptosis on Active FGF23 Levels in MLO-Y4 Cells: The Protective Role of 17-β-Estradiol

 , , ,
, , ,
Abstract
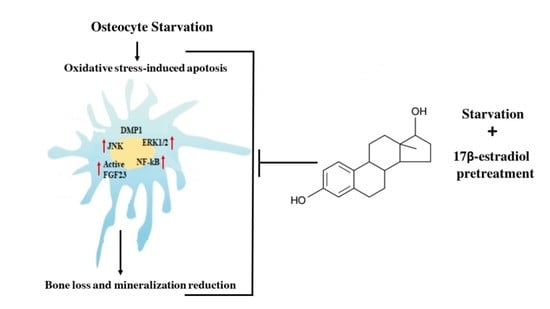
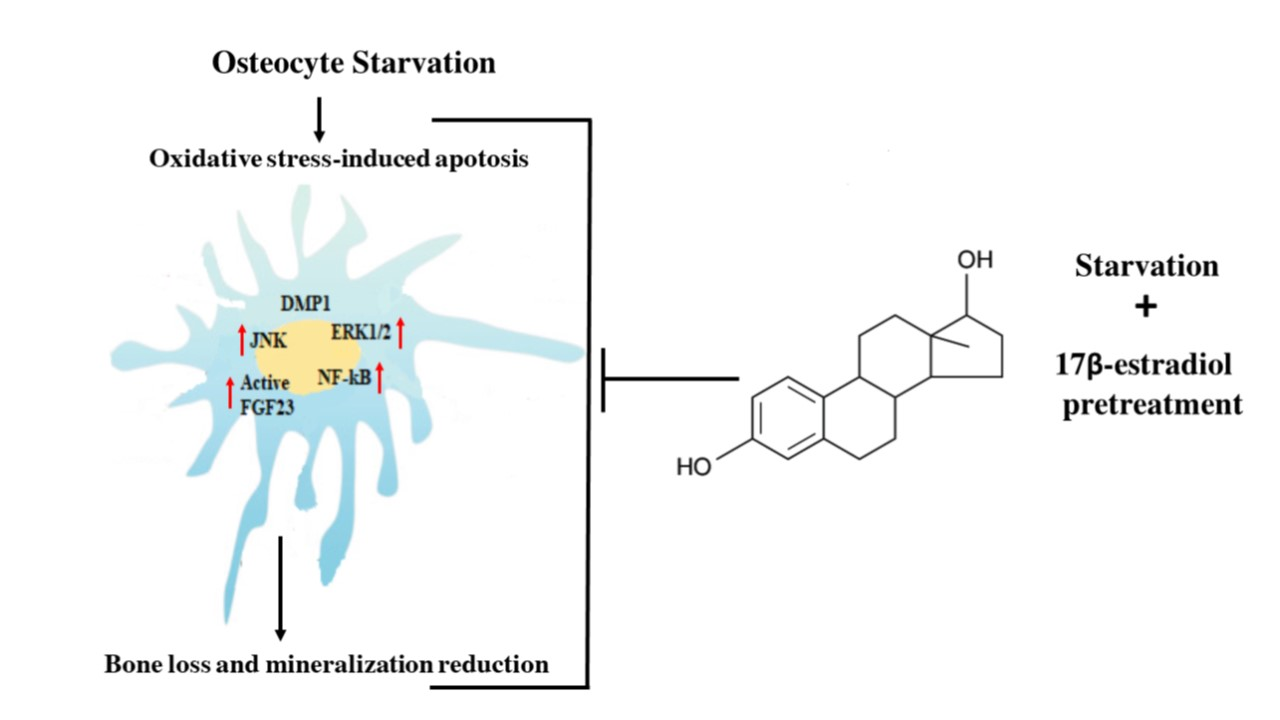
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
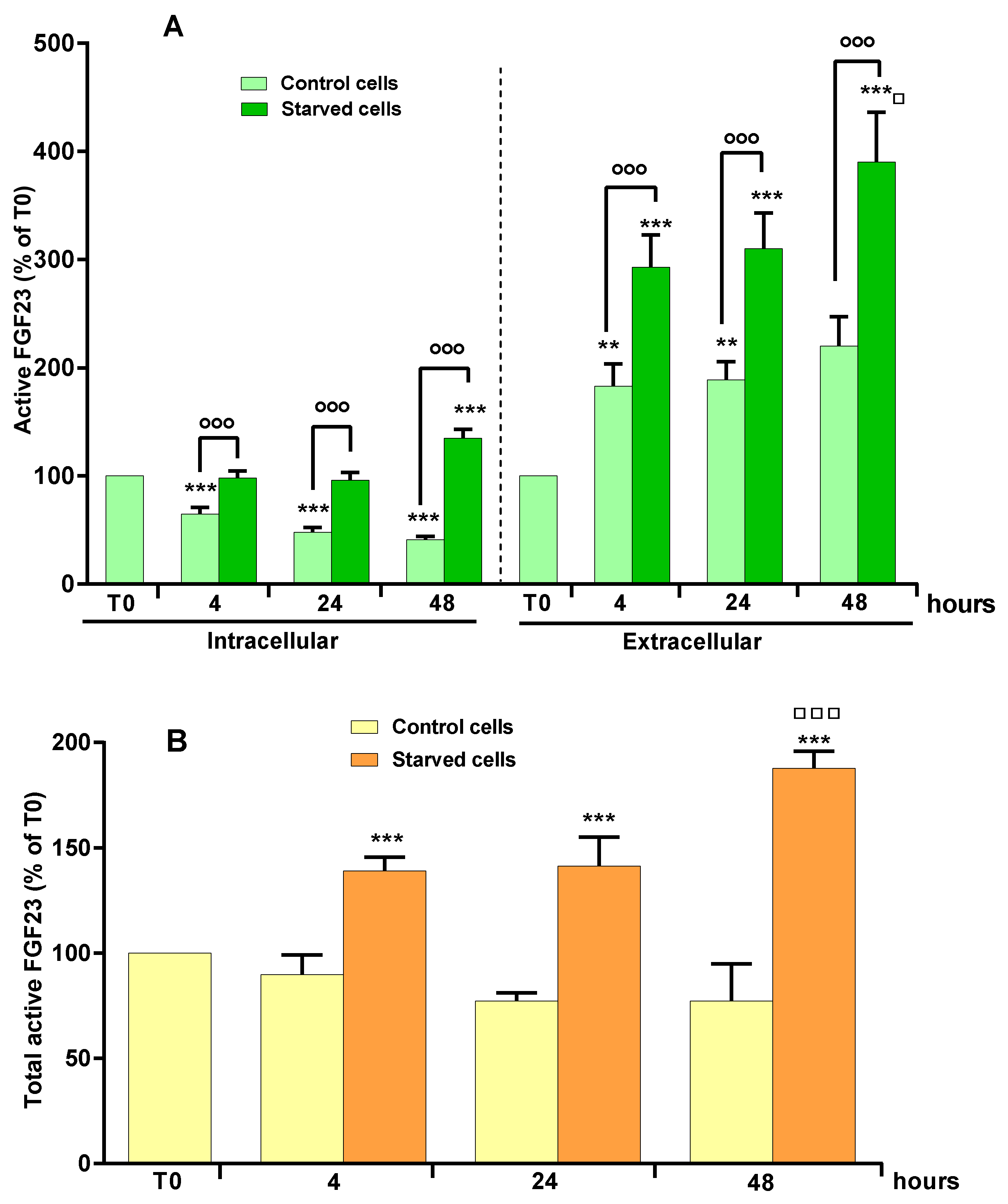
2.1. Effect of OSIA on Intra- and Extracellular Levels of Active FGF23 in Starved MLO-Y4 Cells
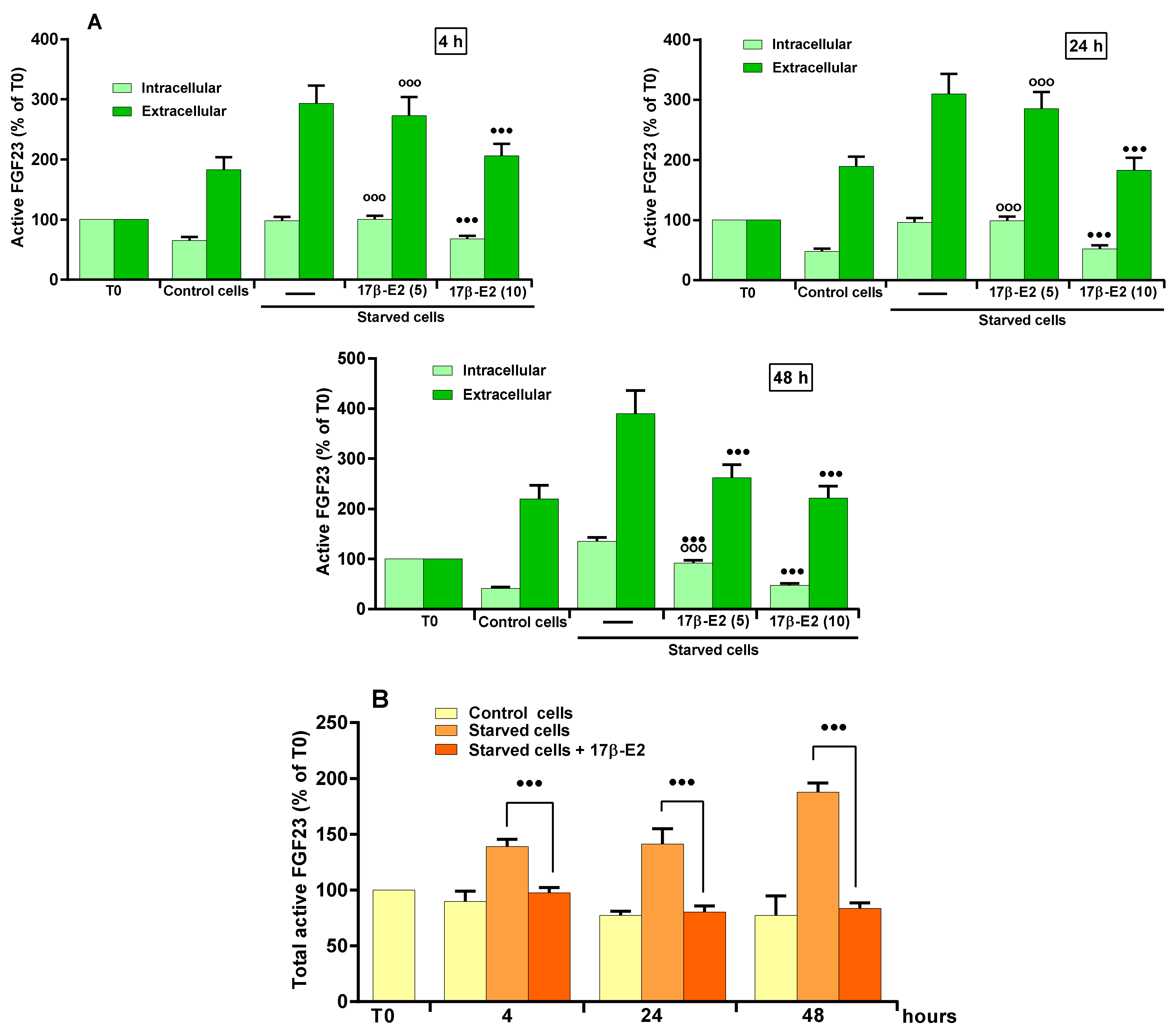
2.2. Role of 17β–E2 on OSIA-Induced Intra- and Extracellular Levels of Active FGF23 in Starved MLO-Y4 Cells
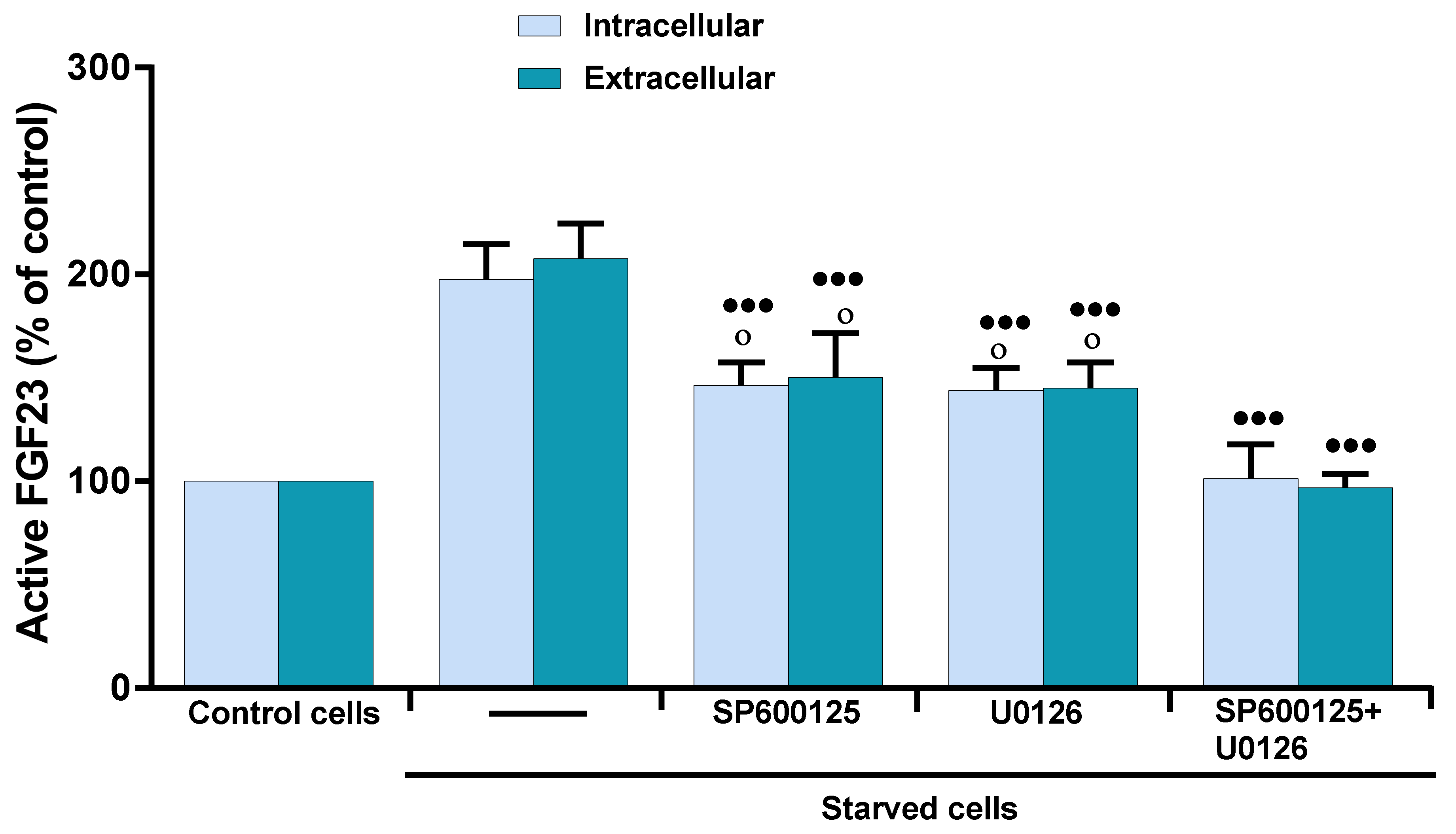
2.3. Role of MAP Kinases on OSIA-Induced Intra- and Extracellular Levels of Active FGF23 in Starved MLO-Y4 Cells
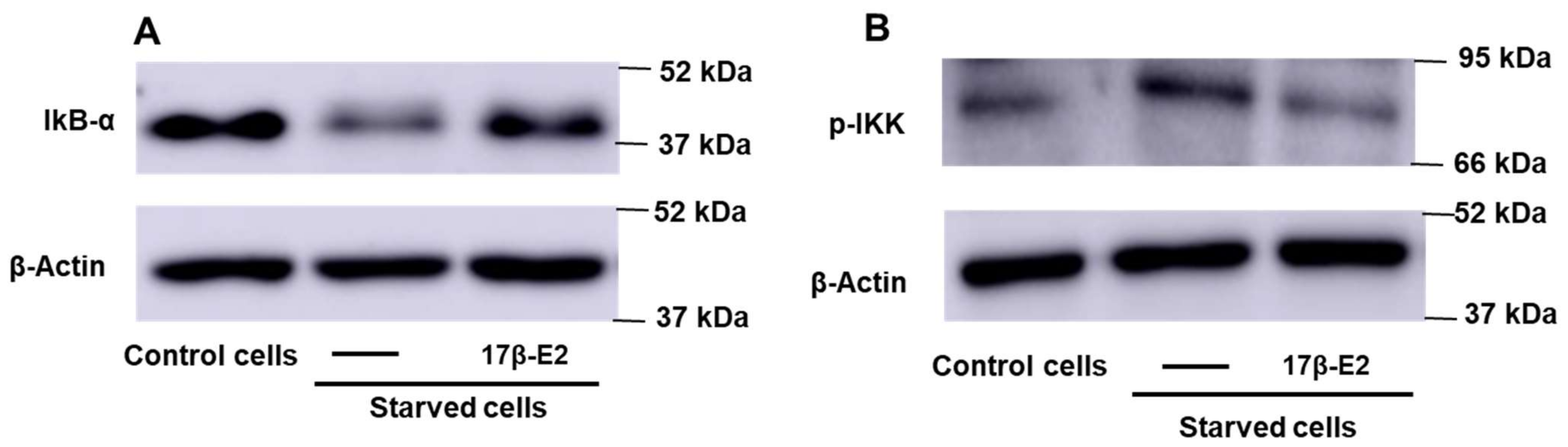
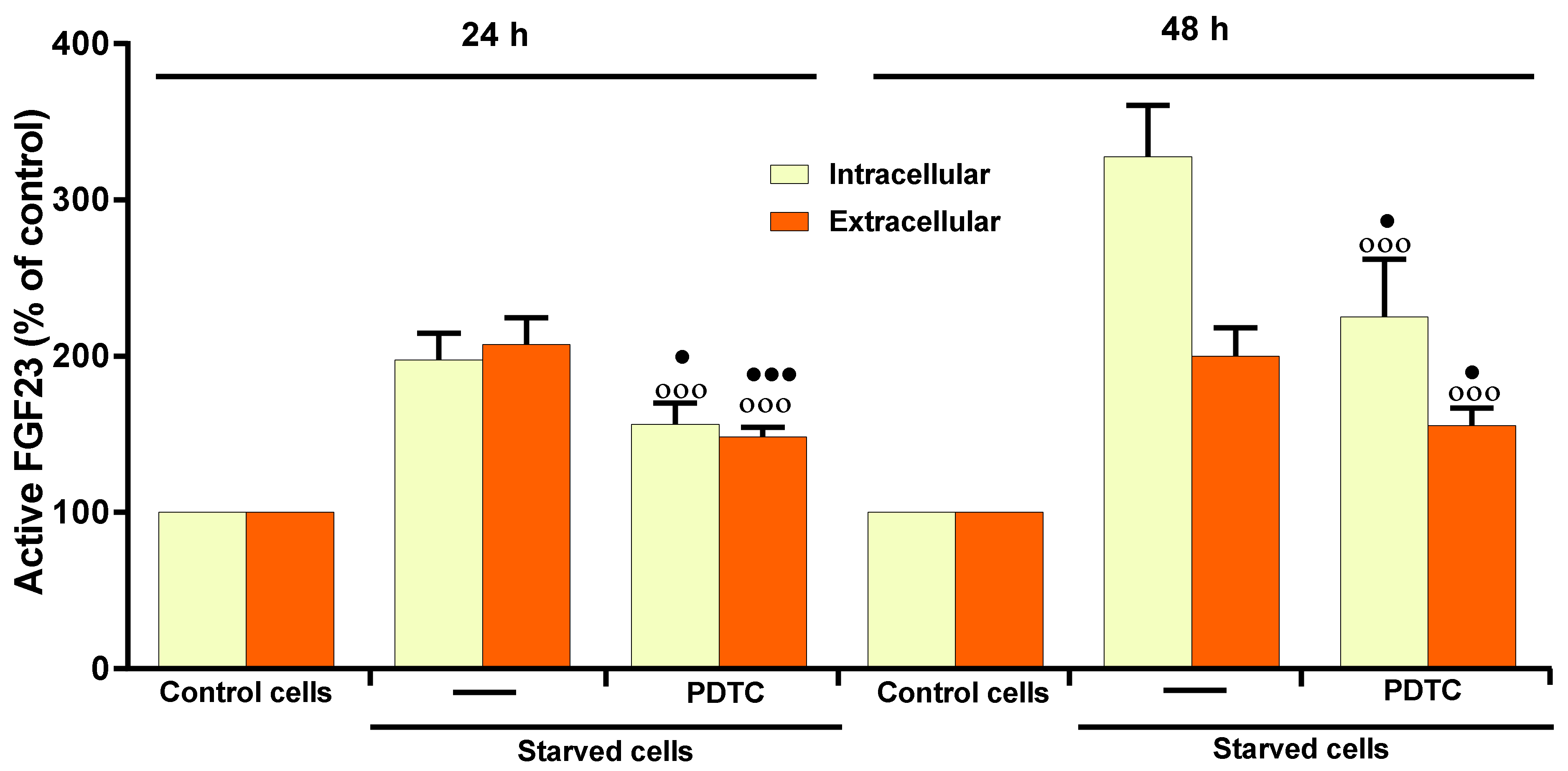
2.4. Role of NF-kB on 17β–E2 Effect on OSIA-Induced Intra- and Extracellular Levels of Active FGF23 in Starved MLO-Y4 Cells
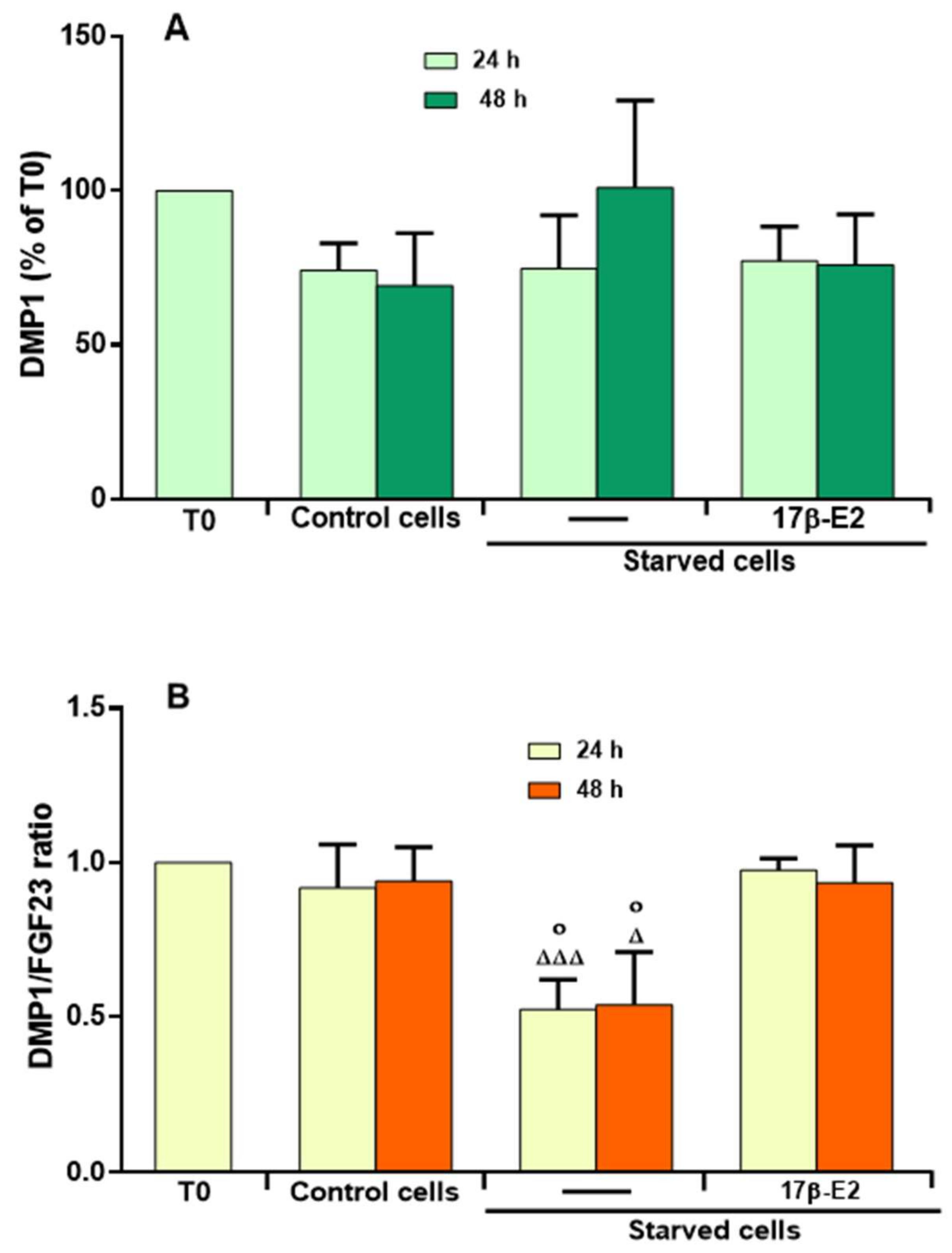
2.5. Effect of 17β–E2 on DMP1 Levels and Relationship with the Levels of Active FGF23 in the Presence of OSIA in Starved MLO-Y4 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. FGF23 and DMP1 Assay
4.3. Western Blot Analysis
4.4. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bonewald, L.F.; Wacker, M.J. FGF23 production by osteocytes. Pediatr. Nephrol. 2013, 28, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Takeshita, A.; Furushima, K.; Miyajima, M.; Hatamura, I.; Kuro-O, M.; Furuta, Y.; Sakaguchi, K. Persistent fibroblast growth factor 23 signalling in the parathyroid glands for secondary hyperparathyroidism in mice with chronic kidney disease. Sci. Rep. 2017, 7, 40534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takashi, Y.; Fukumoto, S. FGF23 beyond Phosphotropic Hormone. Trends Endocrinol. Metab. 2018, 29, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Peacock, M. Phosphate Metabolism in Health and Disease. Calcif. Tissue Int. 2021, 108, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Kuro-O, M. The Klotho proteins in health and disease. Nat. Rev. Nephrol. 2019, 15, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Ewendt, F.; Feger, M.; Föller, M. Role of Fibroblast Growth Factor 23 (FGF23) and αKlotho in Cancer. Front. Cell Dev. Biol. 2021, 8, 601006. [Google Scholar] [CrossRef]
- Ito, N.; Findlay, D.M.; Anderson, P.H.; Bonewald, L.F.; Atkins, G.J. Extracellular phosphate modulates the effect of 1α,25-dihydroxy vitamin D3 (1,25D) on osteocyte like cells. J. Steroid Biochem. Mol. Biol. 2013, 136, 183–186. [Google Scholar] [CrossRef]
- Lang, F.; Leibrock, C.; Pandyra, A.A.; Stournaras, C.; Wagner, C.A.; Föller, M. Phosphate Homeostasis, Inflammation and the Regulation of FGF-23. Kidney Blood Press Res. 2018, 43, 1742–1748. [Google Scholar] [CrossRef]
- Ito, N.; Prideaux, M.; Wijenayaka, A.R.; Yang, D.; Ormsby, R.T.; Bonewald, L.F.; Atkins, G.J. Sclerostin Directly Stimulates Osteocyte Synthesis of Fibroblast Growth Factor-23. Calcif. Tissue Int. 2021, 109, 66–76. [Google Scholar] [CrossRef]
- Courbebaisse, M.; Lanske, B. Biology of Fibroblast Growth Factor 23: From Physiology to Pathology. Cold Spring Harb. Perspect. Med. 2018, 8, a031260. [Google Scholar] [CrossRef]
- Kitaura, H.; Marahleh, A.; Ohori, F.; Noguchi, T.; Shen, W.R.; Qi, J.; Nara, Y.; Pramusita, A.; Kinjo, R.; Mizoguchi, I. Osteocyte-Related Cytokines Regulate Osteoclast Formation and Bone Resorption. Int. J. Mol. Sci. 2020, 21, 5169. [Google Scholar] [CrossRef] [PubMed]
- Pathak, J.L.; Bravenboer, N.; Klein-Nulend, J. The Osteocyte as the New Discovery of Therapeutic Options in Rare Bone Diseases. Front. Endocrinol. 2020, 11, 405. [Google Scholar] [CrossRef] [PubMed]
- Bonewald, L.F. The Role of the Osteocyte in Bone and Nonbone Disease. Endocrinol. Metab. Clin. North Am. 2017, 46, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Li, S.; Pathak, J.L. Pro-inflammatory Cytokines and Osteocytes. Curr. Osteoporos. Rep. 2019, 17, 97–104. [Google Scholar] [CrossRef]
- Jilka, R.L.; O’Brien, C.A. The Role of Osteocytes in Age-Related Bone Loss. Curr. Osteoporos. Rep. 2016, 14, 16–25. [Google Scholar] [CrossRef]
- Sapir-Koren, R.; Livshits, G. Bone mineralization is regulated by signaling cross talk between molecular factors of local and systemic origin: The role of fibroblast growth factor 23. Biofactors 2014, 40, 555–568. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, H.; Jani, P.; Wang, X.; Lu, Y.; Li, N.; Xiao, J.; Qin, C. FAM20C regulates osteoblast behaviors and intracellular signaling pathways in a cell-autonomous manner. J. Cell Physiol. 2018, 233, 3476–3486. [Google Scholar] [CrossRef]
- Masi, L.; Beltrami, G.; Ottanelli, S.; Franceschelli, F.; Gozzini, A.; Zonefrati, R.; Galli, G.; Ciuffi, S.; Mavilia, C.; Giusti, F.; et al. Human Preosteoblastic Cell Culture from a Patient with Severe Tumoral Calcinosis-Hyperphosphatemia Due to a New GALNT3 Gene Mutation: Study of In Vitro Mineralization. Calcif. Tissue Int. 2015, 96, 438–452. [Google Scholar] [CrossRef]
- Fukumoto, S. FGF23-related hypophosphatemic rickets/osteomalacia: Diagnosis and new treatment. J. Mol. Endocrinol. 2021, 66, R57–R65. [Google Scholar] [CrossRef]
- Domazetovic, V.; Marcucci, G.; Iantomasi, T.; Brandi, M.L.; Vincenzini, M.T. Oxidative stress in bone remodeling: Role of antioxidants. Clin. Cases Miner. Bone Metab. 2017, 14, 209–216. [Google Scholar] [CrossRef]
- Cao, X.; Luo, D.; Li, T.; Huang, Z.; Zou, W.; Wang, L.; Lian, K.; Lin, D. MnTBAP inhibits bone loss in ovariectomized rats by reducing mitochondrial oxidative stress in osteoblasts. J. Bone Miner. Metab. 2020, 38, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, C.; Marcucci, G.; Favilli, F.; Zonefrati, R.; Mavilia, C.; Galli, G.; Tanini, A.; Iantomasi, T.; Brandi, M.L.; Vincenzini, M.T. Role of GSH/GSSG redox couple in osteogenic activity and osteoclastogenic markers of human osteoblast-like SaOS-2 cells. FEBS J. 2013, 280, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Callaway, D.A.; Jiang, J.X. Reactive oxygen species and oxidative stress in osteoclastogenesis, skeletal aging and bone diseases. J. Bone Miner. Metab. 2015, 33, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Domazetovic, V.; Marcucci, G.; Falsetti, I.; Bilia, A.R.; Vincenzini, M.T.; Brandi, M.L.; Iantomasi, T. Blueberry Juice Antioxidants Protect Osteogenic Activity against Oxidative Stress and Improve Long-Term Activation of the Mineralization Process in Human Osteoblast-Like SaOS-2 Cells: Involvement of SIRT1. Antioxidants 2020, 9, 125. [Google Scholar] [CrossRef] [Green Version]
- Bellido, T. Osteocyte-driven bone remodeling. Calcif. Tissue Int. 2014, 94, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Wein, M.N.; Kronenberg, H.M. Regulation of Bone Remodeling by Parathyroid Hormone. Cold Spring Harb. Perspect. Med. 2018, 8, a031237. [Google Scholar] [CrossRef] [Green Version]
- Werner, S.L.; Sharma, R.; Woodruff, K.; Horn, D.; Harris, S.E.; Gorin, Y.; Lee, D.Y.; Hua, R.; Gu, S.; Fajardo, R.J.; et al. CSF-1 in Osteocytes Inhibits Nox4-mediated Oxidative Stress and Promotes Normal Bone Homeostasis. JBMR Plus 2019, 4, e10080. [Google Scholar] [CrossRef]
- Zhu, X.; Zhao, Z.; Zeng, C.; Chen, B.; Huang, H.; Chen, Y.; Zhou, Q.; Yang, L.; Lv, J.; Zhang, J.; et al. HNGF6A Inhibits Oxidative Stress-Induced MC3T3-E1 Cell Apoptosis and Osteoblast Phenotype Inhibition by Targeting Circ_0001843/miR-214 Pathway. Calcif. Tissue Int. 2020, 106, 518–532. [Google Scholar] [CrossRef]
- Cao, Z.; Geng, X.; Jiang, X.; Gao, X.; Liu, K.; Li, Y. Melatonin Attenuates AlCl 3-Induced Apoptosis and Osteoblastic Differentiation Suppression by Inhibiting Oxidative Stress in MC3T3-E1 Cells. Biol. Trace Elem. Res. 2020, 196, 214–222. [Google Scholar] [CrossRef]
- Fontani, F.; Marcucci, G.; Iantomasi, T.; Brandi, M.L.; Vincenzini, M.T. Glutathione, N-acetylcysteine and lipoic acid down-regulate starvation-induced apoptosis, RANKL/OPG ratio and sclerostin in osteocytes: Involvement of JNK and ERK1/2 signalling. Calcif. Tissue Int. 2015, 96, 335–346. [Google Scholar] [CrossRef]
- Davis, H.M.; Aref, M.W.; Aguilar-Perez, A.; Pacheco-Costa, R.; Allen, K.; Valdez, S.; Herrera, C.; Atkinson, E.G.; Mohammad, A.; Lopez, D.; et al. Cx43 overexpression in osteocytes prevents osteocyte apoptosis and preserves cortical bone quality in aging mice. JBMR Plus 2018, 2, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Han, L.; Cong, W. Alpinumisoflavone rescues glucocorticoid-induced apoptosis of osteocytes via suppressing Nox2-dependent ROS generation. Pharmacol. Rep. 2018, 70, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Chai, S.; Wan, L.; Wang, J.L.; Huang, J.C.; Huang, H.X. Gushukang inhibits osteocyte apoptosis and enhances BMP-2/Smads signaling pathway in ovariectomized rats. Phytomedicine 2019, 64, 153063. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.A.B.; Sousa-Pereira, A.P.; Lucisano, M.P.; Romualdo, P.C.; Paula-Silva, F.W.G.; Consolaro, A.; Silva, L.A.B.; Nelson-Filho, P. Alendronate inhibits osteocyte apoptosis and inflammation via IL-6, inhibiting bone resorption in periapical lesions of ovariectomized rats. Int. Endod. J. 2020, 53, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Nahian, A.; AlEssa, A.M. Histology, Osteocytes; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar] [PubMed]
- Huidrom, S.; Beg, M.A.; Masood, T. Post-menopausal Osteoporosis and Probiotics. Curr. Drug Targets 2021, 22, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Stepan, J.J.; Hruskova, H.; Kverka, M. Update on Menopausal Hormone Therapy for Fracture Prevention. Curr. Osteoporos. Rep. 2019, 17, 465–473. [Google Scholar] [CrossRef] [Green Version]
- Hori, M.; Kinoshita, Y.; Taguchi, M.; Fukumoto, S. Phosphate enhances Fgf23 expression through reactive oxygen species in UMR-106 cells. J. Bone Miner. Metab. 2016, 34, 132–139. [Google Scholar] [CrossRef]
- Wang, T.; Yu, X.; He, C. Pro-inflammatory Cytokines: Cellular and Molecular Drug Targets for Glucocorticoid-induced-osteoporosis via Osteocyte. Curr. Drug Targets 2019, 20, 1–15. [Google Scholar] [CrossRef]
- Domazetovic, V.; Marcucci, G.; Pierucci, F.; Bruno, G.; Di Cesare Mannelli, L.; Ghelardini, C.; Brandi, M.L.; Iantomasi, T.; Meacci, E.; Vincenzini, M.T. Blueberry juice protects osteocytes and bone precursor cells against oxidative stress partly through SIRT1. FEBS OpenBio 2019, 9, 1082–1096. [Google Scholar] [CrossRef]
- Austermann, K.; Baecker, N.; Stehle, P.; Heer, M. Putative Effects of Nutritive Polyphenols on Bone Metabolism In Vivo-Evidence from Human Studies. Nutrients 2019, 11, 871. [Google Scholar] [CrossRef] [Green Version]
- Mohamad, N.V.; Ima-Nirwana, S.; Chin, K.Y. Are Oxidative Stress and Inflammation Mediators of Bone Loss Due to Estrogen Deficiency? A Review of Current Evidence. Endocr. Metab. Immune Disord. Drug Targets 2020, 20, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Domazetovic, V.; Fontani, F.; Marcucci, G.; Iantomasi, T.; Brandi, M.L.; Vincenzini, M.T. Estrogen inhibits starvation-induced apoptosis in osteocytes by a redox-independent process involving association of JNK and glutathione S-transferase P1-1. FEBS Open Bio 2017, 7, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Kalajzic, I.; Matthews, B.G.; Torreggiani, E.; Harris, M.A.; Divieti Pajevic, P.; Harris, S.E. In vitro and in vivo approaches to study osteocyte biology. Bone 2013, 54, 296–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCutcheon, S.; Majeska, R.J.; Spray, D.C.; Schaffler, M.B.; Vazquez, M. Apoptotic Osteocytes Induce RANKL Production in Bystanders via Purinergic Signaling and Activation of Pannexin Channels. J. Bone Miner. Res. 2020, 35, 966–977. [Google Scholar] [CrossRef]
- Hu, B.; Sun, X.; Yang, Y.; Ying, Z.; Meng, J.; Zhou, C.; Jiang, G.; Li, S.; Wu, F.; Zhao, X.; et al. Tomatidine suppresses osteoclastogenesis and mitigates estrogen deficiency-induced bone mass loss by modulating TRAF6-mediated signaling. FASEB J. 2019, 33, 2574–2586. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Yan, Z.; Wang, Y.; Yang, Y.; Cai, M.; Huang, C.; Li, B.; Yang, M.; Zhou, X.; Wei, X.; et al. Shikonin mitigates ovariectomy-induced bone loss and RANKL-induced osteoclastogenesis via TRAF6-mediated signaling pathways. Biomed. Pharmacother. 2020, 126, 110067. [Google Scholar] [CrossRef]
- Chen, J.; Yu, M.; Li, X.; Sun, Q.F.; Yang, C.Z.; Yang, P.S. Progranulin promotes osteogenic differentiation of human periodontal ligament stem cells via tumor necrosis factor receptors to inhibit TNF-α sensitized NF-kB and activate ERK/JNK signaling. J. Periodontal. Res. 2020, 55, 363–373. [Google Scholar] [CrossRef]
- Ito, N.; Wijenayaka, A.R.; Prideaux, M.; Kogawa, M.; Ormsby, R.T.; Evdokiou, A.; Bonewald, L.F.; Findlay, D.M.; Atkins, G.J. Regulation of FGF23 expression in IDG-SW3 osteocytes and human bone by pro-inflammatory stimuli. Mol. Cell Endocrinol. 2015, 399, 208–218. [Google Scholar] [CrossRef]
- Quarles, L.D. Skeletal secretion of FGF-23 regulates phosphate and vitamin D metabolism. Nat. Rev. Endocrinol. 2012, 8, 276–286. [Google Scholar] [CrossRef]
- Francis, C.; David, V. Inflammation regulates fibroblast growth factor 23 production. Curr. Opin. Nephrol. Hypertens. 2016, 25, 325–332. [Google Scholar] [CrossRef] [Green Version]
- David, V.; Martin, A.; Isakova, T.; Spaulding, C.; Qi, L.; Ramirez, V.; Zumbrennen-Bullough, K.B.; Sun, C.C.; Lin, H.Y.; Babitt, J.L.; et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016, 89, 135–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, B.; Haller, J.; Haffner, D.; Leifheit-Nestler, M. Klotho modulates FGF23-mediated NO synthesis and oxidative stress in human coronary artery endothelial cells. Pflugers Arch. 2016, 468, 1621–1635. [Google Scholar] [CrossRef] [PubMed]
- Daenen, K.; Andries, A.; Mekahli, D.; Schepdael, A.V.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dussold, C.; Gerber, C.; White, S.; Wang, X.; Qi, L.; Francis, C.; Capella, M.; Courbon, G.; Wang, J.; Li, C.; et al. DMP1 prevents osteocyte alterations, FGF23 elevation and left ventricular hypertrophy in mice with chronic kidney disease. Bone Res. 2019, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S. Protective effect of Polygonatum sibiricum Polysaccharide on D-galactose-induced aging rats model. Sci. Rep. 2020, 10, 2246. [Google Scholar] [CrossRef] [Green Version]
- Ru, J.Y.; Wang, Y.F. Osteocyte apoptosis: The roles and key molecular mechanisms in resorption-related bone diseases. Cell Death Dis. 2020, 11, 846. [Google Scholar] [CrossRef]
- Ye, T.; Cao, P.; Qi, J.; Zhou, Q.; Rao, D.S.; Qiu, S. Protective effect of low-dose risedronate against osteocyte apoptosis and bone loss in ovariectomized rats. PLoS ONE 2017, 12, e0186012. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.Y.; Lv, Z.D.; Wang, K.; Qian, L.; Song, X.X.; Li, X.F.; Shen, H.X. Estradiol Alleviates Intervertebral Disc Degeneration through Modulating the Antioxidant Enzymes and Inhibiting Autophagy in the Model of Menopause Rats. Oxid. Med. Cell Longev. 2018, 2018, 7890291. [Google Scholar] [CrossRef] [Green Version]
- Aguilar-Castro, J.; Cervantes-Candelas, L.A.; Buendía-González, F.O.; de Jesús Nolasco-Pérez, T.; López-Padilla, M.S.; Fernández-Rivera, O.; Cervantes-Sandoval, A.; Legorreta-Herrera, M. Dimorphic effect of 17β-oestradiol on pathology and oxidative stress in experimental malaria. Immunobiology 2020, 225, 151873. [Google Scholar] [CrossRef]
- Son, H.J.; Kim, N.; Song, C.H.; Lee, S.M.; Lee, H.N.; Surh, Y.J. 17β-Estradiol reduces inflammation and modulates antioxidant enzymes in colonic epithelial cells. Korean J. Intern. Med. 2020, 35, 310–319. [Google Scholar] [CrossRef]
- Takashi, Y.; Kosako, H.; Sawatsubashi, S.; Kinoshita, Y.; Ito, N.; Tsoumpra, M.K.; Nangaku, M.; Abe, M.; Matsuhisa, M.; Kato, S.; et al. Activation of unliganded FGF receptor by extracellular phosphate potentiates proteolytic protection of FGF23 by its O-glycosylation. Proc. Natl. Acad. Sci. USA 2019, 116, 11418–11427. [Google Scholar] [CrossRef] [Green Version]
- Ghisletti, S.; Meda, C.; Maggi, A.; Vegeto, E. 17beta-estradiol inhibits inflammatory gene expression by controlling NF-kappaB intracellular localization. Mol. Cell Biol. 2005, 25, 2957–2968. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Gan, Y.; Xu, Y.; Wang, L.; Ouyang, B.; Zhang, C.; Luo, L.; Zhao, C.; Zhou, Q. 17beta-estradiol Attenuates TNF-α-Induced Premature Senescence of Nucleus Pulposus Cells through Regulating the ROS/NF-κB Pathway. Int. J. Biol. Sci. 2017, 13, 145–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Liao, E.; Xiang, G.; Dai, R.; Xiao, X.; Luo, X. Effects of 17beta-estradiol on the expression of IL-6, IL-11 and NF-kappaB in human MG-63 osteoblast-like cell line. J. Huazhong Univ. Sci. Technolog. Med. Sci. 2006, 26, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Bär, L.; Hase, P.; Föller, M. PKC regulates the production of fibroblast growth factor 23 (FGF23). PLoS ONE 2019, 14, e0211309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Yan, J.; Umbach, A.T.; Fakhri, H.; Fajol, A.; Schmidt, S.; Salker, M.S.; Chen, H.; Alexander, D.; Spichtig, D.; et al. NFκB-sensitive Orai1 expression in the regulation of FGF23 release. J. Mol. Med. 2016, 94, 557–566. [Google Scholar] [CrossRef]
- Mazière, C.; Salle, V.; Gomila, C.; Mazière, J.C. Oxidized low density lipoprotein enhanced RANKL expression in human osteoblast-like cells. Involvement of ERK, NFkappaB and NFAT. Biochim. Biophys. Acta 2013, 1832, 1756–1764. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.c.; Lu, D.; Bai, J.; Zheng, H.; Ke, Z.y.; Li, X.m.; Luo, S.q. Oxidative stress inhibits osteoblastic differentiation of bone cells by ERK and NF-kappaB. Biochem. Biophys. Res. Commun. 2004, 314, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, B.; Sun, J.; Jiang, Y.; Zhang, H.; Zhang, P.; Fei, B.; Xu, Y. Iron-induced oxidative stress stimulates osteoclast differentiation via NF-κB signaling pathway in mouse model. Metabolism 2018, 83, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Zha, L.; He, L.; Liang, Y.; Qin, H.; Yu, B.; Chang, L.; Xue, L. TNF-α contributes to postmenopausal osteoporosis by synergistically promoting RANKL-induced osteoclast formation. Biomed. Pharmacother. 2018, 102, 369–374. [Google Scholar] [CrossRef]
- Wu, G.; Pan, L.; Sun, J.; Chen, G.; Wang, S. Hydrogen gas protects against ovariectomy-induced osteoporosis by inhibiting NF-κB activation. Menopause 2019, 26, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, P.; Goswami, K.; Nanjaiah, N.D.; Srinivas, D.; Prasad, C. TNF-α mediated MEK-ERK signaling in invasion with putative network involving NF-κB and STAT-6: A new perspective in glioma. Cell Biol. Int. 2019, 43, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Figurek, A.; Rroji, M.; Spasovski, G. Sclerostin: A new biomarker of CKD-MBD. Int. Urol. Nephrol. 2020, 52, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Sapir-Koren, R.; Livshits, G. Osteocyte control of bone remodeling: Is sclerostin a key molecular coordinator of the balanced bone resorption-formation cycles? Osteoporos. Int. 2014, 25, 2685–2700. [Google Scholar] [CrossRef] [PubMed]
- Ryan, Z.C.; Ketha, H.; McNulty, M.S.; McGee-Lawrence, M.; Craig, T.A.; Grande, J.P.; Westendorf, J.J.; Singh, R.J.; Kumar, R. Sclerostin alters serum vitamin D metabolite and fibroblast growth factor 23 concentrations and the urinary excretion of calcium. Proc. Natl. Acad. Sci. USA 2013, 110, 6199–6204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramli, F.F.; Chin, K.Y. A Review of the Potential Application of Osteocyte-Related Biomarkers, Fibroblast Growth Factor-23, Sclerostin, and Dickkopf-1 in Predicting Osteoporosis and Fractures. Diagnostics 2020, 10, 145. [Google Scholar] [CrossRef] [Green Version]
- Feger, M.; Ewendt, F.; Strotmann, J.; Schäffler, H.; Kempe-Teufel, D.; Glosse, P.; Stangl, G.I.; Föller, M. Glucocorticoids dexamethasone and prednisolone suppress fibroblast growth factor 23 (FGF23). J. Mol. Med. 2021, 99, 699–711. [Google Scholar] [CrossRef]
- Christov, M.; Jüppner, H. Phosphate homeostasis disorders. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 685–706. [Google Scholar] [CrossRef]
- Lindberg, I.; Pang, H.W.; Stains, J.P.; Clark, D.; Yang, A.J.; Bonewald, L.; Li, K.Z. FGF23 is endogenously phosphorylated in bone cells. J. Bone Miner. Res. 2015, 30, 449–454. [Google Scholar] [CrossRef] [Green Version]
- Domazetovic, V.; Bonanomi, A.G.; Stio, M.; Vincenzini, M.T.; Iantomasi, T. Resveratrol decreases TNFα-induced ICAM-1 expression and release by Sirt-1-independent mechanism in intestinal myofibroblasts. Exp. Cell Res. 2019, 382, 111479. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domazetovic, V.; Falsetti, I.; Ciuffi, S.; Iantomasi, T.; Marcucci, G.; Vincenzini, M.T.; Brandi, M.L. Effect of Oxidative Stress-Induced Apoptosis on Active FGF23 Levels in MLO-Y4 Cells: The Protective Role of 17-β-Estradiol. Int. J. Mol. Sci. 2022, 23, 2103. https://doi.org/10.3390/ijms23042103
Domazetovic V, Falsetti I, Ciuffi S, Iantomasi T, Marcucci G, Vincenzini MT, Brandi ML. Effect of Oxidative Stress-Induced Apoptosis on Active FGF23 Levels in MLO-Y4 Cells: The Protective Role of 17-β-Estradiol. International Journal of Molecular Sciences. 2022; 23(4):2103. https://doi.org/10.3390/ijms23042103
Chicago/Turabian StyleDomazetovic, Vladana, Irene Falsetti, Simone Ciuffi, Teresa Iantomasi, Gemma Marcucci, Maria Teresa Vincenzini, and Maria Luisa Brandi. 2022. "Effect of Oxidative Stress-Induced Apoptosis on Active FGF23 Levels in MLO-Y4 Cells: The Protective Role of 17-β-Estradiol" International Journal of Molecular Sciences 23, no. 4: 2103. https://doi.org/10.3390/ijms23042103