
Scaffolding of Mitogen-Activated Protein Kinase Signaling by β-Arrestins


School of Pharmacy, Sungkyunkwan University, 2066 Seoburo, Suwon 16419, Korea
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2022, 23(2), 1000; https://doi.org/10.3390/ijms23021000
Submission received: 28 December 2021
/
Revised: 14 January 2022
/
Accepted: 15 January 2022
/
Published: 17 January 2022
(This article belongs to the Special Issue Arrestins: Structure, Function, and Biology)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:β-arrestins were initially identified to desensitize and internalize G-protein-coupled receptors (GPCRs). Receptor-bound β-arrestins also initiate a second wave of signaling by scaffolding mitogen-activated protein kinase (MAPK) signaling components, MAPK kinase kinase, MAPK kinase, and MAPK. In particular, β-arrestins facilitate ERK1/2 or JNK3 activation by scaffolding signal cascade components such as ERK1/2-MEK1-cRaf or JNK3-MKK4/7-ASK1. Understanding the precise molecular and structural mechanisms of β-arrestin-mediated MAPK scaffolding assembly would deepen our understanding of GPCR-mediated MAPK activation and provide clues for the selective regulation of the MAPK signaling cascade for therapeutic purposes. Over the last decade, numerous research groups have attempted to understand the molecular and structural mechanisms of β-arrestin-mediated MAPK scaffolding assembly. Although not providing the complete mechanism, these efforts suggest potential binding interfaces between β-arrestins and MAPK signaling components and the mechanism for MAPK signal amplification by β-arrestin-mediated scaffolding. This review summarizes recent developments of cellular and molecular works on the scaffolding mechanism of β-arrestin for MAPK signaling cascade.
1. Introduction
G-protein-coupled receptors (GPCRs) constitute the largest membrane receptor family that mediates various physiological and pathological functions [1]. The canonical downstream signaling molecule for GPCRs is the heterotrimeric G-protein, which regulates second messenger (e.g., cAMP and Ca2+) generation [2]. Arrestins were first discovered to desensitize GPCR-mediated G-protein signaling [3]. A total of four arrestins (arrestin-1–4) have been identified in humans; arrestin-1 and -4 (visual and cone arrestins, respectively) are expressed exclusively in the visual system, whereas arrestin-2 and -3 (also known as β-arrestin 1 and 2, respectively) are ubiquitously expressed [3]. GPCR kinases (GRKs) phosphorylate agonist-activated GPCRs at their C-tail or intracellular loops, which provide binding sites for arrestins (Figure 1A). The arrestin binding sites on receptors are similar to the G-protein binding sites (Figure 1A,B); therefore, arrestin binding precludes receptor coupling to G-proteins, resulting in the desensitization of G-protein signaling [3,4]. However, recently, it was found that the GPCR–G-protein–β-arrestin megacomplex can exist at the endosome where arrestin does not bind to the receptor cytosolic core but interacts only at the phosphorylated C-tail [5,6].
Arrestin binding also induces clathrin-mediated endocytosis of receptors [3,4]. In this process, arrestin acts as a scaffolding protein, linking the receptor with internalization machinery molecules such as clathrin and AP-2 [4,7,8,9,10]. Likewise, arrestins have been reported to interact with numerous other proteins including signaling proteins, adapter proteins, and transcriptional regulators [11,12,13]. Interaction with arrestins may alter the cellular localization or activation status of the binding partners [11,12]. The interaction occurs either with the basal or GPCR-induced active state of arrestins [4,10,14]. Among these binding partners, the mitogen-activated protein kinase (MAPK) family is a well-characterized binding partner of arrestins.
MAPKs are the most important signaling protein families that respond to diverse external stresses such as heat, mitogens, and cytokines [15]. MAPKs regulate various cellular functions such as gene expression, cell survival, differentiation, proliferation, and apoptosis [15]. MAPK activation occurs through cascades of phosphorylation by three components: MAPK kinase kinase (MAP3K), MAPK kinase (MAP2K), and MAPK. MAP3K is an upstream kinase for MAP2K; MAP2K is an upstream kinase for MAPK; and MAPK phosphorylates the downstream effector proteins to regulate their active status [15,16].
In mammals, there are four distinct MAPK groups: ERKs (ERK1 and 2), JNKs (JNK1, 2, and 3), p38 (p38α–δ), and ERK5, each of which has its own upstream MAP2Ks and MAP3Ks [16,17]. How the MAPK signaling components accomplish signaling fidelity has been a long-standing mystery. Many studies have suggested that the scaffolding proteins co-localize MAPK signaling components which either facilitate the signaling cascade or modulate the negative feedback of a specific MAPK signaling pathway [18,19]. MAPK scaffolding proteins also regulate the subcellular localization of MAPK signaling components. Thus, MAPK scaffolding proteins organize specific MAPK signaling components to link the input to proper biological outcomes [18,19,20,21,22]. Therefore, MAPK scaffolding proteins have been considered as potential therapeutic targets [20,23,24,25].
Several MAPK scaffolding proteins have been identified in mammals including JIP, KSR, MP1, IQGAP1, paxillin, MORG1, JSAP1, DUSP22, and arrestins [21,22,26,27]. Depending on the scaffolding proteins, their functional consequences and molecular mechanisms are different [18]. For example, KSR recruits Raf, MEK1, and ERK1/2 at the plasma membrane promoting the ERK1/2 activation while PEB (phosphatidyl-ethanolamine-binding protein 1/RKIP) binds to Raf and MEK1 preventing their physical interaction and MEK1 activation by Raf [22]. Therefore, to precisely understand and regulate a specific MAPK signaling pathway, it is important to study the detailed scaffolding mechanism of each MAPK scaffolding protein. In this review, we discuss the functional consequences of arrestin scaffolding of MAPK signaling components and the structural mechanism of scaffolding.
2. Scaffolding ERK1/2 Signaling by β-Arrestin
Many previous studies have reported that GPCR activation induces ERK1/2 phosphorylation, which is either G-protein-dependent or -independent [28,29,30]. Gi or Gq activation has been shown to activate the ERK1/2 signaling cascades with unknown precise mechanisms. G-protein-mediated ERK1/2 activation occurs quickly after GPCR stimulation with a transient nature [28,29,30]. The G-protein-activated ERK1/2 tends to move into the nucleus where it phosphorylates downstream nuclear effector proteins [28].
G-protein-independent ERK1/2 activation after GPCR stimulation was first suggested by Lefkowitz group in 1999 [31]; β-arrestin was shown to be involved in c-Src recruitment to the receptor to activate ERK1/2. In this report, the authors claimed that β-arrestin initiates a second wave of receptor signal transduction. Soon after, the same research group published a paper showing that β-arrestin acts as a “scaffold” for cRaf, MEK1, and ERK1/2 to facilitate ERK1/2 activation [32]. ERK1/2 activation mediated by β-arrestin is distinct from that mediated by G-protein. β-arrestin-mediated ERK1/2 activation tends to occur slowly after GPCR stimulation in a sustained manner; moreover, the activated ERK1/2 either stays in the cytosol or moves to the nucleus depending on the stimulated receptor [28,29,30,33,34,35,36]. All these studies agree that β-arrestin activation by GPCRs is essential for ERK1/2 activation [29,32,33,37,38,39] and that the molecular mechanism of β-arrestin-mediated ERK1/2 activation is via β-arrestin’s scaffolding action of cRaf, MEK1, and ERK1/2 [32,35].
3. Structural Mechanism of Scaffolding ERK1/2 Signaling by β-Arrestin
The molecular mechanism of β-arrestin-mediated scaffolding of the ERK1/2 signaling components is a long-standing mystery. Several studies have suggested the structural mechanism of scaffolding using biochemical and biophysical tools such as mutagenesis, peptide array, molecular simulation, and hydrogen/deuterium exchange mass spectrometry (HDX-MS) [40,41,42,43,44]. Although not completely understood, these studies provide insight into how β-arrestins scaffold ERK1/2 signaling components.
Arrestins are composed of N- and C-domains (Figure 2A), which are connected by two linkers with the second linker bringing the C-terminal tail, including the β-strand XX (Figure 2A, orange), to the N-domain. The agonist-activated phosphorylated receptor binding induces conformational changes in arrestin (Figure 2B). The phosphorylated C-tail of the receptor interacts at the N-domain of arrestin, which displaces the β-strand XX; the loops between the two domains (finger loop, middle loop, C-loop, and lariat loop) undergo conformational changes; the activation also induces disruption of the ionic interaction between the two domains (i.e., polar core) and interdomain rotation [45,46,47,48,49,50]. Thus, the removal of the C-terminal tail or a disruption of the polar core could mimic the active conformation of arrestins [51,52]. As receptor-induced activation of β-arrestin is suggested to be required for ERK1/2 activation, the scaffolding mechanism of β-arrestin for ERK1/2 activation has been studied using these active state-mimicking constructs.
The direct interaction between β-arrestin and cRaf was suggested by an early study that proposed β-arrestin as a scaffolding protein for ERK1/2 signaling components [32]. A mutation and co-immunoprecipitation study suggested the cRaf binding site on β-arrestin, in which the β-arrestin 1 R307A mutant failed to interact with cRaf [42]. Later, a molecular simulation study suggested that a region near R307 (i.e., back loop) of β-arrestin 1 interacts with a region near K84 of cRaf [43]. cRaf is composed of a Ras-binding domain (RBD), C1 domain (CRD), hinge region, and kinase domain (Figure 2C, left) [53,54], and K84 is located within the RBD. A recent hydrogen/deuterium exchange mass spectrometry (HDX-MS) study with purified β-arrestin 1 and cRaf RBD confirmed that the back loop of β-arrestin 1 (i.e., near R307) interacts with cRaf RBD (Figure 2D) [44]. It is interesting to note that the kinase activity of the Raf family is auto-inhibited by the N-terminal domains, including RBD and CRD, and the binding of Ras to RBD releases the N-terminal domain, leading to exposure of the kinase domain (Figure 2C, right) [53]. The observation that β-arrestin 1 interacts with the RBD of cRaf prompted the hypothesis that β-arrestin 1 binding would release the N-terminal domains from the kinase domain, resulting in kinase domain activation (Figure 2D). This hypothesis was confirmed by a recent study in which the kinase activity of cRaf increased upon β-arrestin 1 binding [55].
Whether β-arrestin 1 needs to be activated by a phosphorylated receptor for cRaf binding remains controversial. Our results, with HDX-MS and in cell BRET assays, suggest that activated receptors are not required for β-arrestin 1 and cRaf interaction, while a recent study using a GST pull-down assay showed an increased β-arrestin 1 and cRaf interaction when V2Rpp, a phosphorylated C-terminal peptide of the V2 vasopressin receptor, was co-incubated [44,55]. Therefore, further studies are needed to fully understand whether receptor-mediated β-arrestin 1 activation is required for cRaf binding. Moreover, the involvement of β-arrestin’s scaffolding action for A-Raf or B-Raf needs to be investigated.
The binding interface between β-arrestin and MEK1 has been suggested in a few studies, and these studies have shown different results [40,41,43,44]. Meng et al. suggested that residues 46–70 in the N-terminal region of MEK1 and D26 and D29 in the N-domain of β-arrestin 1 are critical for the interaction [40], and a molecular simulation study suggested that D29 and E35 of β-arrestin 1 interact with a region near E272 of MEK1 [43]. However, our recent study using HDX-MS and fluorescence quenching analysis suggested that the N-lobe of MEK1 interacts with the interdomain loop I of β-arrestin 1 (Figure 2D) [56]. The discrepancies between different studies may be due to the different experimental techniques that were used; Meng et al. used peptide arrays, while our HDX-MS study used whole proteins to define the binding interfaces. Nevertheless, the requirement of GPCR-mediated β-arrestin activation for MEK1 binding is consistent between the previous studies (Figure 2D) [43,44,57].
The interaction between β-arrestin and ERK1/2 is also increased by receptor-mediated β-arrestin activation [43,44,57]. Our recent HDX-MS and fluorescence quenching analysis study suggested that activated β-arrestin interacts with ERK1/2 between the gate loop of β-arrestin 1 and the N-lobe of ERK1/2 and between the interdomain loop II of β-arrestin 1 and the C-lobe of ERK1/2 (Figure 2D) [44]. The interaction at the interdomain loop II of β-arrestin was also suggested by Cassier et al. who observed that phosphorylation of threonine 383 on the interdomain loop II of β-arrestin 2 induced a conformational change in this region, which facilitated phosphorylated receptor binding and subsequent ERK interaction [37]. The interaction at the gate loop of β-arrestin was also proposed in previous studies; Xu et al. suggested that ERK2 interacts at the lariat loop through the gate loop of β-arrestin 2 [58], and Bourquard et al. predicted that the gate loop of β-arrestin 1 interacts with the N-lobe of ERK1 [43].
Whether the interaction of the three ERK1/2 signaling components with β-arrestin is cooperative has been comprehensively studied. On the one hand, it was previously reported that the cRaf interaction with β-arrestin 2 is not affected by simultaneous expression of either MEK1 or ERK2 and that the simultaneous expression of MEK1 and ERK2 does not affect the interaction of either kinase with β-arrestin 2 [32]. On the other hand, in the same study, the binding of MEK1 or ERK2 to β-arrestin 2 was enhanced by the co-expression of cRaf [32], and the authors suggested that the binding of cRaf to β-arrestin 2 facilitates the assembly of other ERK1/2 signaling components on β-arrestin. Further studies are needed to understand how cRaf binding facilitates MEK1 or ERK2 interactions to β-arrestin.
In summary, these studies propose that the assembly of the ERK1/2 signaling components on β-arrestin locates each upstream kinase to phosphorylate the downstream kinase (Figure 2D). Our recent study also suggested that cRaf and MEK1 bind to β-arrestin when the two kinases are in an active state. However, ERK1/2 may dissociate from β-arrestin when it is activated so that phosphorylated active ERK1/2 can leave the β-arrestin-mediated scaffolding assembly to phosphorylate downstream effector proteins and free up the space for the recruitment of inactive ERK1/2 into the scaffolding assembly [44].
4. Scaffolding JNK3 Signaling by β-Arrestin
The first clue for the scaffolding role of β-arrestin in the JNK signaling was also suggested by the Lefkowitz group in 2000 [59]; one year after the β-arrestin-mediated scaffolding of ERK1/2 signaling was suggested by the same research group [31]. In this study, the interaction between β-arrestin 2 and JNK3 was identified by a yeast-2-hybrid screening study, and the result was confirmed by co-immunoprecipitation. In the same study, β-arrestin 2 was co-immunoprecipitated with ASK1 and MKK4, the MAP3K and MAP2K of JNK3, respectively. Later, the direct interactions between β-arrestin 2 and JNK3 [41,56,60], β-arrestin 2 and MKK4 or MKK7 [61,62,63], and β-arrestin 2 and ASK1 [36,51] were confirmed by various experimental systems, suggesting that the assembly of three JNK3 signaling components on β-arrestin 2 facilitates the kinase cascade. Interestingly, most studies have shown that β-arrestin 2, but not β-arrestin 1, is the major subtype of the JNK3 signaling component scaffolding and activation. The β-arrestin-mediated scaffolding and activation of JNK1 or JNK2 is controversial; in the early study, β-arrestin failed to facilitate JNK1 activation [59] while in another study β-arrestin 2 facilitated the activation of JNK1 or JNK2 via scaffolding them [64].
Similar to the ERK1/2 scaffolding, the interaction between JNK3 and β-arrestin 2 results in cytosolic localization of JNK3 [59,60]. Unlike ERK1/2, however, whether receptor-mediated β-arrestin activation is required for the interaction between β-arrestin and JNK3 remains controversial. Early studies have suggested that receptor-mediated β-arrestin activation is not required [41,59,65] while more recent studies suggest that β-arrestin activation either by a receptor or other molecule, such as IP6, facilitates β-arrestin 2 and JNK3 interaction [63,66,67].
5. Structural Mechanism of Scaffolding JNK3 Signaling by Arrestins
Several studies have suggested binding interfaces between β-arrestin and JNK3 [41,56,59,63,67,68,69,70]. Earlier studies with mutation or domain swapping proposed that the C-terminal half of β-arrestin 2 is the binding site for JNK3 [59,68,71]. However, recent studies suggest that the first 25 residues of β-arrestin 2 are the key binding site for JNK3 (Figure 3A), although it is possible that other parts of β-arrestin 2 are also involved in JNK3 binding [56,63,67,70]. In the basal state, this region is blocked by the β-strand XX (Figure 2A and Figure 3A); therefore, the β-strand XX should be released for JNK3 interaction. This is consistent with previous reports in which a receptor or IP6-mediated β-arrestin 2 activation facilitates JNK3 interaction [63,66,67].
JNK3 is activated by phosphorylation at two residues, Thr221 and Tyr223, which are phosphorylated by MMK7 and MKK4, respectively. The binding interface between β-arrestin 2 and MKK4 or NKK7 has been less extensively studied than that between β-arrestin 2 and JNK3. Nevertheless, a few studies have suggested that MKK4 and MKK7 compete for the same binding sites on β-arrestin 2 [56,62]. A recent study by the Gurevich group observed that the first 25 residues of β-arrestin 2, the same region where JNK3 interacts, is the binding site for MKK4 and NKK7 (Figure 3A) [56]. In this study, the interaction between β-arrestin 2 and MKK4/7 increased when MKK4/7 was occupied with ATP, and the interaction between β-arrestin 2 and MKK7 was stronger than that between β-arrestin 2 and MKK4. Interestingly, the phosphorylation-induced activation of MKK4 and MKK7 resulted in a change in the binding site within the first 25 residues of β-arrestin 2. The same study also showed that the binding affinity between JNK3 and β-arrestin 2 was reduced when JNK3 was activated so that the active JNK3 was released from β-arrestin 2. Taken together, the authors suggested a “conveyor belt” model for the JNK3 signal amplification by β-arrestin 2 (Figure 3B); ATP-bound MKK4 and MKK7 compete for the similar binding site on β-arrestin 2 to phosphorylate JNK3; in turn, the dual-phosphorylated JNK3 leaves the β-arrestin-mediated scaffolding assembly so that unphosphorylated inactive JNK3 can bind to the assembly.
The interaction mechanism between β-arrestin 2 and ASK1 has been reported in a few studies [41,56,69]. These studies found that the C-terminal half, including the kinase domain of ASK1, is the binding site for the first 25 residues of β-arrestin 2 (Figure 3A). As all four components of JNK3 activation (i.e., ASK1, MKK4, MKK7, and JNK3) interact at a similar region on β-arrestin 2, more studies are needed to precisely identify the binding interfaces.
6. Phosphorylation Barcode and MAPK Scaffolding by β-Arrestin
Different receptors have different phosphorylation patterns, and, even for the same receptor, they can have distinct phosphorylation patterns depending on the kinases. β-arrestins have several of phosphate-binding sites (Figure 4A) [72,73], and it has been suggested that binding of differently phosphorylated receptors induces different active conformations of β-arrestins, resulting in distinct downstream signaling pathways [46,47,72,74,75,76]. This sophisticated regulation of β-arrestin conformation and function by the phosphorylation pattern of the receptor is named the “barcode theory” (Figure 4B) [74].
Long before the barcode theory was suggested, it was reported that β-arrestin-mediated ERK1/2 activation is dependent on the GRK isoforms that phosphorylate the receptor [29,77]. GRK2 and GRK3 phosphorylate the V2 vasopressin receptor and β2-adrenergic receptor to be desensitized by β-arrestin recruitment. However, GRK5 and GRK6 phosphorylate receptors to induce β-arrestin-mediated ERK1/2 activation. Moreover, different ligands induced different receptor conformations and different phosphorylation patterns, resulting in different β-arrestin activation [78,79]. The phosphorylation sites on the receptors by different kinases have been identified [74], and more detailed studies with mutation studies, NMR, or X-ray crystallography have deepened our understanding of the correlation between the phosphorylation pattern of the receptor, β-arrestin conformation, and ERK1/2 activation [47,76,80,81]. For example, carefully designed phospho-peptides or phosphorylation site-deleting mutants showed distinct ERK1/2 activation properties [76,81]. However, a specific phosphorylation pattern for JNK3 activation has not yet been studied. Moreover, as there are no high-resolution structures of β-arrestin-MAPK, MAP2K, or MAP3K mega-complexes, the precise structural mechanism of biased downstream signaling by β-arrestin needs to be elucidated.
7. Summary
In this review, we discussed the molecular and structural mechanisms of β-arrestin-mediated scaffolding of ERK1/2 and JNK3 signaling components. In both cases, β-arrestin must be activated to complete the scaffolding assembly. Furthermore, it has been suggested that, upon activation, both ERK1/2 and JNK3, the most downstream kinase of MAPK signaling cascade, leave the β-arrestin-mediated scaffolding assembly so that inactive MAPK can be recruited, which results in signal amplification.
However, the binding interfaces within the β-arrestin-mediated scaffolding assemblies seem to differ; cRaf, MEK1, and ERK1/2 have distinct binding interfaces to interact with β-arrestin, while ASK1, MKK4/7, and JNK3 share a similar binding interface to interact with β-arrestin. The binding interfaces with β-arrestin are different between cRaf and ASK1, MEK1 and MKK4/7, and ERK1/2 and JNK3. Thus, defining the precise interacting residues in the different β-arrestin-mediated scaffolding assemblies could pave the way for the selective regulation of arrestin-mediated ERK or JNK signaling.
Author Contributions
Conceptualization, K.Y.C.; Writing—Original Draft Preparation, K.Y.C.; Writing—Review & Editing, K.K., Y.H. and L.D.; Supervision, K.Y.C.; Funding Acquisition, K.Y.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Research Foundation of Korea and funded by the Ministry of Science and ICT of Korea (NRF-2021R1A2C3003518).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milligan, G.; Kostenis, E. Heterotrimeric G-proteins: A short history. Br. J. Pharmacol. 2006, 147 (Suppl. S1), S46–S55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohse, M.J.; Hoffmann, C. Arrestin interactions with G protein-coupled receptors. Handb. Exp. Pharmacol. 2014, 219, 15–56. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Rajagopal, S. The beta-Arrestins: Multifunctional Regulators of G Protein-coupled Receptors. J. Biol. Chem. 2016, 291, 8969–8977. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, A.R.B.; Plouffe, B.; Cahill, T.J., 3rd; Shukla, A.K.; Tarrasch, J.T.; Dosey, A.M.; Kahsai, A.W.; Strachan, R.T.; Pani, B.; Mahoney, J.P.; et al. GPCR-G Protein-beta-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell 2016, 166, 907–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, A.H.; Thomsen, A.R.B.; Cahill, T.J., 3rd; Huang, R.; Huang, L.Y.; Marcink, T.; Clarke, O.B.; Heissel, S.; Masoudi, A.; Ben-Hail, D.; et al. Structure of an endosomal signaling GPCR-G protein-beta-arrestin megacomplex. Nat. Struct. Mol. Biol. 2019, 26, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Benovic, J.L.; Kuhn, H.; Weyand, I.; Codina, J.; Caron, M.G.; Lefkowitz, R.J. Functional desensitization of the isolated beta-adrenergic receptor by the beta-adrenergic receptor kinase: Potential role of an analog of the retinal protein arrestin (48-kDa protein). Proc. Natl. Acad. Sci. USA 1987, 84, 8879–8882. [Google Scholar] [CrossRef] [Green Version]
- Lohse, M.J.; Benovic, J.L.; Codina, J.; Caron, M.G.; Lefkowitz, R.J. beta-Arrestin: A protein that regulates beta-adrenergic receptor function. Science 1990, 248, 1547–1550. [Google Scholar] [CrossRef]
- Vishnivetskiy, S.A.; Sullivan, L.S.; Bowne, S.J.; Daiger, S.P.; Gurevich, E.V.; Gurevich, V.V. Molecular Defects of the Disease-Causing Human Arrestin-1 C147F Mutant. Investig. Ophthalmol. Vis. Sci. 2018, 59, 13–20. [Google Scholar] [CrossRef] [Green Version]
- DeFea, K.A. Beta-arrestins as regulators of signal termination and transduction: How do they determine what to scaffold? Cell Signal. 2011, 23, 621–629. [Google Scholar] [CrossRef]
- Xiao, K.; McClatchy, D.B.; Shukla, A.K.; Zhao, Y.; Chen, M.; Shenoy, S.K.; Yates, J.R., 3rd; Lefkowitz, R.J. Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proc. Natl. Acad. Sci. USA 2007, 104, 12011–12016. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, V.V.; Gurevich, E.V.; Cleghorn, W.M. Arrestins as multi-functional signaling adaptors. Handb. Exp. Pharmacol. 2008, 15–37. [Google Scholar] [CrossRef]
- Crepieux, P.; Poupon, A.; Langonne-Gallay, N.; Reiter, E.; Delgado, J.; Schaefer, M.H.; Bourquard, T.; Serrano, L.; Kiel, C. A Comprehensive View of the beta-Arrestinome. Front. Endocrinol. 2017, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, E.V.; Gurevich, V.V. Arrestins: Ubiquitous regulators of cellular signaling pathways. Genome Biol. 2006, 7, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R., Jr. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.K. MAP kinase pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef]
- Peti, W.; Page, R. Molecular basis of MAP kinase regulation. Protein Sci. 2013, 22, 1698–1710. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.D.; Sacks, D.B. Protein scaffolds in MAP kinase signalling. Cell. Signal. 2009, 21, 462–469. [Google Scholar] [CrossRef] [Green Version]
- Kolch, W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 2005, 6, 827–837. [Google Scholar] [CrossRef]
- Liang, Y.; Sheikh, F. Scaffold Proteins Regulating Extracellular Regulated Kinase Function in Cardiac Hypertrophy and Disease. Front. Pharmacol. 2016, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Ju, A.; Cho, Y.C.; Kim, B.R.; Park, S.G.; Kim, J.H.; Kim, K.; Lee, J.; Park, B.C.; Cho, S. Scaffold Role of DUSP22 in ASK1-MKK7-JNK Signaling Pathway. PLoS ONE 2016, 11, e0164259. [Google Scholar] [CrossRef]
- Witzel, F.; Maddison, L.; Bluthgen, N. How scaffolds shape MAPK signaling: What we know and opportunities for systems approaches. Front. Physiol. 2012, 3, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.H.; Neela, P.H.; Kent, M.S.; Zehnder, A.M. IQGAP1 is an oncogenic target in canine melanoma. PLoS ONE 2017, 12, e0176370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Kablaoui, N.; Little, J.; Timofeevski, S.; Tschantz, W.R.; Chen, P.; Feng, J.; Charlton, M.; Stanton, R.; Bauer, P. Identification of small-molecule inhibitors of the JIP-JNK interaction. Biochem. J. 2009, 420, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Neilsen, B.K.; Frodyma, D.E.; Lewis, R.E.; Fisher, K.W. KSR as a therapeutic target for Ras-dependent cancers. Expert. Opin. Ther. Targets 2017, 21, 499–509. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Kashef, K.; Lee, C.M.; Xu, H.; Reddy, E.P. Scaffold proteins of MAP-kinase modules. Oncogene 2007, 26, 3185–3202. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, K. Scaffold proteins in mammalian MAP kinase cascades. J. Biochem. 2004, 135, 657–661. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.; Shenoy, S.K.; Wei, H.; Lefkowitz, R.J. Differential kinetic and spatial patterns of beta-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J. Biol. Chem. 2004, 279, 35518–35525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy, S.K.; Drake, M.T.; Nelson, C.D.; Houtz, D.A.; Xiao, K.; Madabushi, S.; Reiter, E.; Premont, R.T.; Lichtarge, O.; Lefkowitz, R.J. Beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J. Biol. Chem. 2006, 281, 1261–1273. [Google Scholar] [CrossRef] [Green Version]
- Gesty-Palmer, D.; Chen, M.; Reiter, E.; Ahn, S.; Nelson, C.D.; Wang, S.; Eckhardt, A.E.; Cowan, C.L.; Spurney, R.F.; Luttrell, L.M.; et al. Distinct beta-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J. Biol. Chem. 2006, 281, 10856–10864. [Google Scholar] [CrossRef] [Green Version]
- Luttrell, L.M.; Ferguson, S.S.; Daaka, Y.; Miller, W.E.; Maudsley, S.; Della Rocca, G.J.; Lin, F.; Kawakatsu, H.; Owada, K.; Luttrell, D.K.; et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science 1999, 283, 655–661. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Roudabush, F.L.; Choy, E.W.; Miller, W.E.; Field, M.E.; Pierce, K.L.; Lefkowitz, R.J. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc. Natl. Acad. Sci. USA 2001, 98, 2449–2454. [Google Scholar] [CrossRef] [Green Version]
- Tohgo, A.; Pierce, K.L.; Choy, E.W.; Lefkowitz, R.J.; Luttrell, L.M. Beta-Arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J. Biol. Chem. 2002, 277, 9429–9436. [Google Scholar] [CrossRef] [Green Version]
- DeFea, K.A.; Zalevsky, J.; Thoma, M.S.; Dery, O.; Mullins, R.D.; Bunnett, N.W. Beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol. 2000, 148, 1267–1281. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lu, R.; Zhao, J.; Limbird, L.E. Arrestin serves as a molecular switch, linking endogenous alpha2-adrenergic receptor to SRC-dependent, but not SRC-independent, ERK activation. J. Biol. Chem. 2006, 281, 25948–25955. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Loh, H.H.; Law, P.Y. Beta-arrestin-dependent mu-opioid receptor-activated extracellular signal-regulated kinases (ERKs) Translocate to Nucleus in Contrast to G protein-dependent ERK activation. Mol. Pharmacol. 2008, 73, 178–190. [Google Scholar] [CrossRef] [Green Version]
- Cassier, E.; Gallay, N.; Bourquard, T.; Claeysen, S.; Bockaert, J.; Crepieux, P.; Poupon, A.; Reiter, E.; Marin, P.; Vandermoere, F. Phosphorylation of beta-arrestin2 at Thr(383) by MEK underlies beta-arrestin-dependent activation of Erk1/2 by GPCRs. eLife 2017, 6, e23777. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.R.; Kushmerick, C.; Seo, J.B.; Koh, D.S.; Hille, B. Muscarinic receptor regulates extracellular signal regulated kinase by two modes of arrestin binding. Proc. Natl. Acad. Sci. USA 2017, 114, E5579–E5588. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, H.; Narita, Y.; Nishida, M.; Kurose, H. Beta-arrestin2 enhances beta2-adrenergic receptor-mediated nuclear translocation of ERK. Cell. Signal. 2005, 17, 1248–1253. [Google Scholar] [CrossRef]
- Meng, D.; Lynch, M.J.; Huston, E.; Beyermann, M.; Eichhorst, J.; Adams, D.R.; Klussmann, E.; Houslay, M.D.; Baillie, G.S. MEK1 binds directly to betaarrestin1, influencing both its phosphorylation by ERK and the timing of its isoprenaline-stimulated internalization. J. Biol. Chem. 2009, 284, 11425–11435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Coffa, S.; Fu, H.; Gurevich, V.V. How does arrestin assemble MAPKs into a signaling complex? J. Biol. Chem. 2009, 284, 685–695. [Google Scholar] [CrossRef] [Green Version]
- Coffa, S.; Breitman, M.; Spiller, B.W.; Gurevich, V.V. A single mutation in arrestin-2 prevents ERK1/2 activation by reducing c-Raf1 binding. Biochemistry 2011, 50, 6951–6958. [Google Scholar] [CrossRef] [Green Version]
- Bourquard, T.; Landomiel, F.; Reiter, E.; Crepieux, P.; Ritchie, D.W.; Aze, J.; Poupon, A. Unraveling the molecular architecture of a G protein-coupled receptor/beta-arrestin/Erk module complex. Sci. Rep. 2015, 5, 10760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, C.; Park, J.Y.; Yun, M.W.; He, Q.T.; Yang, F.; Kim, K.; Ham, D.; Li, R.R.; Iverson, T.M.; Gurevich, V.V.; et al. Scaffolding mechanism of arrestin-2 in the cRaf/MEK1/ERK signaling cascade. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Masureel, M.; Qu, Q.; Janetzko, J.; Inoue, A.; Kato, H.E.; Robertson, M.J.; Nguyen, K.C.; Glenn, J.S.; Skiniotis, G.; et al. Structure of the neurotensin receptor 1 in complex with beta-arrestin 1. Nature 2020, 579, 303–308. [Google Scholar] [CrossRef]
- Zhou, X.E.; He, Y.; de Waal, P.W.; Gao, X.; Kang, Y.; Van Eps, N.; Yin, Y.; Pal, K.; Goswami, D.; White, T.A.; et al. Identification of Phosphorylation Codes for Arrestin Recruitment by G Protein-Coupled Receptors. Cell 2017, 170, 457–469.e413. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Yu, X.; Liu, C.; Qu, C.X.; Gong, Z.; Liu, H.D.; Li, F.H.; Wang, H.M.; He, D.F.; Yi, F.; et al. Phospho-selective mechanisms of arrestin conformations and functions revealed by unnatural amino acid incorporation and (19)F-NMR. Nat. Commun. 2015, 6, 8202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staus, D.P.; Hu, H.; Robertson, M.J.; Kleinhenz, A.L.W.; Wingler, L.M.; Capel, W.D.; Latorraca, N.R.; Lefkowitz, R.J.; Skiniotis, G. Structure of the M2 muscarinic receptor-beta-arrestin complex in a lipid nanodisc. Nature 2020, 579, 297–302. [Google Scholar] [CrossRef]
- Scheerer, P.; Sommer, M.E. Structural mechanism of arrestin activation. Curr. Opin. Struct. Biol. 2017, 45, 160–169. [Google Scholar] [CrossRef]
- Shukla, A.K.; Manglik, A.; Kruse, A.C.; Xiao, K.; Reis, R.I.; Tseng, W.C.; Staus, D.P.; Hilger, D.; Uysal, S.; Huang, L.Y.; et al. Structure of active beta-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature 2013, 497, 137–141. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.K.; Yun, Y.; Kim, H.R.; Seo, M.D.; Chung, K.Y. Different conformational dynamics of various active states of beta-arrestin1 analyzed by hydrogen/deuterium exchange mass spectrometry. J. Struct. Biol. 2015, 190, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Hofmann, K.P.; Ernst, O.P.; Scheerer, P.; Choe, H.W.; Sommer, M.E. Crystal structure of pre-activated arrestin p44. Nature 2013, 497, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Rawson, S.; Li, K.; Kim, B.W.; Ficarro, S.B.; Pino, G.G.; Sharif, H.; Marto, J.A.; Jeon, H.; Eck, M.J. Architecture of autoinhibited and active BRAF-MEK1-14-3-3 complexes. Nature 2019, 575, 545–550. [Google Scholar] [CrossRef]
- Matallanas, D.; Birtwistle, M.; Romano, D.; Zebisch, A.; Rauch, J.; von Kriegsheim, A.; Kolch, W. Raf family kinases: Old dogs have learned new tricks. Genes Cancer 2011, 2, 232–260. [Google Scholar] [CrossRef] [Green Version]
- Zang, Y.; Kahsai, A.W.; Pakharukova, N.; Huang, L.Y.; Lefkowitz, R.J. The GPCR-beta-arrestin complex allosterically activates C-Raf by binding its amino terminus. J. Biol. Chem. 2021, 297, 101369. [Google Scholar] [CrossRef]
- Perry, N.A.; Kaoud, T.S.; Ortega, O.O.; Kaya, A.I.; Marcus, D.J.; Pleinis, J.M.; Berndt, S.; Chen, Q.; Zhan, X.; Dalby, K.N.; et al. Arrestin-3 scaffolding of the JNK3 cascade suggests a mechanism for signal amplification. Proc. Natl. Acad. Sci. USA 2019, 116, 810–815. [Google Scholar] [CrossRef] [Green Version]
- Coffa, S.; Breitman, M.; Hanson, S.M.; Callaway, K.; Kook, S.; Dalby, K.N.; Gurevich, V.V. The effect of arrestin conformation on the recruitment of c-Raf1, MEK1, and ERK1/2 activation. PLoS ONE 2011, 6, e28723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, T.R.; Baillie, G.S.; Bhari, N.; Houslay, T.M.; Pitt, A.M.; Adams, D.R.; Kolch, W.; Houslay, M.D.; Milligan, G. Mutations of beta-arrestin 2 that limit self-association also interfere with interactions with the beta2-adrenoceptor and the ERK1/2 MAPKs: Implications for beta2-adrenoceptor signalling via the ERK1/2 MAPKs. Biochem. J. 2008, 413, 51–60. [Google Scholar] [CrossRef]
- McDonald, P.H.; Chow, C.W.; Miller, W.E.; Laporte, S.A.; Field, M.E.; Lin, F.T.; Davis, R.J.; Lefkowitz, R.J. Beta-arrestin 2: A receptor-regulated MAPK scaffold for the activation of JNK3. Science 2000, 290, 1574–1577. [Google Scholar] [CrossRef]
- Song, X.; Raman, D.; Gurevich, E.V.; Vishnivetskiy, S.A.; Gurevich, V.V. Visual and both non-visual arrestins in their “inactive” conformation bind JNK3 and Mdm2 and relocalize them from the nucleus to the cytoplasm. J. Biol. Chem. 2006, 281, 21491–21499. [Google Scholar] [CrossRef] [Green Version]
- Zhan, X.; Kaoud, T.S.; Dalby, K.N.; Gurevich, V.V. Nonvisual arrestins function as simple scaffolds assembling the MKK4-JNK3alpha2 signaling complex. Biochemistry 2011, 50, 10520–10529. [Google Scholar] [CrossRef] [Green Version]
- Zhan, X.; Kaoud, T.S.; Kook, S.; Dalby, K.N.; Gurevich, V.V. JNK3 enzyme binding to arrestin-3 differentially affects the recruitment of upstream mitogen-activated protein (MAP) kinase kinases. J. Biol. Chem. 2013, 288, 28535–28547. [Google Scholar] [CrossRef] [Green Version]
- Zhan, X.; Perez, A.; Gimenez, L.E.; Vishnivetskiy, S.A.; Gurevich, V.V. Arrestin-3 binds the MAP kinase JNK3alpha2 via multiple sites on both domains. Cell Signal. 2014, 26, 766–776. [Google Scholar] [CrossRef] [Green Version]
- Kook, S.; Zhan, X.; Kaoud, T.S.; Dalby, K.N.; Gurevich, V.V.; Gurevich, E.V. Arrestin-3 binds c-Jun N-terminal kinase 1 (JNK1) and JNK2 and facilitates the activation of these ubiquitous JNK isoforms in cells via scaffolding. J. Biol. Chem. 2013, 288, 37332–37342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitman, M.; Kook, S.; Gimenez, L.E.; Lizama, B.N.; Palazzo, M.C.; Gurevich, E.V.; Gurevich, V.V. Silent scaffolds: Inhibition OF c-Jun N-terminal kinase 3 activity in cell by dominant-negative arrestin-3 mutant. J. Biol. Chem. 2012, 287, 19653–19664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Perry, N.A.; Vishnivetskiy, S.A.; Berndt, S.; Gilbert, N.C.; Zhuo, Y.; Singh, P.K.; Tholen, J.; Ohi, M.D.; Gurevich, E.V.; et al. Structural basis of arrestin-3 activation and signaling. Nat. Commun. 2017, 8, 1427. [Google Scholar] [CrossRef] [Green Version]
- Park, J.Y.; Qu, C.X.; Li, R.R.; Yang, F.; Yu, X.; Tian, Z.M.; Shen, Y.M.; Cai, B.Y.; Yun, Y.; Sun, J.P.; et al. Structural Mechanism of the Arrestin-3/JNK3 Interaction. Structure 2019, 27, 1162–1170.e1163. [Google Scholar] [CrossRef]
- Guo, C.; Whitmarsh, A.J. The beta-arrestin-2 scaffold protein promotes c-Jun N-terminal kinase-3 activation by binding to its nonconserved N terminus. J. Biol. Chem. 2008, 283, 15903–15911. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; MacLeod, R.; Dunlop, A.J.; Edwards, H.V.; Advant, N.; Gibson, L.C.; Devine, N.M.; Brown, K.M.; Adams, D.R.; Houslay, M.D.; et al. A scanning peptide array approach uncovers association sites within the JNK/beta arrestin signalling complex. FEBS Lett. 2009, 583, 3310–3316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, X.; Stoy, H.; Kaoud, T.S.; Perry, N.A.; Chen, Q.; Perez, A.; Els-Heindl, S.; Slagis, J.V.; Iverson, T.M.; Beck-Sickinger, A.G.; et al. Peptide mini-scaffold facilitates JNK3 activation in cells. Sci. Rep. 2016, 6, 21025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, W.E.; McDonald, P.H.; Cai, S.F.; Field, M.E.; Davis, R.J.; Lefkowitz, R.J. Identification of a motif in the carboxyl terminus of beta -arrestin2 responsible for activation of JNK3. J. Biol. Chem. 2001, 276, 27770–27777. [Google Scholar] [CrossRef] [Green Version]
- Latorraca, N.R.; Masureel, M.; Hollingsworth, S.A.; Heydenreich, F.M.; Suomivuori, C.M.; Brinton, C.; Townshend, R.J.L.; Bouvier, M.; Kobilka, B.K.; Dror, R.O. How GPCR Phosphorylation Patterns Orchestrate Arrestin-Mediated Signaling. Cell 2020, 183, 1813–1825.e18. [Google Scholar] [CrossRef] [PubMed]
- Min, K.; Yoon, H.J.; Park, J.Y.; Baidya, M.; Dwivedi-Agnihotri, H.; Maharana, J.; Chaturvedi, M.; Chung, K.Y.; Shukla, A.K.; Lee, H.H. Crystal Structure of beta-Arrestin 2 in Complex with CXCR7 Phosphopeptide. Structure 2020, 28, 1014–1023.e4. [Google Scholar] [CrossRef]
- Nobles, K.N.; Xiao, K.; Ahn, S.; Shukla, A.K.; Lam, C.M.; Rajagopal, S.; Strachan, R.T.; Huang, T.Y.; Bressler, E.A.; Hara, M.R.; et al. Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci. Signal. 2011, 4, ra51. [Google Scholar] [CrossRef] [Green Version]
- Mayer, D.; Damberger, F.F.; Samarasimhareddy, M.; Feldmueller, M.; Vuckovic, Z.; Flock, T.; Bauer, B.; Mutt, E.; Zosel, F.; Allain, F.H.T.; et al. Distinct G protein-coupled receptor phosphorylation motifs modulate arrestin affinity and activation and global conformation. Nat. Commun. 2019, 10, 1261. [Google Scholar] [CrossRef]
- He, Q.T.; Xiao, P.; Huang, S.M.; Jia, Y.L.; Zhu, Z.L.; Lin, J.Y.; Yang, F.; Tao, X.N.; Zhao, R.J.; Gao, F.Y.; et al. Structural studies of phosphorylation-dependent interactions between the V2R receptor and arrestin-2. Nat. Commun. 2021, 12, 2396. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.R.; Reiter, E.; Ahn, S.; Kim, J.; Chen, W.; Lefkowitz, R.J. Different G protein-coupled receptor kinases govern G protein and beta-arrestin-mediated signaling of V2 vasopressin receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 1448–1453. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, B.; Beautrait, A.; Aguila, B.; Charles, R.; Escher, E.; Claing, A.; Bouvier, M.; Laporte, S.A. Differential beta-arrestin-dependent conformational signaling and cellular responses revealed by angiotensin analogs. Sci. Signal. 2012, 5, ra33. [Google Scholar] [CrossRef]
- Mann, A.; Liebetrau, S.; Klima, M.; Dasgupta, P.; Massotte, D.; Schulz, S. Agonist-induced phosphorylation bar code and differential post-activation signaling of the delta opioid receptor revealed by phosphosite-specific antibodies. Sci. Rep. 2020, 10, 8585. [Google Scholar] [CrossRef]
- Kaya, A.I.; Perry, N.A.; Gurevich, V.V.; Iverson, T.M. Phosphorylation barcode-dependent signal bias of the dopamine D1 receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 14139–14149. [Google Scholar] [CrossRef] [PubMed]
- Baidya, M.; Kumari, P.; Dwivedi-Agnihotri, H.; Pandey, S.; Chaturvedi, M.; Stepniewski, T.M.; Kawakami, K.; Cao, Y.; Laporte, S.A.; Selent, J.; et al. Key phosphorylation sites in GPCRs orchestrate the contribution of beta-Arrestin 1 in ERK1/2 activation. EMBO Rep. 2020, 21, e49886. [Google Scholar] [CrossRef] [PubMed]
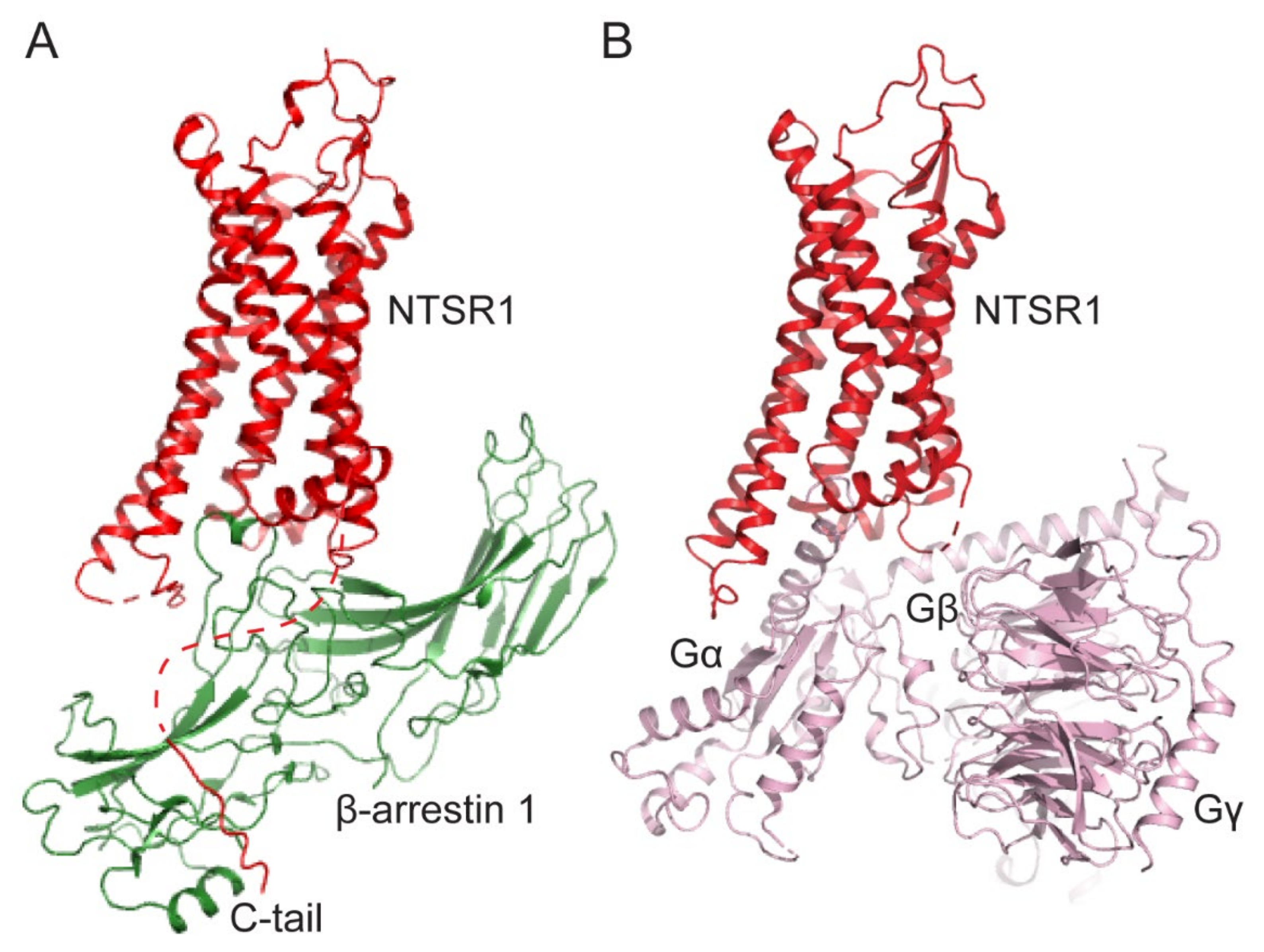
Figure 1.
Comparison of GPCR-arrestin and GPCR-G-protein complexes. (A) The structure of the neurotensin receptor-1 (NTSR1, red) and β-arrestin-1 (green) complex (PDB: 6UP7). (B) The structure of the NTSR1 (red) and heterotrimeric Gi1(light pink) complex (PDB: 7L0Q).
Figure 1.
Comparison of GPCR-arrestin and GPCR-G-protein complexes. (A) The structure of the neurotensin receptor-1 (NTSR1, red) and β-arrestin-1 (green) complex (PDB: 6UP7). (B) The structure of the NTSR1 (red) and heterotrimeric Gi1(light pink) complex (PDB: 7L0Q).

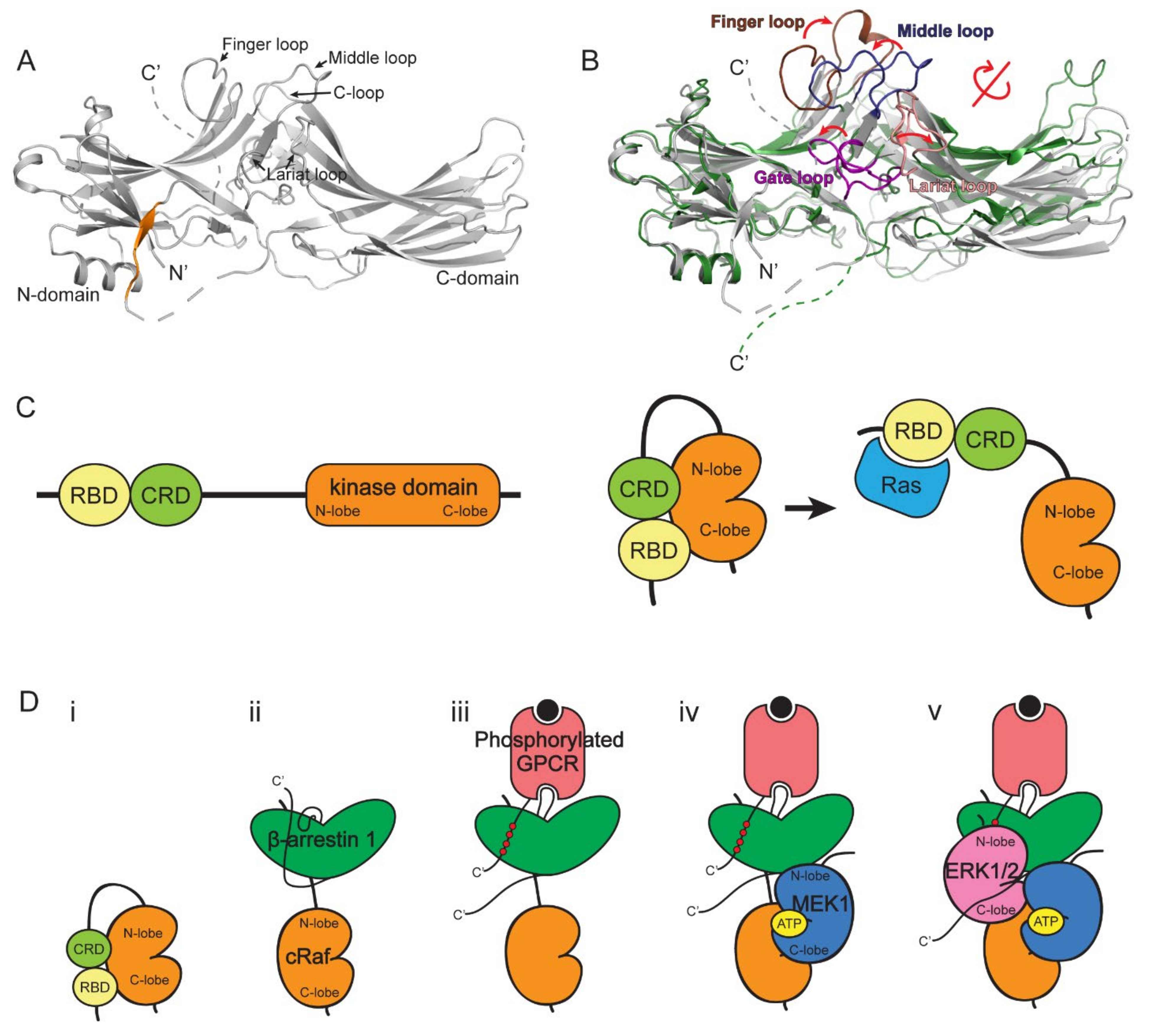
Figure 2.
The structures of β-arrestin and the scaffolding mechanism of ERK1/2 signaling cascade. (A) The structure of β-arrestin 1 in the basal state (PDB: 1G4R). The C-terminal βXX strand is colored in orange. (B) Comparison of β-arrestin in the basal and active states. The structures of β-arrestin 1 in basal (grey, PDB: 1G4R) and NTSR1-bound active (green, PDB: 6UP7) states are compared. The loops on β-arrestin 1 are colored as follows: finger loop, brown; middle loop, dark blue; gate loop, purple; lariat loop, salmon. (C) The representation of cRaf domains (left) and conformational changes of cRaf upon Ras binding (right). (D) The proposed model of the scaffolding mechanism of ERK1/2 signaling cascade by β-arrestin 1. (i) cRaf is auto-inhibited by the N-terminal domains, including RBD and CRD. (ii) cRaf RBD interacts with the back loop of β-arrestin 1. (iii) The phosphorylated GPCR may bind to β-arrestin 1. (iv) The N-lobe of MEK1 interacts with the interdomain loop I of β-arrestin 1. (v) The activated β-arrestin 1 interacts with ERK1/2 between the gate loop of β-arrestin 1 and the N-lobe of ERK1/2 and the interdomain loop II of β-arrestin 1 and the C-lobe of ERK1/2.
Figure 2.
The structures of β-arrestin and the scaffolding mechanism of ERK1/2 signaling cascade. (A) The structure of β-arrestin 1 in the basal state (PDB: 1G4R). The C-terminal βXX strand is colored in orange. (B) Comparison of β-arrestin in the basal and active states. The structures of β-arrestin 1 in basal (grey, PDB: 1G4R) and NTSR1-bound active (green, PDB: 6UP7) states are compared. The loops on β-arrestin 1 are colored as follows: finger loop, brown; middle loop, dark blue; gate loop, purple; lariat loop, salmon. (C) The representation of cRaf domains (left) and conformational changes of cRaf upon Ras binding (right). (D) The proposed model of the scaffolding mechanism of ERK1/2 signaling cascade by β-arrestin 1. (i) cRaf is auto-inhibited by the N-terminal domains, including RBD and CRD. (ii) cRaf RBD interacts with the back loop of β-arrestin 1. (iii) The phosphorylated GPCR may bind to β-arrestin 1. (iv) The N-lobe of MEK1 interacts with the interdomain loop I of β-arrestin 1. (v) The activated β-arrestin 1 interacts with ERK1/2 between the gate loop of β-arrestin 1 and the N-lobe of ERK1/2 and the interdomain loop II of β-arrestin 1 and the C-lobe of ERK1/2.

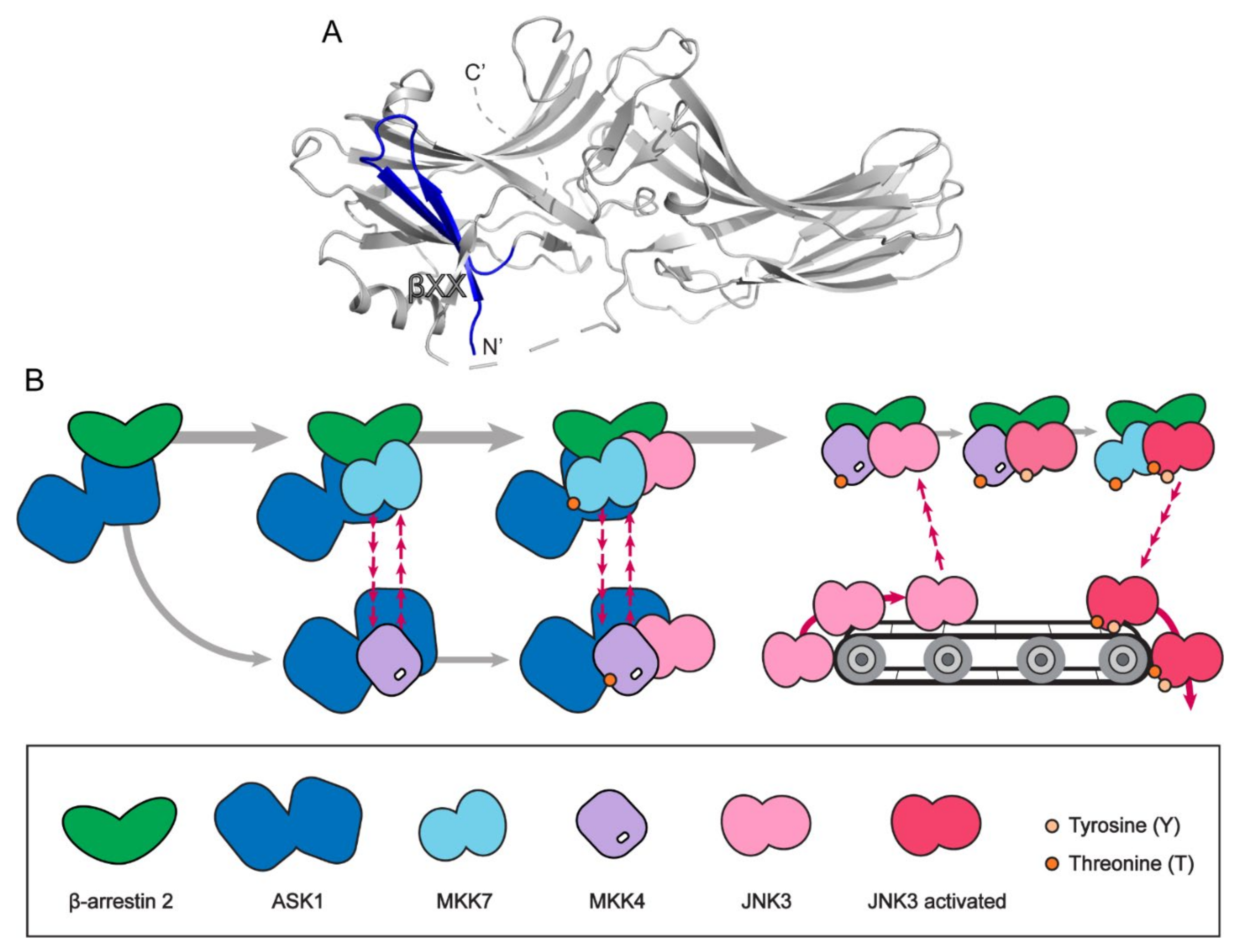
Figure 3.
The JNK3 signaling scaffolding mechanism of β-arrestin 2. (A) The β-arrestin 2 structure with first 25 residues highlighted with blue (PDB: 3P2D). (B) The conveyor belt model of JNK3 signaling scaffolding mechanism of β-arrestin 2.
Figure 3.
The JNK3 signaling scaffolding mechanism of β-arrestin 2. (A) The β-arrestin 2 structure with first 25 residues highlighted with blue (PDB: 3P2D). (B) The conveyor belt model of JNK3 signaling scaffolding mechanism of β-arrestin 2.

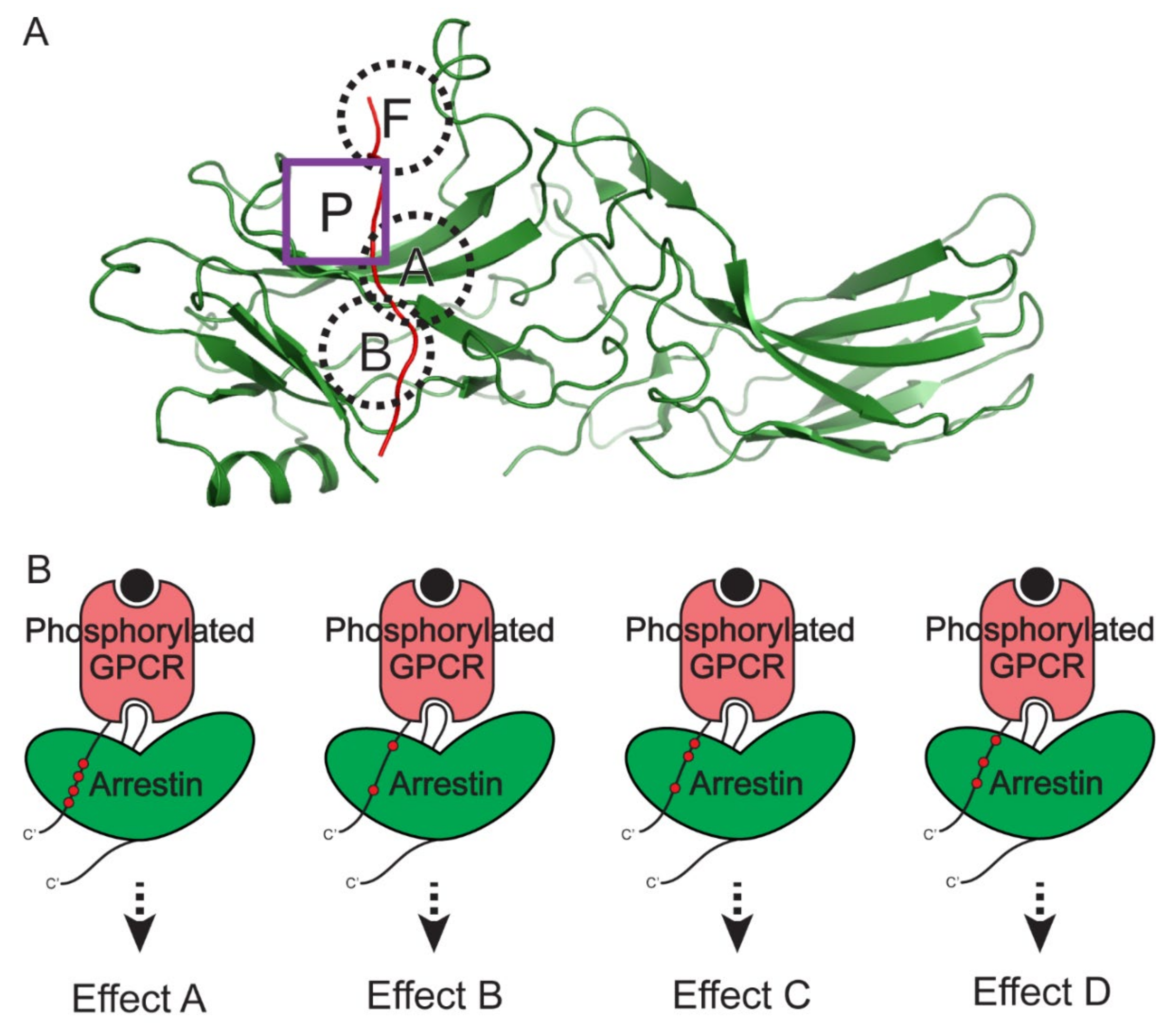
Figure 4.
Distinct downstream signaling effects that are mediated by different phosphorylation patterns of GPCRs. (A) The four known phosphate-binding pockets on arrestins. The structure of CXCR7 phosphopeptide (red) and the activated β-arrestin 2 (green) complex (PDB: 6K3F) is shown as a representative structure. The conserved positively charged pockets on arrestins (pocket A, B, and F) are shown in the dashed black circles. The newly identified phosphate-binding pocket P on β-arrestin 2 is shown in the purple square. (B) Differently phosphorylated GPCRs induce differently activated β-arrestins, leading to distinct downstream signaling effects.
Figure 4.
Distinct downstream signaling effects that are mediated by different phosphorylation patterns of GPCRs. (A) The four known phosphate-binding pockets on arrestins. The structure of CXCR7 phosphopeptide (red) and the activated β-arrestin 2 (green) complex (PDB: 6K3F) is shown as a representative structure. The conserved positively charged pockets on arrestins (pocket A, B, and F) are shown in the dashed black circles. The newly identified phosphate-binding pocket P on β-arrestin 2 is shown in the purple square. (B) Differently phosphorylated GPCRs induce differently activated β-arrestins, leading to distinct downstream signaling effects.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kim, K.; Han, Y.; Duan, L.; Chung, K.Y. Scaffolding of Mitogen-Activated Protein Kinase Signaling by β-Arrestins. Int. J. Mol. Sci. 2022, 23, 1000. https://doi.org/10.3390/ijms23021000
AMA Style
Kim K, Han Y, Duan L, Chung KY. Scaffolding of Mitogen-Activated Protein Kinase Signaling by β-Arrestins. International Journal of Molecular Sciences. 2022; 23(2):1000. https://doi.org/10.3390/ijms23021000
Chicago/Turabian StyleKim, Kiae, Yeonjin Han, Longhan Duan, and Ka Young Chung. 2022. "Scaffolding of Mitogen-Activated Protein Kinase Signaling by β-Arrestins" International Journal of Molecular Sciences 23, no. 2: 1000. https://doi.org/10.3390/ijms23021000
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.