
Disease Outcome and Brain Metabolomics of Cyclophilin-D Knockout Mice in Sepsis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
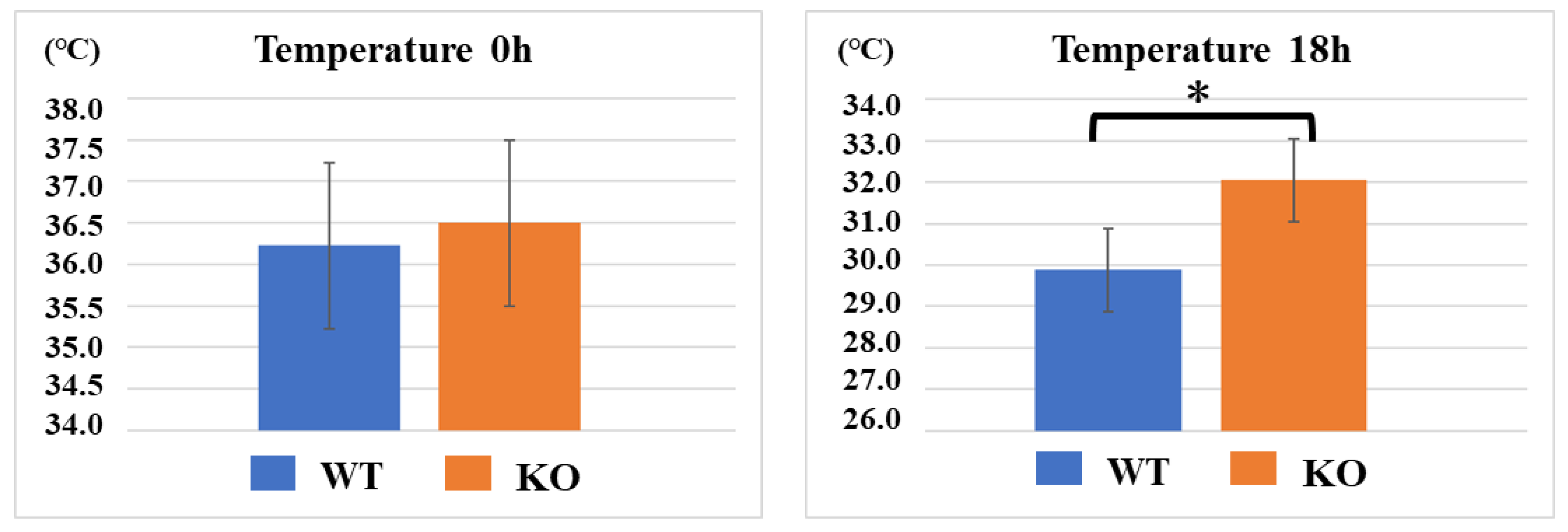
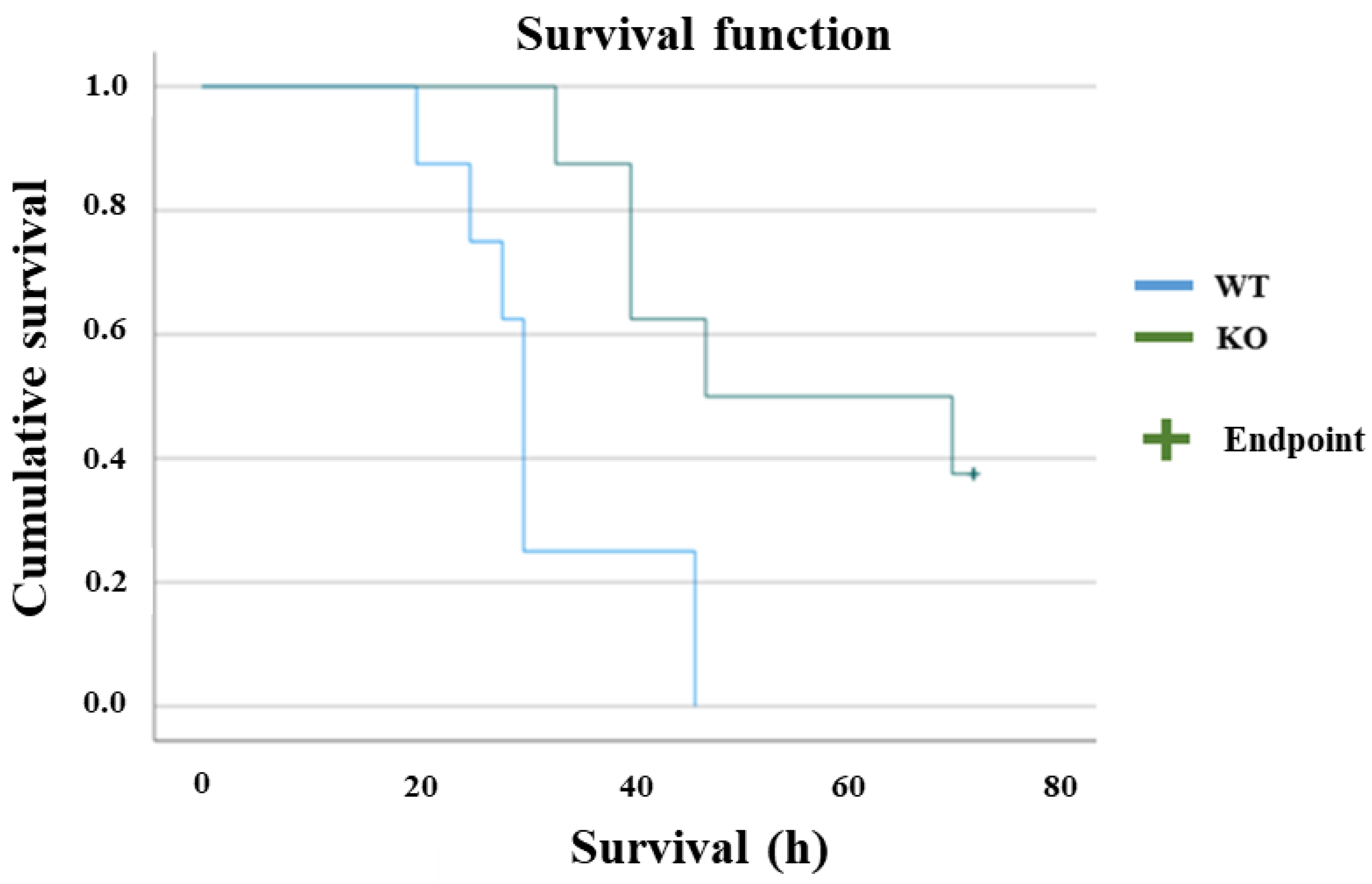
2.1. Sepsis-Induced Hypothermia and Mortality
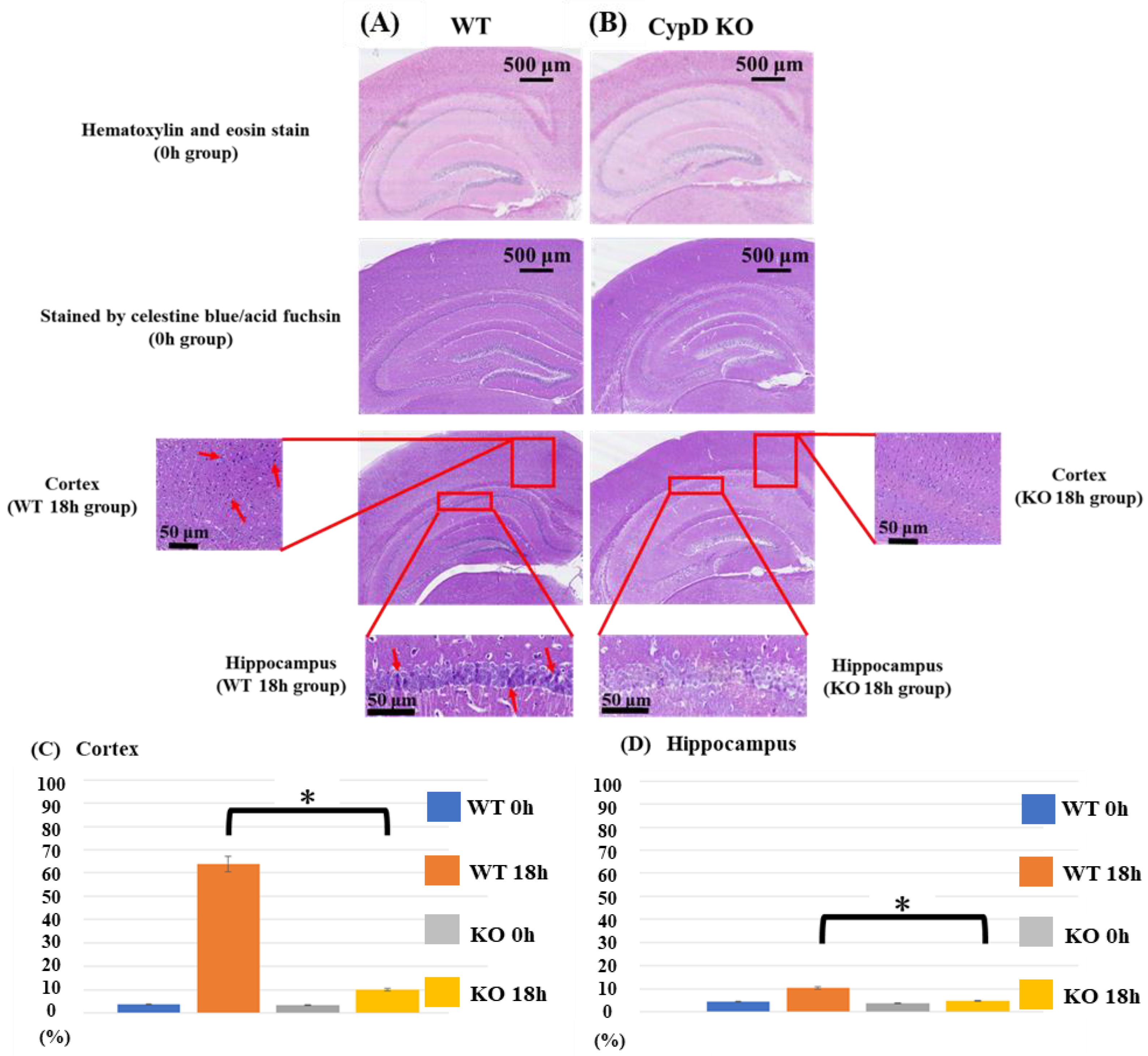
2.2. Effects of Cyclophilin D KO on Neuronal Cell Death
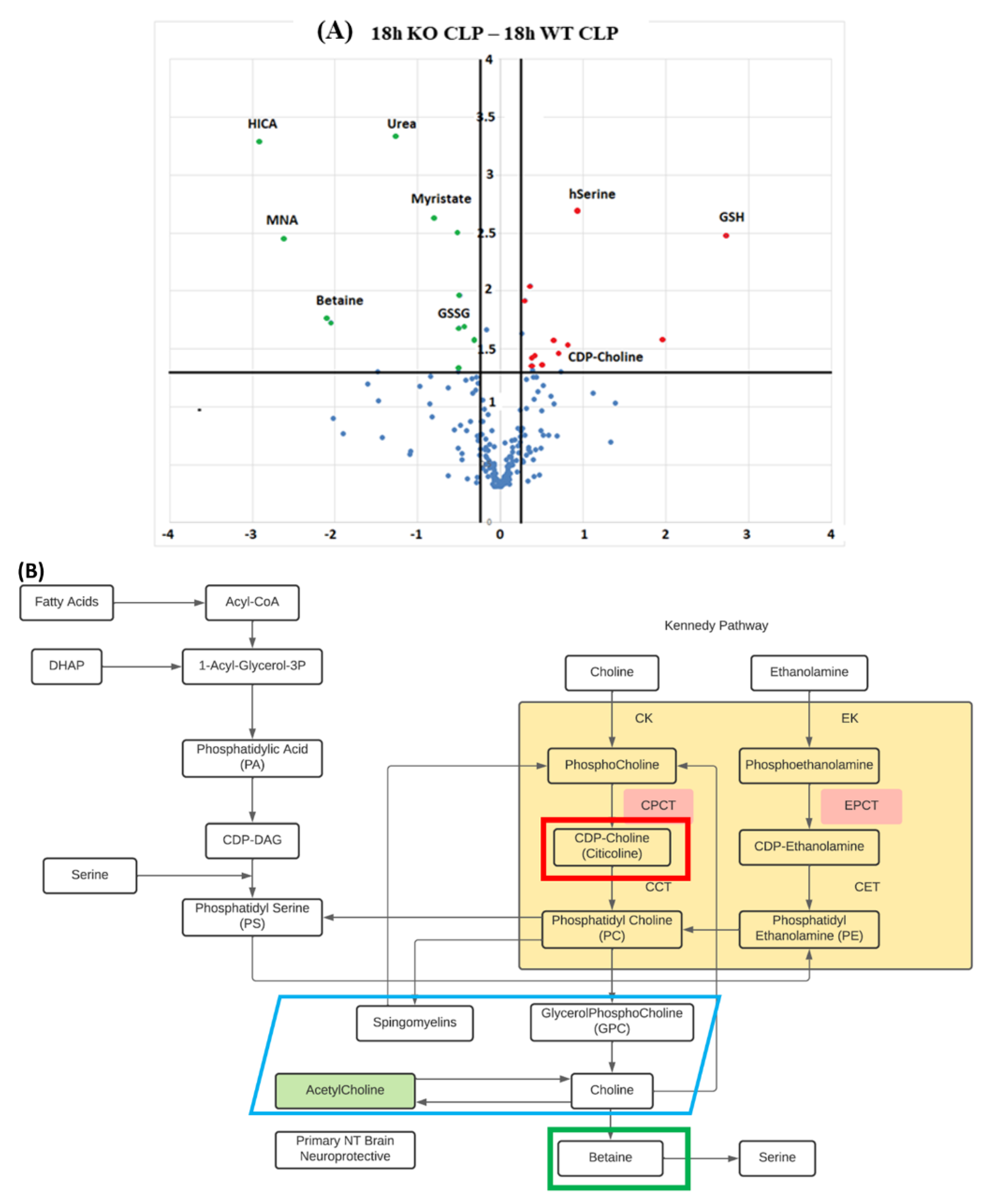
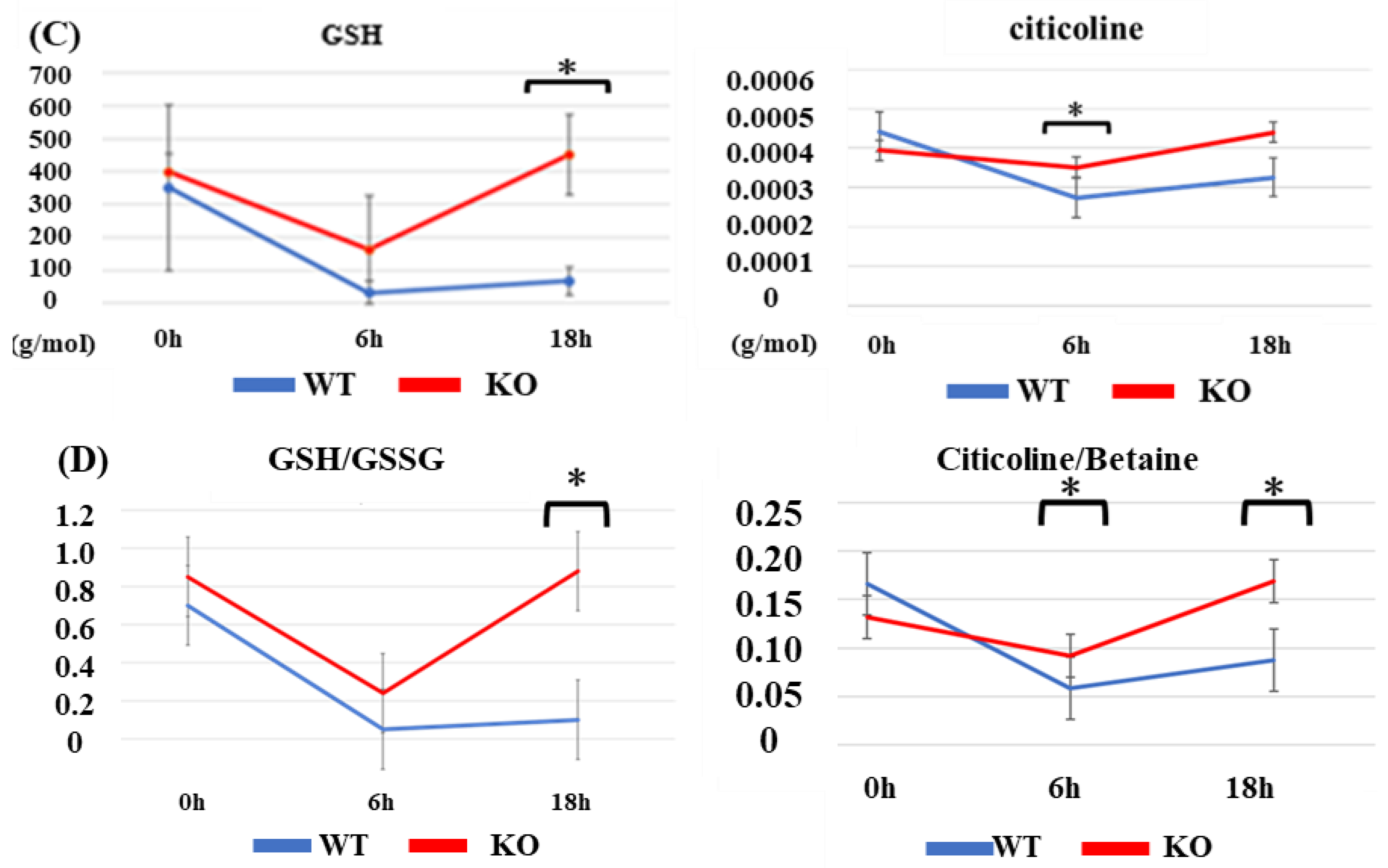
2.3. Metabolomics of Brain Tissue
3. Discussion
4. Materials and Methods
4.1. Sepsis Model
4.2. Measurement of Body Temperature and Survival Rate
4.3. Histological Analysis
4.4. Metabolomic Analysis
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Angus, D.C.; Linde-Zwirble, W.T.; Lidicker, J.; Clermont, G.; Carcillo, J.; Pinsky, M.R. Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Crit. Care Med. 2001, 29, 1303–1310. [Google Scholar] [CrossRef]
- Frost, M.T.; Wang, Q.; Moncada, S.; Singer, M. Hypoxia accelerates nitric oxide-dependent inhibition of mitochondrial complex I in activated macrophages. Am. J. Physiol. Integr. Comp. Physiol. 2005, 288, R394–R400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perl, T.M.; Dvorak, L. Long-term survival and function after suspected gram-negative sepsis. JAMA 1995, 274, 338–345. [Google Scholar] [CrossRef]
- Eidelman, L.A.; Putterman, D.; Putterman, C.; Sprung, C.L. The Spectrum of Septic Encephalopathy. JAMA 1996, 275, 470–473. [Google Scholar] [CrossRef]
- Sprung, C.L.; Peduzzi, P.N. Impact of encephalopathy on mortality in the sepsis syndrome. The Veterans Administration Systemic Sepsis Cooperative Study Group. Crit. Care Med. 1990, 18, 801–806. [Google Scholar] [CrossRef]
- Shi, J.; Xu, H. Blocking HMGB1/RAGE Signaling by Berberine Alleviates A1 Astrocyte and Attenuates Sepsis-Associated Encephalopathy. Front. Pharmacol. 2021, 12, 3012. [Google Scholar] [CrossRef] [PubMed]
- Dal-Pizzol, F.; Tomasi, C.D. Septic encephalopathy: Does inflammation drive the brain crazy? Rev. Bras. Psiquiatr. 2014, 36, 251–258. [Google Scholar] [CrossRef] [Green Version]
- D’Avila, J.C.; Santiago, A.P. Sepsis induces brain mitochondrial dysfunction. Crit. Care Med. 2008, 36, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- Iacobone, E.; Bailly-Salin, J. Sepsis-associated encephalopathy and its differential diagnosis. Crit. Care Med. 2009, 37, S331–S336. [Google Scholar] [CrossRef]
- Pandharipande, P.; Cotton, B.A. Motoric subtypes of delirium in mechanically ventilated surgical and trauma intensive care unit patients. Intensive Care Med. 2007, 33, 1726–1731. [Google Scholar] [CrossRef]
- Zhan, R.-Z.; Fujiwara, N.; Shimoji, K. Regionally different elevation of intracellular free calcium in hippocampus of septic rat brain. Shock 1996, 6, 293–297. [Google Scholar] [CrossRef]
- Haileselassie, B.; Joshi, A.U.; Minhas, P.S.; Mukherjee, R.; Andreasson, K.I.; Mochly-Rosen, D. Mitochondrial dysfunction mediated through dynamin-related protein 1 (Drp1) propagates impairment in blood brain barrier in septic encephalopathy. J. Neuroinflamm. 2020, 17, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Secades, J.J.; Lorenzo, J.L. Citicoline: Pharmacological and clinical review, 2006 update. Methods Find. Exp. Clin. Pharmacol. 2006, 28, 1–56. [Google Scholar] [PubMed]
- Alessandri, B.; Rice, A.C.; Levasseur, J.; DeFord, M.; Hamm, R.J.; Bullock, M.R. Cyclosporin A improves brain tissue oxygen consumption and learning/memory performance after lateral fluid percussion injury in rats. J. Neurotrauma 2002, 19, 829–841. [Google Scholar] [CrossRef]
- Uchino, H.; Minamikawa, R. Differential neuroprotection by cyclosporin A and FK506 following ischemia corresponds with differing abilities to inhibit calcineurin and the mitochondrial permeability transition. Neurobiol. Dis. 2002, 10, 219–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Wang, Y.; Yan, S.; Du, F.; Yan, S.S. NR2B-dependent cyclophilin D translocation suppresses the recovery of synaptic transmission after oxygen-glucose deprivation. Biochim. Biophys. Acta 2015, 1852, 2225–2234. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.; Lechleiter, J.D. Mitochondrial targeted cyclophilin D protects cells from cell death by peptidyl prolyl isomerization. J. Biol. Chem. 2002, 277, 31134–31141. [Google Scholar] [CrossRef] [Green Version]
- Gordan, R.; Fefelova, N.; Gwathmey, J.K.; Xie, L.-H. Iron Overload, Oxidative Stress and Calcium Mishandling in Cardiomyocytes: Role of the Mitochondrial Permeability Transition Pore. Antioxidants 2020, 9, 758. [Google Scholar] [CrossRef]
- Readnower, R.D.; Hubbard, W.B.; Kalimon, O.J.; Geddes, J.W.; Sullivan, P.G. Genetic Approach to Elucidate the Role of Cyclophilin D in Traumatic Brain Injury Pathology. Cells 2021, 10, 199. [Google Scholar] [CrossRef] [PubMed]
- Fiskum, G.; Starkov, A. Mitochondrial mechanisms of neural cell death and neuroprotective interventions in Parkinson’s disease. Ann. N. Y. Acad. Sci. 2003, 991, 111–119. [Google Scholar] [CrossRef]
- Lin, T.; Cheng, C. Mitochondrial dysfunction and oxidative stress promote apoptotic cell death in the striatum via cytochrome c/caspase-3 signaling cascade following chronic rotenone intoxication in rats. Int. J. Mol. Sci. 2012, 13, 8722–8739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elrod, J.W.; Wong, R. Cyclophilin D controls mitochondrial pore-dependent Ca2+ exchange, metabolic flexibility, and propensity for heart failure in mice. J. Clin. Investig. 2010, 120, 3680–3687. [Google Scholar] [CrossRef]
- Basso, E.; Fante, L.; Fowlkes, J.; Petronilli, V.; Forte, M.A.; Bernardi, P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J. Biol. Chem. 2005, 280, 18558–18561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latus, A.; Noël, J.-M.; Volanschi, E.; Lagrost, C.; Hapiot, P. Scanning Electrochemical Microscopy Studies of Glutathione-Modified Surfaces. An Erasable and Sensitive-to-Reactive Oxygen Species Surface. Langmuir 2011, 27, 11206–11211. [Google Scholar] [CrossRef]
- Narayanankutty, A.; Job, J.T.; Narayanankutty, V. Dual Roles in Carcinogenesis and Chemoprevention. Curr. Protein Pept. Sci. 2019, 20, 907–917. [Google Scholar] [CrossRef]
- Dringen, R.; Hirrlinger, J. Glutathione pathways in the brain. Biol. Chem. 2003, 384, 505–516. [Google Scholar] [CrossRef]
- Liang, Y.; Yeligar, S.M.; Brown, L.A.S. Chronic-alcohol-abuse-induced oxidative stress in the development of acute respiratory distress syndrome. Sci. World J. 2012, 2012, 740308. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Checa, J.C.; Kaplowitz, N. Hepatic mitochondrial glutathione: Transport and role in disease and toxicity. Toxicol. Appl. Pharmacol. 2005, 204, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Checa, J.C. Redox regulation and signaling lipids in mitochondrial apoptosis. Biochem. Biophys. Res. Commun. 2003, 304, 471–479. [Google Scholar] [CrossRef]
- Waisbourd-Zinman, O.; Koh, H.; Tsai, S.; Lavrut, P.; Dang, C.; Zhao, X.; Pack, M.; Cave, J.; Hawes, M.; Koo, K.A.; et al. The toxin biliatresone causes mouse extrahepatic cholangiocyte damage and fibrosis through decreased glutathione and SOX17. Hepatology 2016, 64, 880–893. [Google Scholar] [CrossRef]
- Khan, H.A.; Ahmad, M.Z. Crosstalk of liver immune cells and cell death mechanisms in different murine models of liver injury and its clinical relevance. Hepatobiliary Pancreat Dis. Int. 2017, 16, 245–256. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 14205–14218. [Google Scholar] [CrossRef]
- Faiq, M.A.; Wollstein, G.; Schuman, J.; Chan, K.C. Cholinergic nervous system and glaucoma: From basic science to clinical applications. Prog. Retin. Eye Res. 2019, 72, 100767. [Google Scholar] [CrossRef] [PubMed]
- Adibhatla, R.M.; Hatcher, J.F.; Dempsey, R.J. Effects of citicoline on phospholipid and glutathione levels in transient cerebral ischemia. Stroke 2001, 32, 2376–2381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adibhatla, R.M.; Hatcher, J.F. Citicoline: Neuroprotective mechanisms in cerebral ischemia. J. Neurochem. 2002, 80, 12–23. [Google Scholar] [PubMed] [Green Version]
- Salamah, A.; Mehrez, M. Efficacy of Citicoline as a Neuroprotector in children with post cardiac arrest: A randomized controlled clinical trial. Eur. J. Pediatr. 2021, 180, 1249–1255. [Google Scholar] [CrossRef]
- Alvarez, X.A.; Sampedro, C. Citicoline protects hippocampal neurons against apoptosis induced by brain beta-amyloid deposits plus cerebral hypoperfusion in rats. Methods Find. Exp. Clin. Pharmacol. 1999, 21, 535–540. [Google Scholar] [CrossRef]
- Alvarez-Sabín, J.; Román, G.C. Citicoline in vascular cognitive impairment and vascular dementia after stroke. Stroke 2011, 42, S40–S43. [Google Scholar] [CrossRef] [Green Version]
- Turkkan, A.; Alkan, T.; Goren, B.; Kocaeli, H.; Akar, E.; Korfali, E. Citicoline and postconditioning provides neuroprotection in a rat model of ischemic spinal cord injury. Acta Neurochir. 2010, 152, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Oshitari, T.; Yoshida-Hata, N.; Yamamoto, S. Effect of neurotrophic factors on neuronal apoptosis and neurite regeneration in cultured rat retinas exposed to high glucose. Brain Res. 2010, 1346, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Barrachina, M.; Secades, J.; Lozano, R.; Gómez-Santos, C.; Ambrosio, S.; Ferrer, I. Citicoline increases glutathione redox ratio and reduces caspase-3 activation and cell death in staurosporine-treated SH-SY5Y human neuroblastoma cells. Brain Res. 2002, 957, 84–90. [Google Scholar] [CrossRef]
- Aminzadeh, A.; Salarinejad, A. Citicoline protects against lead-induced oxidative injury in neuronal PC12 cells. Biochem. Cell Biol. 2019, 97, 715–721. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobayashi, T.; Uchino, H.; Elmér, E.; Ogihara, Y.; Fujita, H.; Sekine, S.; Ishida, Y.; Saiki, I.; Shibata, S.; Kawachi, A. Disease Outcome and Brain Metabolomics of Cyclophilin-D Knockout Mice in Sepsis. Int. J. Mol. Sci. 2022, 23, 961. https://doi.org/10.3390/ijms23020961
Kobayashi T, Uchino H, Elmér E, Ogihara Y, Fujita H, Sekine S, Ishida Y, Saiki I, Shibata S, Kawachi A. Disease Outcome and Brain Metabolomics of Cyclophilin-D Knockout Mice in Sepsis. International Journal of Molecular Sciences. 2022; 23(2):961. https://doi.org/10.3390/ijms23020961
Chicago/Turabian StyleKobayashi, Takayuki, Hiroyuki Uchino, Eskil Elmér, Yukihiko Ogihara, Hidetoshi Fujita, Shusuke Sekine, Yusuke Ishida, Iwao Saiki, Shoichiro Shibata, and Aya Kawachi. 2022. "Disease Outcome and Brain Metabolomics of Cyclophilin-D Knockout Mice in Sepsis" International Journal of Molecular Sciences 23, no. 2: 961. https://doi.org/10.3390/ijms23020961