
Novel 1,3,5-Triazinyl Aminobenzenesulfonamides Incorporating Aminoalcohol, Aminochalcone and Aminostilbene Structural Motifs as Potent Anti-VRE Agents, and Carbonic Anhydrases I, II, VII, IX, and XII Inhibitors

 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Activity Evaluation
2.2.1. CA Inhibition
- All tested compounds are weak inhibitors of cytosolic isozyme hCA I with KIs in a range from 8.5 to >10,000 nM. In general, compounds substituted with stilbenes are better inhibitors of hCA I than compounds containing chalcone substituents. Three stilbene derivatives (31, 33, and 42) with the highest activity against hCA I with KIs in the range of 36.9–48.0 nM have at the terminal benzene core (R2) hydrogen (4-H) or hydroxyl functional group (4-OH) in the position para. If the substituents present on the triazine core are very bulky (for example, in a homologous series of compounds 4, 17, and 37), the compounds’ inhibitory activity increases with the increasing number of CH2 groups between the triazine core and the benzenesulfonamide structural moiety. If the methylene or ethylene group between the triazine and benzene core is not present, the compound does not fit into the narrowing cavity of the active site due to the steric inherence.
- All compounds presented in this article are very weak inhibitors of physiologically relevant isoenzyme hCA II. In comparison with previously reported compounds 2, 3, 5–12 and 18–19, chalcone and stilbene derivatives exhibit much lower inhibition activity against hCA II. This is probably caused by the bulkiness of chalcone and stilbene structural moieties. The best inhibition activities of new compounds against hCA II were obtained for chalcone derivatives substituted with 4-OH (16 with KI = 36.0 nM and 27 with KI = 28.8 nM) or 2-OH (14 with KIs = 35.4 nM) at the terminal benzene core (R2). These KIs values were comparable with standard DCP (KI = 38.0 nM). Substitution 3-OH has a strongly negative effect on the affinity to hCA II. On the other hand, most active stilbene derivatives 40, 41, and 17 with KIs in the range of 18.9–46.8 nM are substituted at the terminal benzene core with a hydroxyl functional group in position meta (3-OH).
- Selected compounds were tested as potential inhibitors of isoenzyme hCA VII. Chalcone derivatives are, with KIs in the range from 25.9 to 810.2 nM, rather weak inhibitors of hCA VII. Exceptions of this are compound 15 with the KI = 25.9 nM comparable with the standard DCP (KI = 26.5 nM) and compound 26 (KI = 42.1 nM). Unlike all standards, these compounds show high selectivity over the cytosolic isoenzymes:for compound 15 with KI (hCA II)/KI (hCA VII) = 116.48 and KI (hCA I)/KI (hCA VII) = >386.01, for compound 26 with KI (hCA II)/KI (hCA VII) = 31.71 and KI (hCA I)/KI (hCA VII) = 157.41. All chalcone derivatives with inhibitory activity against hCA VII have common 3-OH substitution at the terminal benzene core of the chalcone structural moiety. From the comparison of the analogue pairs of compounds 15 and 25, and 14 and 22, it can be assumed that biological activity and selectivity of chalcone derivatives are negatively affected by the increasing length of the alkyl chain between the 1,3,5-triazine core and the benzenesulfonamide moiety.
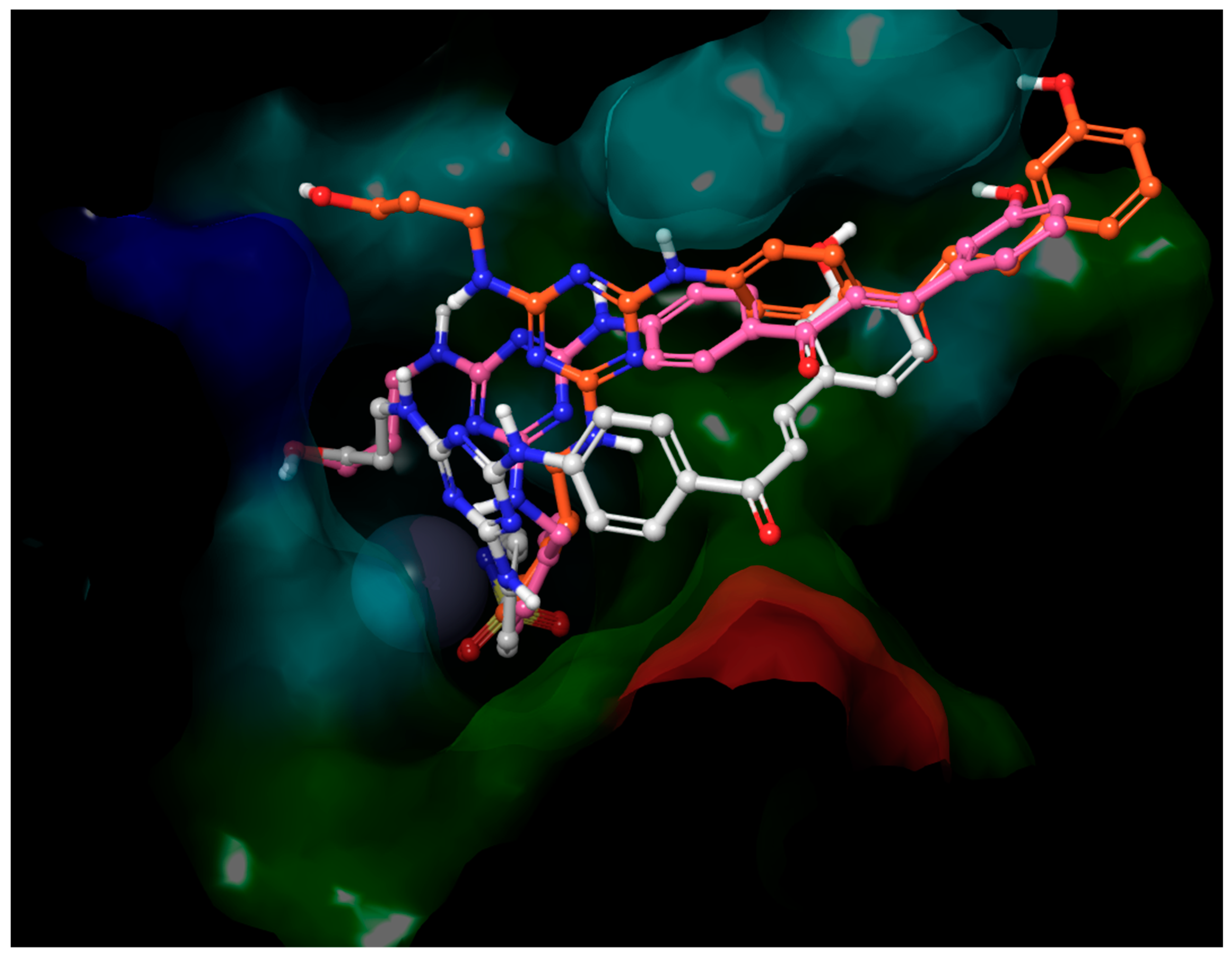
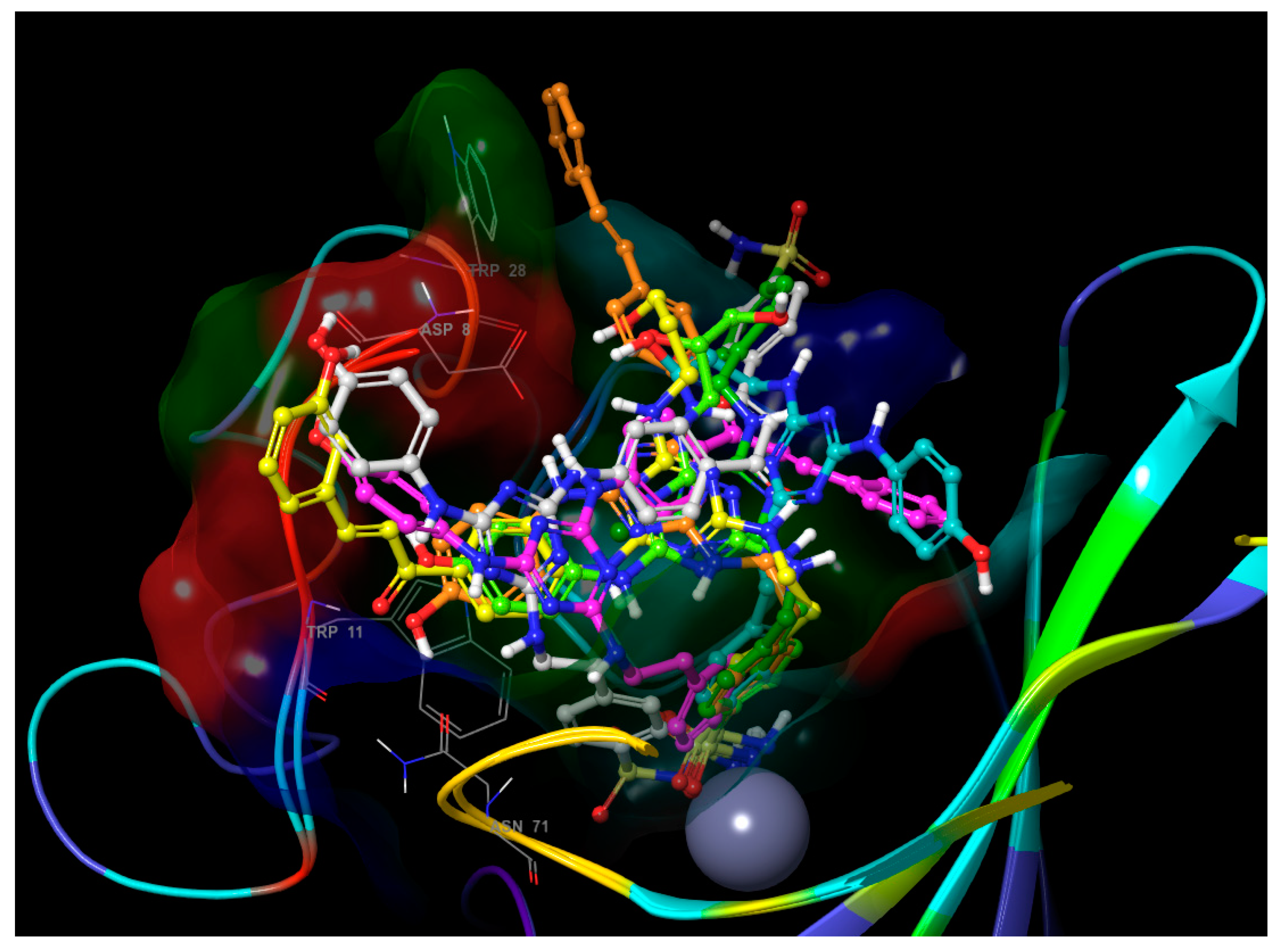
- The inhibitory activities of compounds 2, 3, 5–12 and 18–19 against the isoenzyme hCA IX were published and discussed extensively in [39]. For this reason, only the values of KIs in the inhibition of hCA IX for chalcone and stilbene derivatives were discussed in this part. Although the structures of chalcone derivatives with potential inhibition activity against isoenzyme hCA IX were selected based on docking (please see the Supplementary Materials), none of the tested compounds showed significant biological activity or selectivity against this isoenzyme. Contrarily, four of tested stilbene derivatives (17, 30, 38, and 40) are significantly better inhibitors of hCA IX (KIs =12.1–22.2 nM) than all used standards (KIs = 24.0–52.0 nM), and the inhibitory activities of another four compounds (4, 37, 41, and 43, KIs = 25.7–31.1 nM) are comparable to these standards. Interestingly, most of these compounds have in common the hydroxyl group at the position meta (3-OH) at the terminal benzene of the stilbene structural moiety. The exception is compound 30, with the lowest KI = 12.1 nM, where the hydroxyl group is not present at the stilbene moiety. The three of the best inhibitors are also compounds with a very high selectivity over both cytosolic isoenzymes, with values of selectivity for compound 4 KI (hCA II)/KI (hCA IX) = 132.36, KI (hCA I)/KI (hCA IX) = 273.56; for compound 30 KI (hCA II)/KI (hCA IX) = 424.21, KI (hCA I)/KI (hCA IX) = 470.83; and for compound 38 KI (hCA II)/KI (hCA IX) = 256.82, KI (hCA I)/KI (hCA IX) = 388.79. Comparing the best poses of ligands 25 and 37 from molecular docking provides an explanation for the difference in activity between the chalcone and stilbene derivatives (Figure 1). The only difference in the structure of their molecules is the carbonyl group in the chalcone substituent of inactive ligand 25. Although it is a small structural change in the molecule, the influence on the position and subsequent interactions is significant. While some H-bonds of hydroxyls in distal ends of inactive ligand 25 are made with different residues compared to highly active ligand 37, some H-bonds of the core part disappeared (with residues His-64, Trp-5), and the carbonyl group creates no H-bond itself.
- Stilbene derivatives were also tested as potential inhibitors of tumor-associated isoenzyme hCA XII. Of the tested compounds, eight (30–34, 36, 42, and 44) are excellent inhibitors of hCA XII (KIs = 4.4–10.0 nM), with biological activity better than (DCP, EZA), or comparable to (AAZ, IND, MZA), used standards. The structure of most active compounds is quite diverse, containing different aminoalcohole, aminobenzene sulfonamide, and stilbene structural moieties. It is very interesting that stilbene derivatives containing two aminobenzene sulfonamide substituents (33, 36, and 44), which are very bulky, are among the most active compounds (KIs = 6.2–10.0 nM) with good selectivity (KI (hCA II)/KI (hCA VII) = 9.34–25.21). On the other hand, the comparison of the KIs values of analogue compounds 4, 17, and 37 implies that, with an increasing number of CH2 groups in the aminobenzene sulfonamide structural moiety, the biological activity decreases. Thus, it seems that the bulkiness of the molecule is not the only essential factor that affects the biological activity of the tested substances. However, it does significantly affect their selectivity. These observations suggest that, not only does the interaction of the sulfonamide functional group with the active site influence activity and selectivity, but also that the interactions between the rest of the molecule and amino acid residues of the cavity or amino acid residues close to the cavity must have a major effect on the activity and selectivity. In accordance with this statement, it is remarkable that the compounds with excellent selectivity and, at the same time, inhibitory activities comparable with all standards, contain 4’-H-aminostilbene structural moiety 30 (KI = 7.6 nM; KI (hCA II)/KI (hCA XII) = 675.39; KI (hCA I)/KI (hCA XII) =749.60), 32 (KI = 5.9 nM; KI (hCA II)/KI (hCA XII) = 1 269.15; KI (hCA I)/KI (hCA XII) = 15.36), or 2’-OH-aminostilbene structural moiety 34 (KI = 8.5 nM; KI (hCA II)/KI (hCA XII) = 662.59; KI (hCA I)/KI (hCA XII) = 196.71).
2.2.2. VRE Inhibition
2.2.3. Cytotoxicity Determination against Human Colorectal Tumor Cell Line (HCT116 p53+/+)
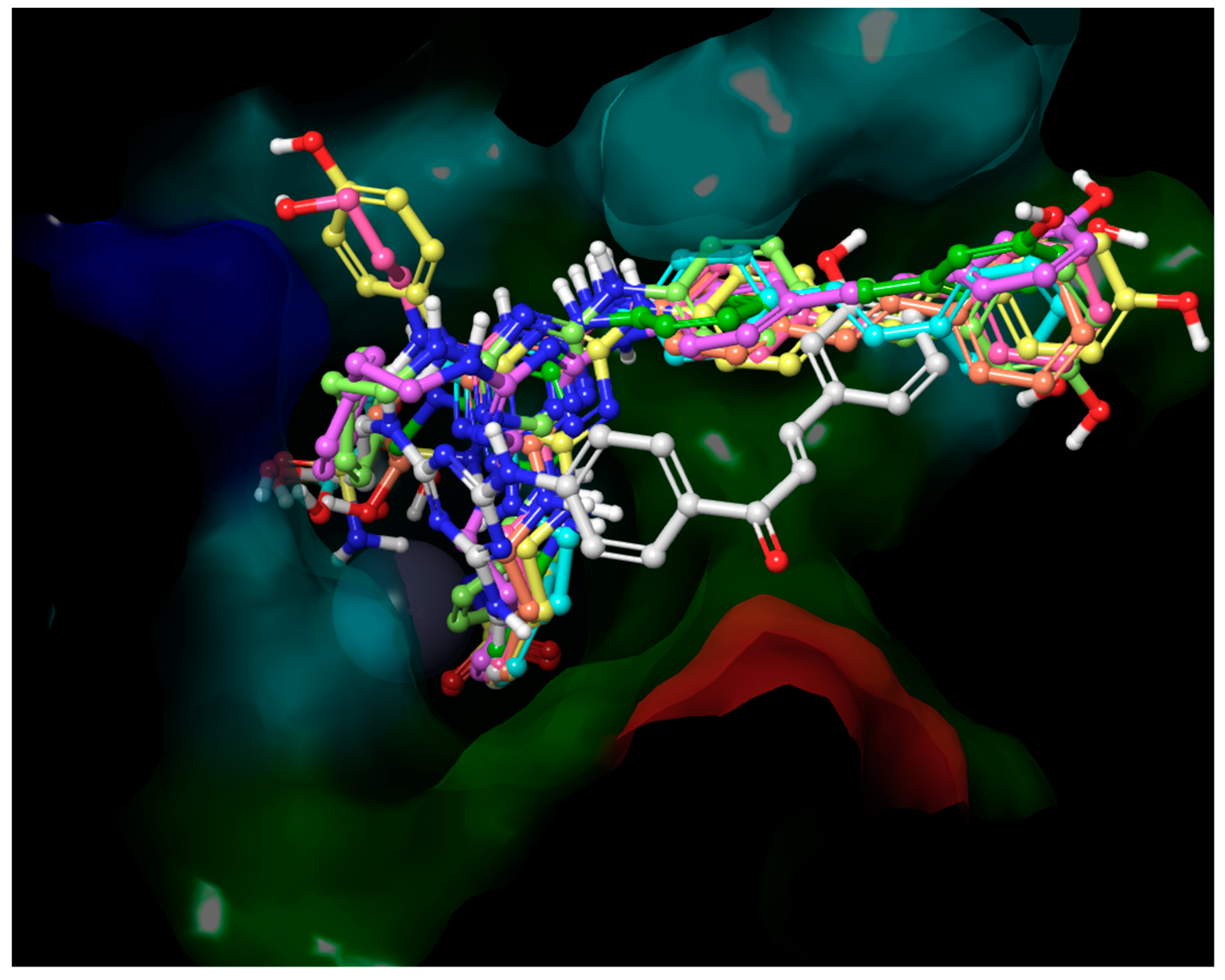
2.3. Molecular Modeling
2.3.1. Molecular Docking into hCA IX
- The first substituent is always the benzenesulfonamide moiety with linkers of three different lengths. The coordination of the sulfonamide group to the metal center usually makes up 60% of the interaction energy of the ligands with the carbonic anhydrase [51].
- The second substituent is either stilbene or chalcone, making hydrophobic interaction, which is the weakest interaction, however is significant because of the big surface covered. Most of these substituents bear phenolic hydroxyl at the end, which usually creates H-bonds with bulk water, but in some cases does so with polar groups of amino acid residues at the edge of the cavity (ligand 37 with the hydroxyl of Ser-20; ligand 4 with the carbonyl of Val-19).
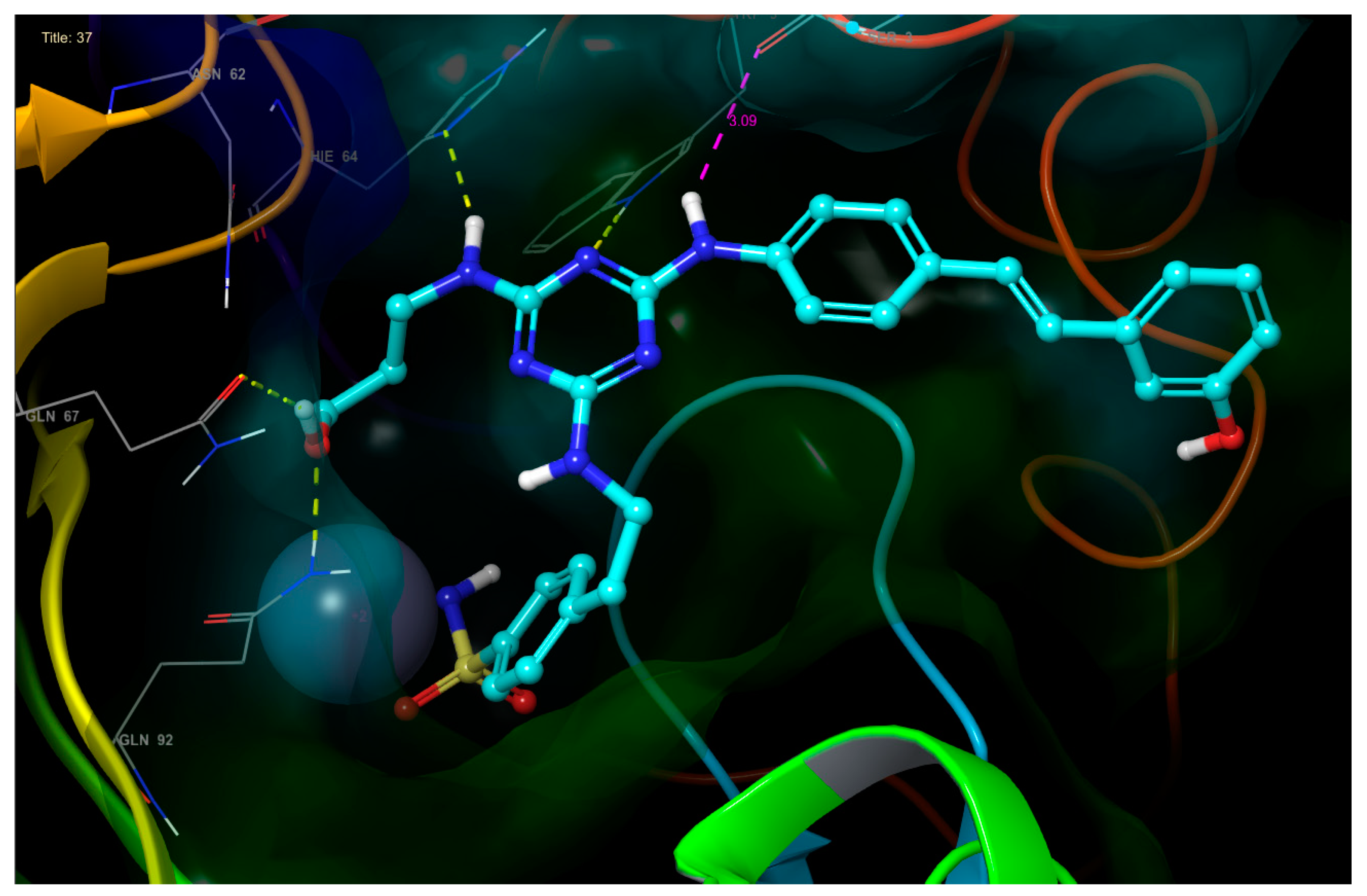
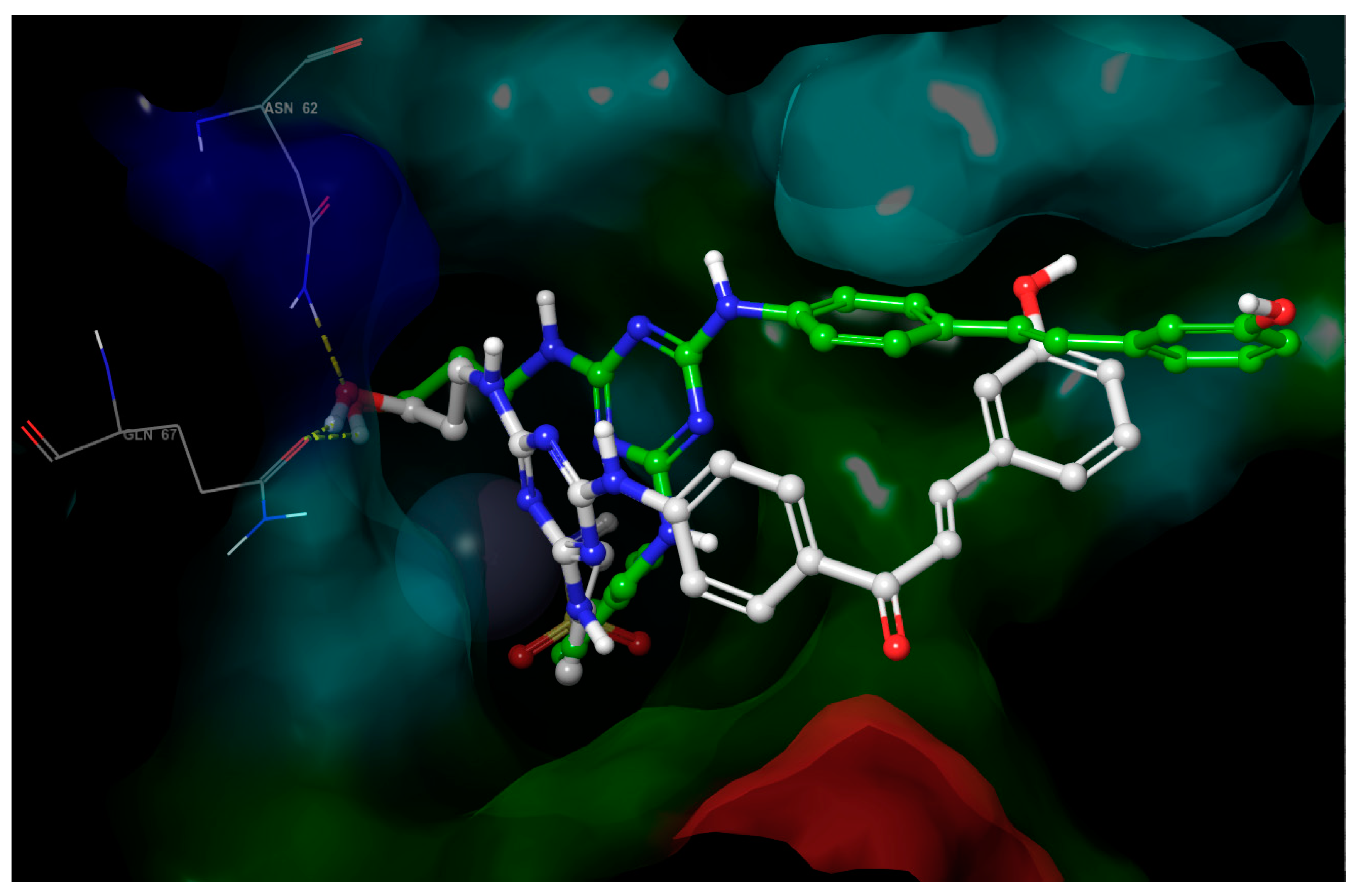
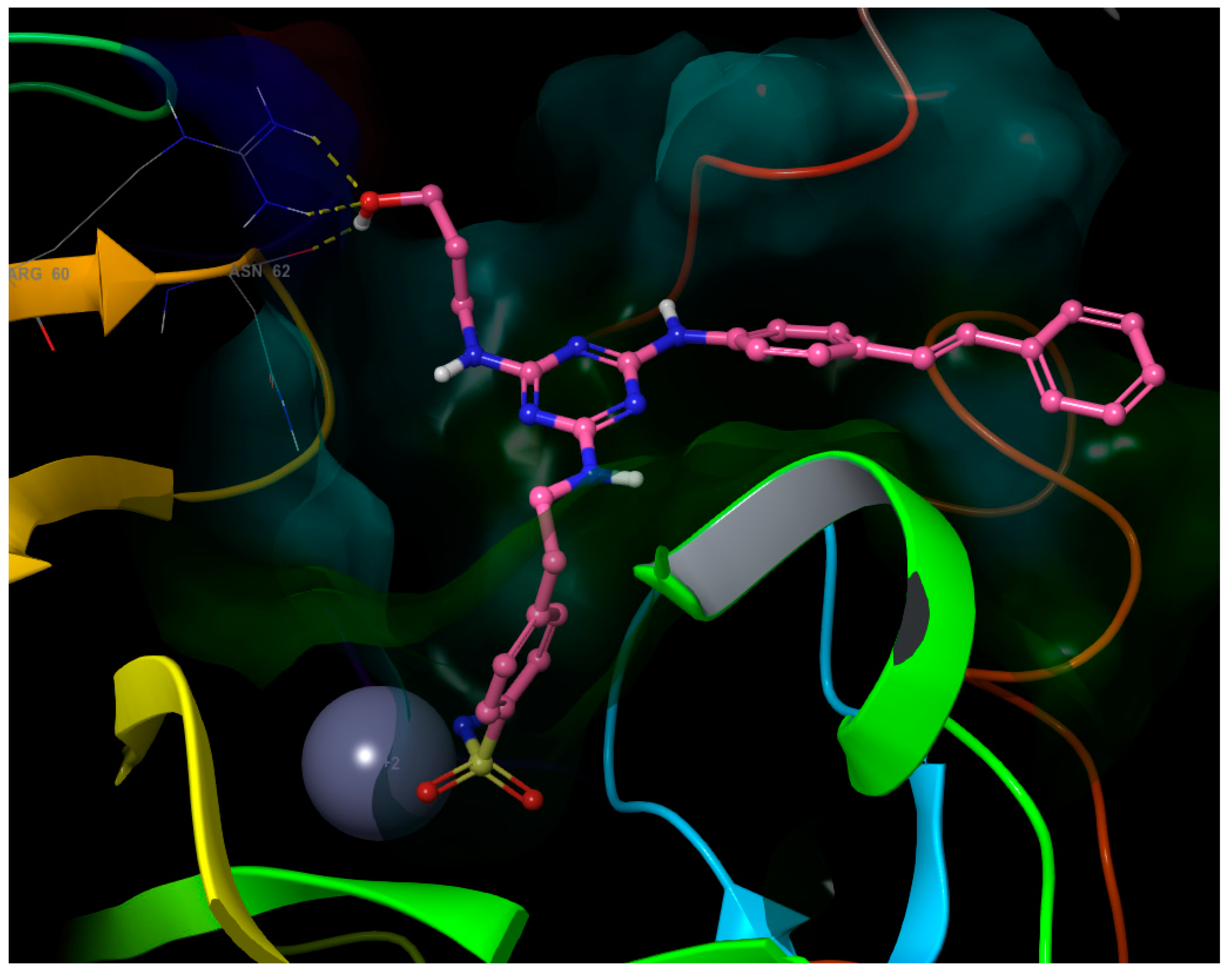
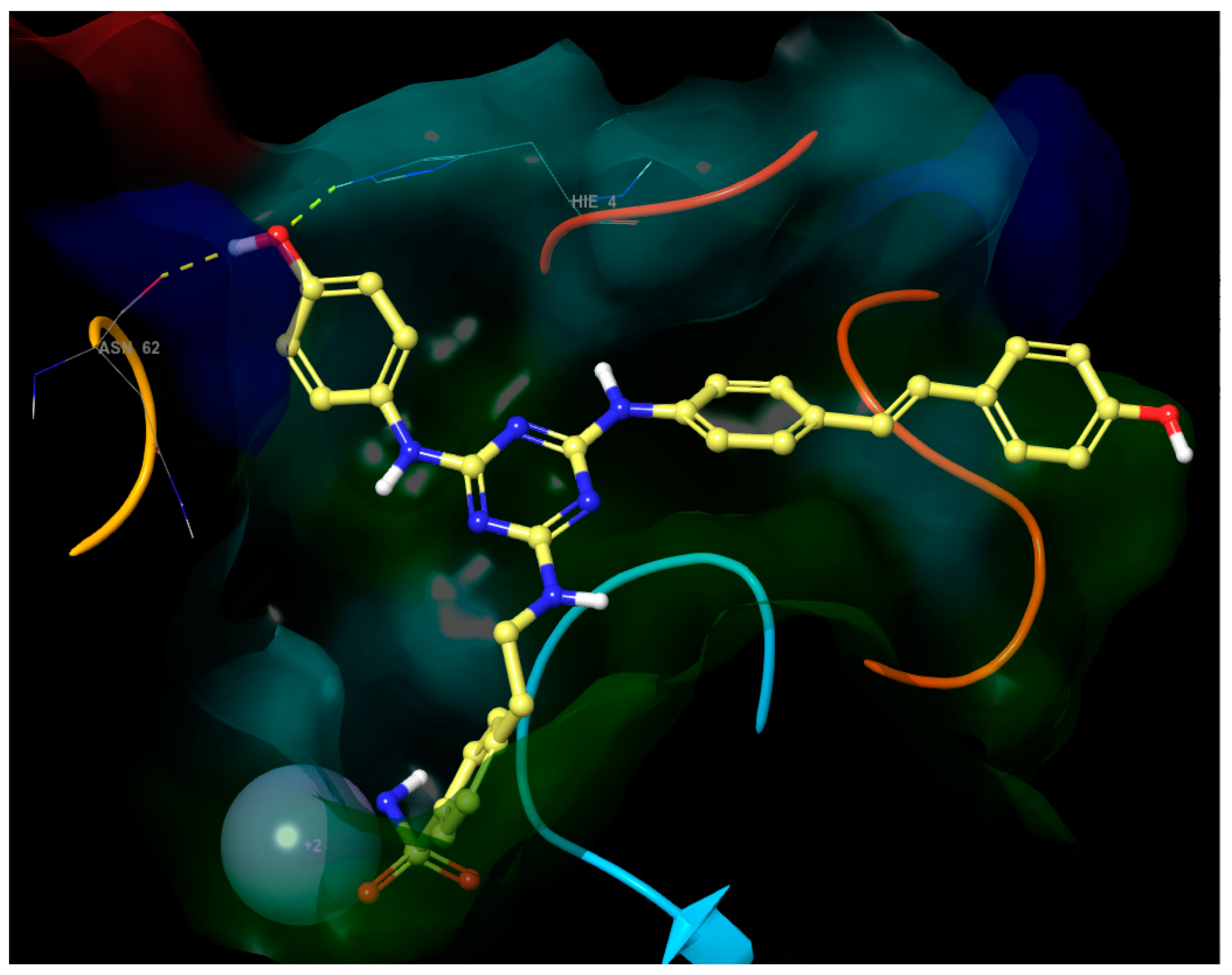
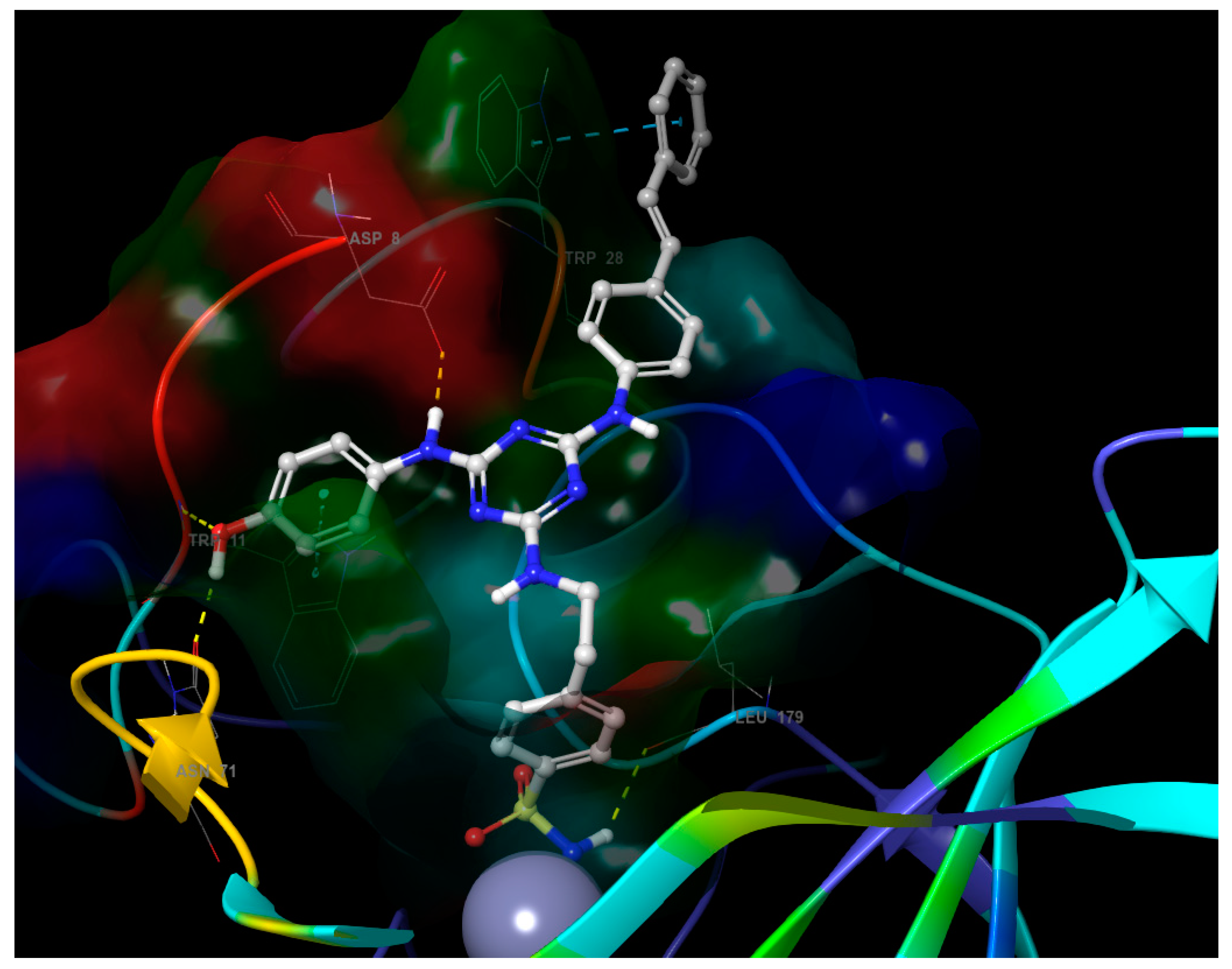
- The third substituent is responsible for the H-bond interactions with polar residues, and the polar side of the active site cavity gives a varied offer of polar residues to interact with them. Ligands 4 and 17 with hydroxypropyl substituent (Figure 5) are H-bond acceptors to the Asn-62 side chain and H-bond donors to the Gln-67 side chain. Ligand 30 (Figure 6) shows that also Arg-60 can serve as a H-bond donor and Asn-62 as a H-bond acceptor to the hydroxypropyl substituent. The hydroxypropyl substituent of ligand 37 (Figure 4) is the H-bond acceptor Gln-67 again, but the H-bond donor is Gln-92. The terminal hydroxyl of the polar substituent on ligand 38 interacts the same way as in ligand 37, and the second hydroxyl on the substituent contributes by accepting the H-bond from Thr-200 (Figure 7). Ligands 40 and 41 with a benzenesulfonamide substituent near the polar side of the cavity use the oxygen of the sulfonamide group as the acceptor of the H-bond from Asn-62, and phenolic hydroxyl on ligand 43 is the H-bond acceptor from the His-4 and H-bond donor to backbone carbonyl of Asn-62 (Figure 8). The aromatic ring of this phenol substituent also allows T-stacking with His-64.
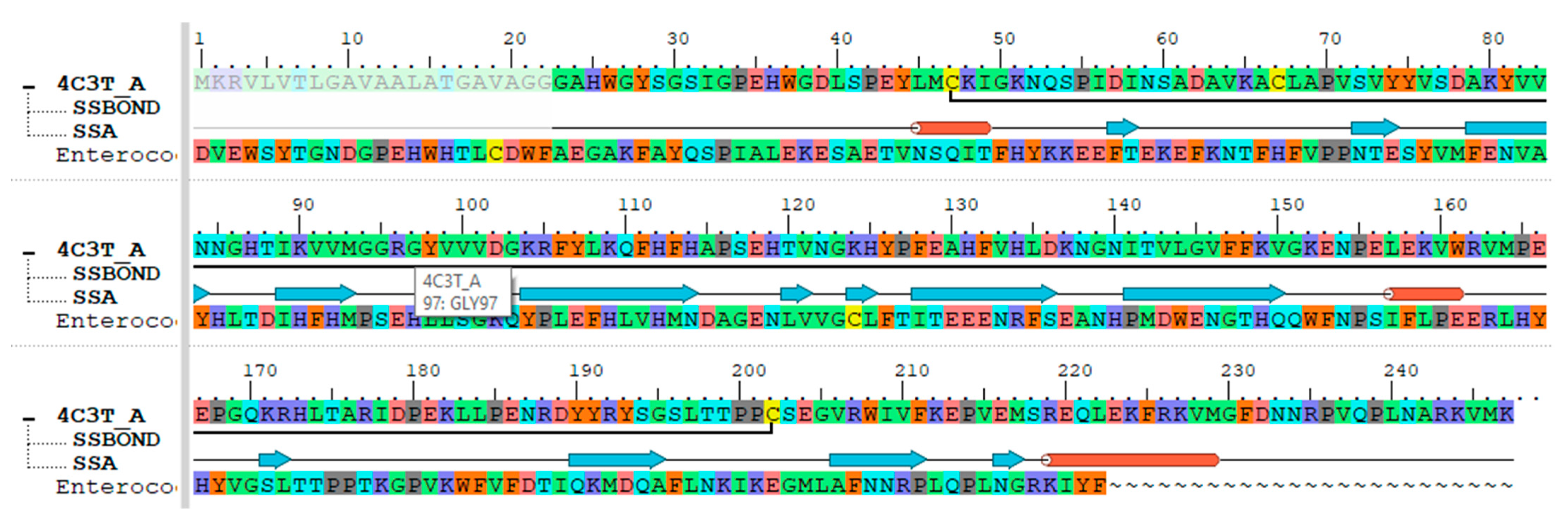
2.3.2. Retrieval of Enterococcal CA Protein Sequence
2.3.3. 3D Homology Modeling
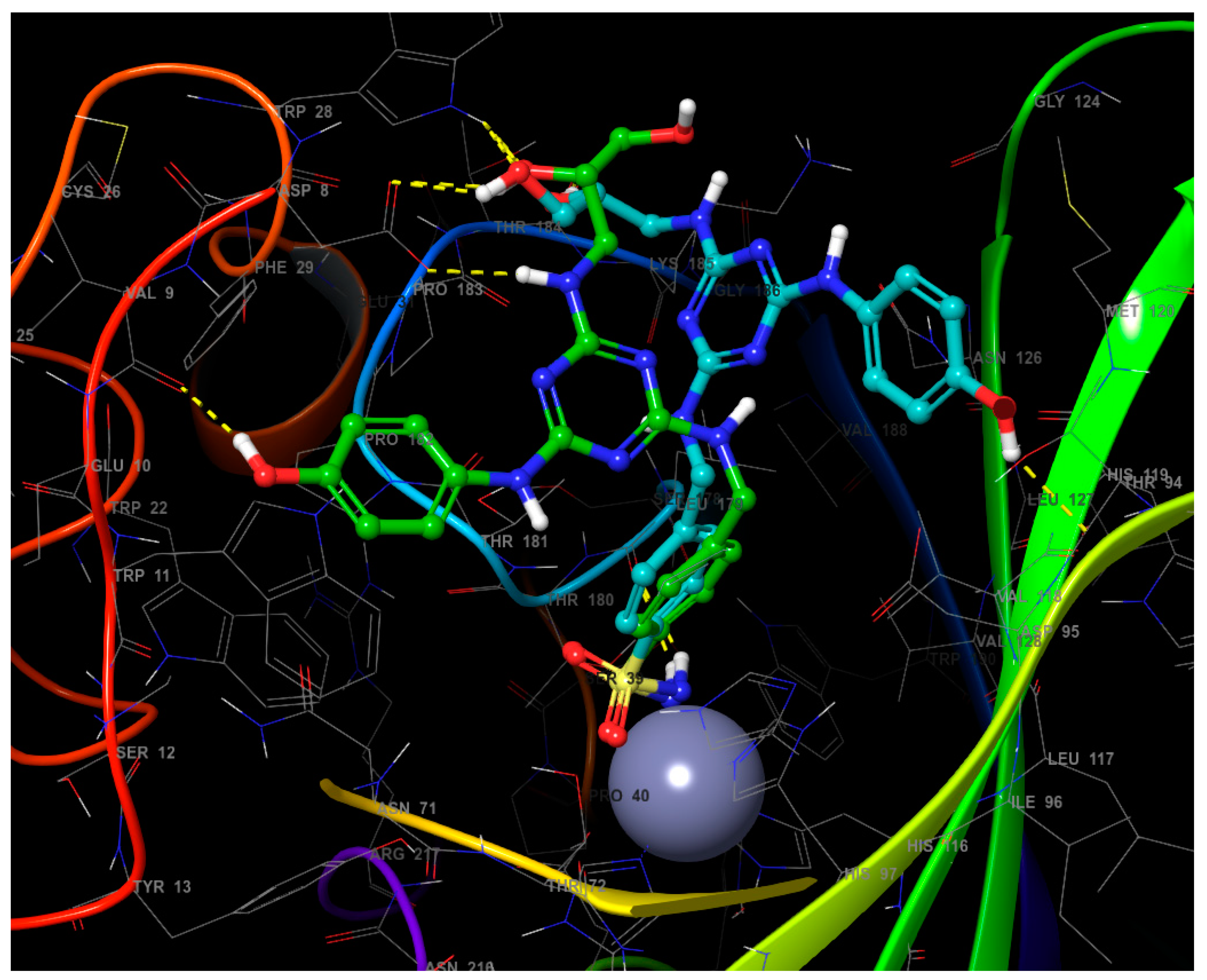
2.3.4. Induced Fit Docking (IFD)
2.3.5. Binding Energy Calculation
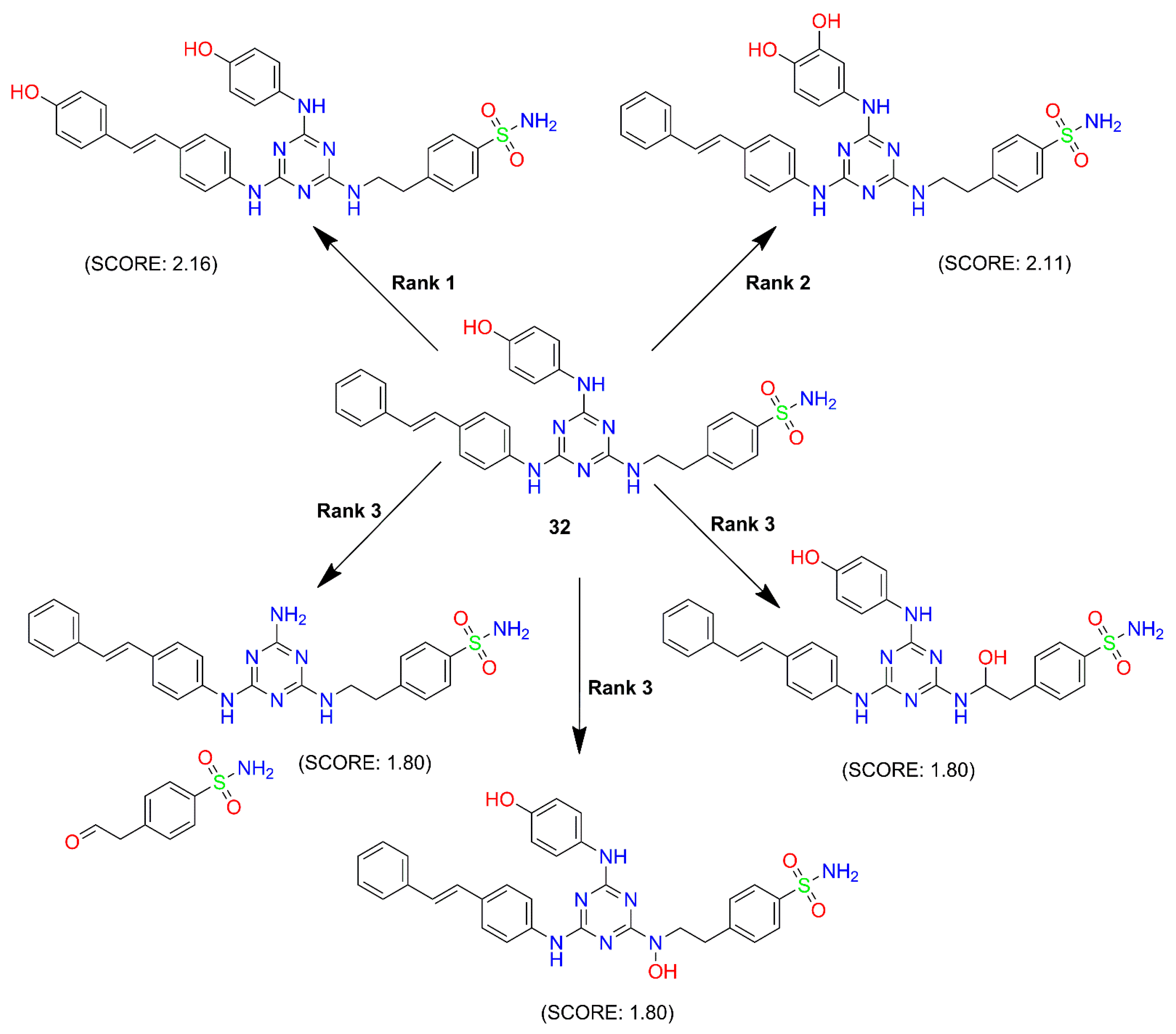
2.3.6. Prediction of ADMET and Fluorescence
3. Conclusions
4. Materials and Methods
4.1. General Informations
4.2. General Synthetic Procedures
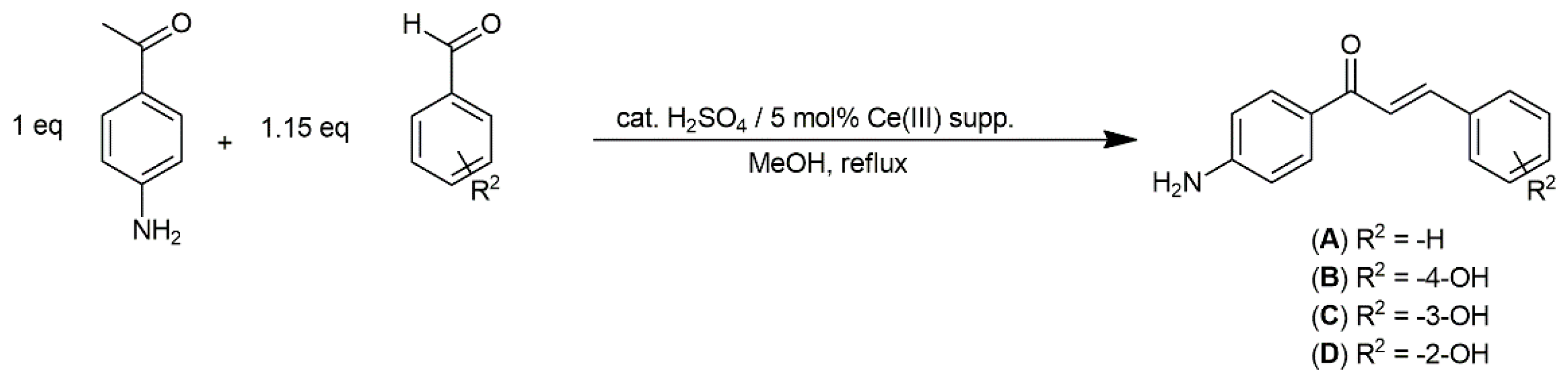
4.2.1. General Method for Synthesis of Chalcones
4.2.2. General Method for Synthesis of Stilbenes
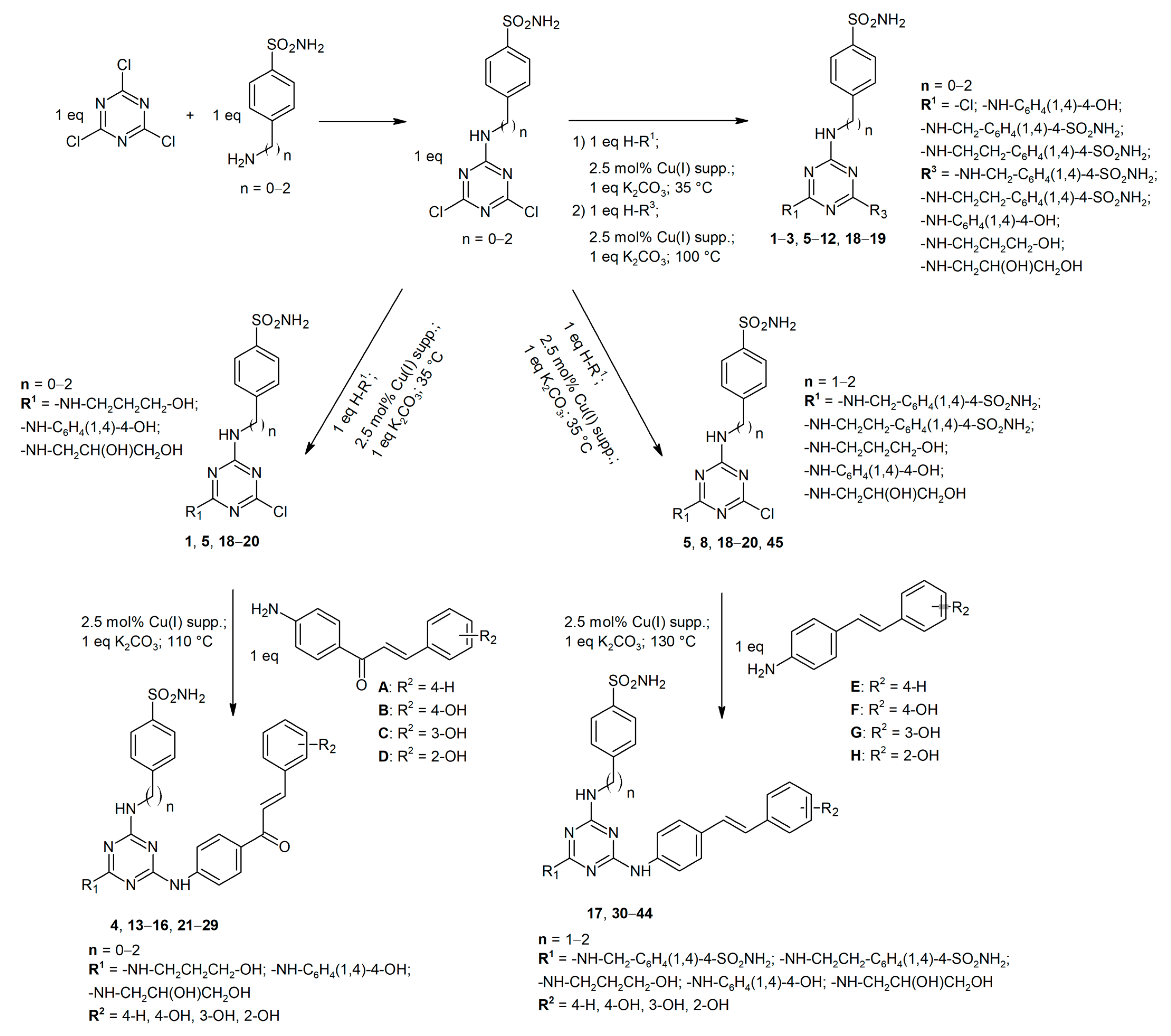
4.2.3. General Method for Synthesis of Starting 4,6-Dichloro-1,3,5-triazin-2-yl Aminobenzenesulfonamides
4.2.4. General Method for Synthesis of Disubstituted Derivatives of 1,3,5-Triazine Containing Aminobenzene Sulfonamide or Aminoalcohol/Phenol Structural Motifs (1, 2, 5–8, 18–20)
4.2.5. General Method for Synthesis of Trisubstituted Derivatives of 1,3,5-Triazine Containing Aminobenzene Sulfonamide or Aminoalcohol/Phenol Structural Motifs (3, 9–12)
4.2.6. General Method for Synthesis of Trisubstituted Derivatives of 1,3,5-Triazine Containing Chalcone Structural Motif (4, 13–16, 21–29)
4.2.7. General Method for Synthesis of Trisubstituted Derivatives of 1,3,5-Triazine Containing Stilbene Structural Motif (17, 30–44)
4.2.8. Characterization of Novel Compounds 4, 16, 21, 25–34, 36, 38, 40–42, 44
4.3. Carbonic Anhydrase Inhibition Assay
4.4. VRE Inhibition Assay
4.5. Cytotoxicity Determination against Human Colorectal Tumor Cell Line (HCT116 p53+/+)
4.5.1. Cell Culture
4.5.2. MTT Assay
4.6. Molecular Modeling
4.6.1. Molecular Docking into hCA IX
4.6.2. Retrieval of Enterococcal CA Protein Sequence
4.6.3. 3D Homology Modeling of Enterococcal CA
4.6.4. Molecular Dynamic (MD) Simulation of Enterococcal CA
4.6.5. Induced Fit Docking to Enterococcal CA
4.6.6. Binding Energy Calculation with Enterococcal CA
4.6.7. Prediction of ADMET and Fluorescence
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAZ | acetazolamide |
| Absorp. | absorption |
| ADMET | absorption, distribution, metabolism, excretion, and toxicity |
| AMP | ampicillin |
| BHI | brain hearth infusion |
| BRZ | brinzolamide |
| CA | carbonic anhydrase |
| cat. | catalyst |
| CLSI | Clinical and Laboratory Standards Institute |
| CNS | central neural system |
| Comp. | compound |
| DCP | dichlorphenamide |
| DMEM | Dulbecco’s modified Eagle medium |
| DMF | N,N-dimethylformamide |
| DMSO | dimethyl sulfoxide |
| DOX | doxorubicin |
| DZA | dorzolamide |
| E. faecalis | Enterococcus faecalis |
| E. faecium | Enterococcus faecium |
| EMA | European Medicines Agency |
| EZA | ethoxzolamide |
| FDA | Food and Drug Administration |
| FTIR | Fourier transform infrared spectroscopy |
| hCA | human carbonic anhydrase |
| HERG | human ether-a-go-go-related gene |
| IFD | induced-fit docking |
| IND | indisulam |
| MD | molecular dynamic |
| MIC | minimum inhibitory concentration |
| MM/GBSA | molecular mechanics generalized Born surface area |
| MRSA | methicillin-resistant S. aureus |
| MTT | methylthiazolyldiphenyl-tetrazolium bromide |
| MZA | methazolamide |
| NMR | nuclear magnetic resonance |
| NT | not tested |
| PBC | periodic boundary conditions |
| PBS | phosphate buffered saline |
| PDB | protein data bank |
| PME | particle mesh Ewald |
| RMSD | root mean square deviation |
| S. aureus | Staphylococcus aureus |
| sp. | species |
| supp. | supported |
| TLC | thin-layer chromatography |
| TMS | tetramethylsilane |
| TOX | toxicity |
| VAN | vancomycin |
| VMD | visual molecular dynamics |
| VRE | vancomycin-resistant enterococci |
| WHO | World Health Organization |
References
- World Health Organization: Fact Sheets–Antibiotic Resistance. Available online: https://www.who.int/news-room/fact-sheets/detail/antibiotic-resistance (accessed on 16 October 2021).
- Supuran, C.T.; Capasso, C. An Overview of the Bacterial Carbonic Anhydrases. Metabolites 2017, 7, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supuran, C.T.; Capasso, C. Antibacterial carbonic anhydrase inhibitors: An update on the recent literature. Expert Opin. Ther. Pat. 2020, 30, 963–982. [Google Scholar] [CrossRef] [PubMed]
- Capasso, C.; Supuran, C.T. Bacterial, fungal and protozoan carbonic anhydrases as drug targets. Expert Opin. Ther. Targets 2015, 19, 1689–1704. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, P.; Seleem, D.M.N.; Supuran, C.T. Bacterial carbonic anhydrases: Underexploited antibacterial therapeutic targets. Future Med. Chem. 2021, 13, 1619–1622. [Google Scholar] [CrossRef] [PubMed]
- Capasso, C.; Supuran, C.T. An overview of the α-, β- and γ-carbonic anhydrases from Bacteria: Can bacterial carbonic anhydrases shed new light on evolution of bacteria? J. Enzyme Inhib. Med. Chem. 2015, 30, 325–332. [Google Scholar] [CrossRef] [Green Version]
- De Luca, V.; Petreni, A.; Nocentini, A.; Scaloni, A.; Supuran, C.T.; Capasso, C. Effect of Sulfonamides and Their Structurally Related Derivatives on the Activity of ι-Carbonic Anhydrase from Burkholderia territorii. Int. J. Mol. Sci. 2021, 22, 571. [Google Scholar] [CrossRef]
- Capasso, C.; Supuran, C.T. An Overview of the Selectivity and Efficiency of the Bacterial Carbonic Anhydrase Inhibitors. Curr. Med. Chem. 2015, 22, 2130–2139. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Capasso, C. Biomedical applications of prokaryotic carbonic anhydrases. Expert Opin. Ther. Pat. 2018, 28, 745–754. [Google Scholar] [CrossRef]
- Bonardi, A.; Nocentini, A.; Osman, S.M.; Alasmary, F.A.; Almutairi, T.M.; Abdullah, D.S.; Gratteri, P.; Supuran, C.T. Inhibition of α-, β- and γ-carbonic anhydrases from the pathogenic bacterium Vibrio cholerae with aromatic sulphonamides and clinically licenced drugs–a joint docking/molecular dynamics study. J. Enzyme Inhib. Med. Chem. 2021, 36, 469–479. [Google Scholar] [CrossRef]
- Del Prete, S.; Isik, S.; Vullo, D.; De Luca, V.; Carginale, V.; Scozzafava, A.; Supuran, C.T.; Capasso, C. DNA Cloning, Characterization, and Inhibition Studies of an α-Carbonic Anhydrase from the Pathogenic Bacterium Vibrio cholerae. J. Med. Chem. 2012, 55, 10742–10748. [Google Scholar] [CrossRef]
- Ceruso, M.; Del Prete, S.; Alothman, Z.; Capasso, C.; Supuran, C.T. Sulfonamides with Potent Inhibitory Action and Selectivity against the α-Carbonic Anhydrase from Vibrio cholerae. ACS Med. Chem. Lett. 2014, 5, 826–830. [Google Scholar] [CrossRef] [Green Version]
- Bua, S.; Berrino, E.; Del Prete, S.; Murthy, V.S.; Vijayakumar, V.; Tamboli, Y.; Capasso, C.; Cerbai, E.; Mugelli, A.; Carta, F.; et al. Synthesis of novel benzenesulfamide derivatives with inhibitory activity against human cytosolic carbonic anhydrase I and II and Vibrio cholerae α- and β-class enzymes. J. Enzyme Inhib. Med. Chem. 2018, 33, 1125–1136. [Google Scholar] [CrossRef] [Green Version]
- Bua, S.; Osman, S.M.; Del Prete, S.; Capasso, C.; Alothman, Z.; Nocentini, A.; Supuran, C.T. Click-tailed benzenesulfonamides as potent bacterial carbonic anhydrase inhibitors for targeting Mycobacterium tuberculosis and Vibrio cholerae. Bioorg. Chem. 2019, 86, 183–186. [Google Scholar] [CrossRef]
- Ceruso, M.; Vullo, D.; Scozzafava, A.; Supuran, C.T. Sulfonamides incorporating fluorine and 1,3,5-triazine moieties are effective inhibitors of three β -class carbonic anhydrases from Mycobacterium tuberculosis. J. Enzyme Inhib. Med. Chem. 2014, 29, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Angeli, A.; Pinteala, M.; Maier, S.S.; Simionescu, B.C.; Milaneschi, A.; Abbas, G.; del Prete, S.; Capasso, C.; Capperucci, A.; Tanini, D.; et al. Evaluation of Thio- and Seleno-Acetamides Bearing Benzenesulfonamide as Inhibitor of Carbonic Anhydrases from Different Pathogenic Bacteria. Int. J. Mol. Sci. 2020, 21, 686–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carta, F.; Maresca, A.; Covarrubias, A.S.; Mowbray, S.L.; Jones, T.A.; Supuran, C.T. Carbonic anhydrase inhibitors. Characterization and inhibition studies of the most active β-carbonic anhydrase from Mycobacterium tuberculosis, Rv3588c. Int. J. Mol. Sci. 2009, 19, 6649–6654. [Google Scholar] [CrossRef]
- Maresca, A.; Carta, F.; Vullo, D.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors. Inhibition of the Rv1284 and Rv3273 β-carbonic anhydrases from Mycobacterium tuberculosis with diazenylbenzenesulfonamides. Int. J. Mol. Sci. 2009, 19, 4929–4932. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Angeli, A.; Bozdag, M.; Carta, F.; Capasso, C.; Farooq, U.; Supuran, C.T. Benzylaminoethylureido-Tailed Benzenesulfonamides Show Potent Inhibitory Activity against Bacterial Carbonic Anhydrases. ChemMedChem 2020, 15, 2444–2447. [Google Scholar] [CrossRef]
- Nocentini, A.; Supuran, C.T.; Capasso, C. An overview on the recently discovered iota-carbonic anhydrases. J. Enzyme Inhib. Med. Chem. 2021, 36, 1988–1995. [Google Scholar] [CrossRef]
- Del Prete, S.; De Luca, V.; Bua, S.; Nocentini, A.; Carginale, V.; Supuran, C.T.; Capasso, C. The Effect of Substituted Benzene-Sulfonamides and Clinically Licensed Drugs on the Catalytic Activity of CynT2, a Carbonic Anhydrase Crucial for Escherichia coli Life Cycle: Characterisation and effects of simple aromatic/heterocyclic sulphonamide inhibitors. Int. J. Mol. Sci. 2020, 21, 4175. [Google Scholar] [CrossRef]
- Del Prete, S.; Bua, S.; Supuran, C.T.; Capasso, C. Escherichia coli γ -carbonic anhydrase: Characterisation and effects of simple aromatic/heterocyclic sulphonamide inhibitors. J. Enzyme Inhib. Med. Chem. 2020, 35, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Cao, X.; Abutaleb, N.S.; Elkashif, A.; Graboski, A.L.; Krabill, A.D.; AbdelKhalek, A.H.; An, W.; Bhardwaj, A.; Seleem, M.N.; et al. Optimization of Acetazolamide-Based Scaffold as Potent Inhibitors of Vancomycin-Resistant Enterococcus: Characterisation and effects of simple aromatic/heterocyclic sulphonamide inhibitors. J. Med. Chem. 2020, 63, 9540–9562. [Google Scholar] [CrossRef]
- Cetinkaya, Y.; Falk, P.; Mayhall, C.G. Vancomycin-Resistant Enterococci: Characterisation and effects of simple aromatic/heterocyclic sulphonamide inhibitors. Clin. Microbiol. Rev. 2000, 13, 686–707. [Google Scholar] [CrossRef]
- Ayobami, O.; Willrich, N.; Reuss, A.; Eckmanns, T.; Markwart, R. The ongoing challenge of vancomycin-resistant Enterococcus faecium and Enterococcus faecalis in Europe: An epidemiological analysis of bloodstream infections. PeerJ 2020, 9, 1180–1193. [Google Scholar] [CrossRef] [PubMed]
- Abutaleb, N.S.; Elhassanny, A.E.M.; Flaherty, D.P.; Seleem, M.N. In vitro and in vivo activities of the carbonic anhydrase inhibitor, dorzolamide, against vancomycin-resistant enterococci: Characterisation and effects of simple aromatic/heterocyclic sulphonamide inhibitors. PeerJ 2021, 9, 686–707. [Google Scholar] [CrossRef]
- Rossolini, G.M.; Arena, F.; Pecile, P.; Pollini, S. Update on the antibiotic resistance crisis: An epidemiological analysis of bloodstream infections. Curr. Opin. Pharmacol. 2014, 18, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.V.; Friedman, D.I. The Idiopathic Intracranial Hypertension Treatment Trial: A Review of the Outcomes. Headache 2017, 57, 1303–1310. [Google Scholar] [CrossRef]
- Supuran, C.T. Emerging role of carbonic anhydrase inhibitors: A Review of the Outcomes. Clin. Sci. 2021, 135, 1233–1249. [Google Scholar] [CrossRef]
- Angeli, A.; Carta, F.; Nocentini, A.; Winum, J.-Y.; Zalubovskis, R.; Akdemir, A.; Onnis, V.; Eldehna, W.M.; Capasso, C.; Simone, G.D.; et al. Carbonic Anhydrase Inhibitors Targeting Metabolism and Tumor Microenvironment: A Review of the Outcomes. Metabolites 2020, 10, 412. [Google Scholar] [CrossRef]
- Kumar, S.; Rulhania, S.; Jaswal, S.; Monga, V. Recent advances in the medicinal chemistry of carbonic anhydrase inhibitors: A Review of the Outcomes. Eur. J. Med. Chem. 2021, 209, 1233–1249. [Google Scholar] [CrossRef]
- Havránková, E.; Csöllei, J.; Pazdera, P. Comparative study for 3, 3´-[(4-X-phenyl)-methanediyl] bis(1H-indoles) synthesis catalyzed by Ce(III) cations. Int. J. Engin. Res. Sci. 2017, 3, 9–14. [Google Scholar]
- Havránková, E.; Pazdera, P. Kabachnik-Fields and Prins-Ritter Synthesis: Application of Ce(III) Supported on a Weakly Acidic Cation-exchanger Resin in Comparative Study. J. Chem. Appl. 2015, 2, 1–6. [Google Scholar]
- Havránková, E.; Pazdera, P. Comparative Studies of Catalytic Application of Cerium(III) Chloride and Resin Supported Cerium(III) in Domino Syntheses of 1,5-Benzodiazepine and 1,3-Diazine Skeletons. J. Chem. Eng. Chem. Res. 2014, 1, 229–237. [Google Scholar]
- Havránková, E.; Pospíšil, P.; Pazdera, P. Synergism of Metal and Organocatalysis in Condensation Reactions of Aromatic Aldehydes with Anilines Affording Imines: Effect of Catalysts on the Base of a Supported Cerium(III) and Proline. Sci. J. Chem. 2014, 2, 1–8. [Google Scholar] [CrossRef]
- Gigante, B.; Esteves, M.A.; Pires, N.; Davies, M.L.; Douglas, P.; Fonseca, S.M.; Burrows, H.D.; Castro, R.A.E.; Pina, J.; Seixas de Melo, J. Synthesis, spectroscopy, photophysics and thermal behaviour of stilbene-based triarylamines with dehydroabietic acid methyl ester moieties. New J. Chem. 2009, 33, 877–885. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.-S.; Chiou, S.-Y.; Liau, K.-L. Fluorescence Enhancement of trans-4-Aminostilbene by N-Phenyl Substitutions: The “Amino Conjugation Effect”. J. Am. Chem. Soc. 2002, 124, 2518–2527. [Google Scholar] [CrossRef]
- Havránková, E.; Csöllei, J.; Pazdera, P. New Approach for the One-Pot Synthesis of 1,3,5-Triazine Derivatives: Application of Cu(I) Supported on a Weakly Acidic Cation-Exchanger Resin in a Comparative Study. Molecules 2019, 24, 3586. [Google Scholar] [CrossRef] [Green Version]
- Havránková, E.; Csöllei, J.; Vullo, D.; Garaj, V.; Pazdera, P.; Supuran, C.T. Novel sulfonamide incorporating piperazine, aminoalcohol and 1,3,5-triazine structural motifs with carbonic anhydrase I, II and IX inhibitory action: Application of Cu(I) Supported on a Weakly Acidic Cation-Exchanger Resin in a Comparative Study. Bioorg. Chem. 2018, 77, 25–37. [Google Scholar] [CrossRef]
- Havránková, E.; Čalkovská, N.; Padrtová, T.; Csöllei, J.; Opatřilová, R.; Pazdera, P. Antioxidative Activity of 1,3,5-Triazine Analogues Incorporating Aminobenzene Sulfonamide, Aminoalcohol/Phenol, Piperazine, Chalcone, or Stilbene Motifs. Molecules 2020, 25, 1787. [Google Scholar] [CrossRef] [Green Version]
- Rivera, C.; Voipio, J.; Kaila, K. Two developmental switches in GABAergic signalling: The K -Cl − cotransporter KCC2 and carbonic anhydrase CAVII. J. Physiol. 2005, 562, 27–36. [Google Scholar] [CrossRef]
- Buonanno, M.; Di Fiore, A.; Langella, E.; D’Ambrosio, K.; Supuran, C.; Monti, S.; De Simone, G. The Crystal Structure of a hCA VII Variant Provides Insights into the Molecular Determinants Responsible for Its Catalytic Behavior: The K -Cl − cotransporter KCC2 and carbonic anhydrase CAVII. Int. J. Mol. Sci. 2018, 19, 1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asiedu, M.; Ossipov, M.H.; Kaila, K.; Price, T.J. Acetazolamide and midazolam act synergistically to inhibit neuropathic pain: The K -Cl − cotransporter KCC2 and carbonic anhydrase CAVII. Pain 2010, 148, 302–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalifah, R.G. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar] [CrossRef]
- Pastorekova, S.; Parkkila, S.; Pastorek, J.; Supuran, C.T. Carbonic anhydrases: Current state of the art, therapeutic applications and future prospects. J. Enzyme Inhib. Med. Chem. 2004, 19, 199–229. [Google Scholar] [CrossRef] [PubMed]
- Garaj, V.; Puccetti, L.; Fasolis, G.; Winum, J.-Y.; Montero, J.-L.; Scozzafava, A.; Vullo, D.; Innocenti, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Synthesis and inhibition of cytosolic/tumor-associated carbonic anhydrase isozymes I, II, and IX with sulfonamides incorporating 1,2,4-triazine moieties. J. Med. Chem 2004, 14, 5427–5433. [Google Scholar] [CrossRef]
- Vullo, D.; Voipio, J.; Innocenti, A.; Rivera, C.; Ranki, H.; Scozzafava, A.; Kaila, K.; Supuran, C.T. Carbonic anhydrase inhibitors. Inhibition of the human cytosolic isozyme VII with aromatic and heterocyclic sulfonamides. Bioorg. Med. Chem. Lett. 2005, 15, 971–976. [Google Scholar] [CrossRef]
- Brzozowski, Z.; Sławiński, J.; Sączewski, F.; Innocenti, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Synthesis and inhibition of the human cytosolic isozymes I and II and transmembrane isozymes IX, XII (cancer-associated) and XIV with 4-substituted 3-pyridinesulfonamides. Eur. J. Med. Chem. 2010, 45, 2396–2404. [Google Scholar] [CrossRef]
- Zadrazilova, I.; Pospisilova, S.; Masarikova, M.; Imramovsky, A.; Ferriz, J.M.; Vinsova, J.; Cizek, A.; Jampilek, J. Salicylanilide carbamates: Promising antibacterial agents with high in vitro activity against methicillin-resistant Staphylococcus aureus (MRSA). Eur. J. Pharm. Sci. 2015, 77, 197–207. [Google Scholar] [CrossRef]
- Oravcova, V.; Zurek, L.; Townsend, A.; Clark, A.B.; Ellis, J.C.; Cizek, A. American crows as carriers of vancomycin-resistant enterococci with vanA gene. Environ. Microbiol. 2014, 16, 939–949. [Google Scholar] [CrossRef]
- Menziani, M.C.; Bendetti, P.G.D.; Richard, E.G. The binding of benzenesulfonamides to carbonic anhydrase enzyme. A molecular mechanics study and quantitative structure−activity relationships. J. Med. Chem. 1989, 32, 951–956. [Google Scholar] [CrossRef]
- Yang, A.-S.; Honig, B. An integrated approach to the analysis and modeling of protein sequences and structures. III. A comparative study of sequence conservation in protein structural families using multiple structural alignments. J. Mol. Biol. 2000, 301, 691–711. [Google Scholar] [CrossRef]
- Abbas, S.H.; Abd El-Hafeez, A.A.; Shoman, M.E.; Montano, M.M.; Hassan, H.A. New quinoline/chalcone hybrids as anti-cancer agents: Design, synthesis, and evaluations of cytotoxicity and PI3K inhibitory activity. Bioorg. Chem. 2019, 82, 360–377. [Google Scholar] [CrossRef]
- Seo, W.D.; Kim, J.H.; Kang, J.E.; Ryu, H.W.; Curtis-Long, M.J.; Lee, H.S.; Yang, M.S.; Park, K.H. Sulfonamide chalcone as a new class of α-glucosidase inhibitors: Design, synthesis, and evaluations of cytotoxicity and PI3K inhibitory activity. Bioorg. Chem. 2005, 15, 5514–5516. [Google Scholar] [CrossRef] [PubMed]
- Moreno, L.; Quiroga, J.; Abonia, R.; Ramírez-Prada, J.; Insuasty, B. Synthesis of New 1,3,5-Triazine-Based 2-Pyrazolines as Potential Anticancer Agents: Design, synthesis, and evaluations of cytotoxicity and PI3K inhibitory activity. Molecules 2018, 23, 1956. [Google Scholar] [CrossRef] [Green Version]
- Leung, S.H.; Angel, S.A. Solvent-Free Wittig Reaction: A Green Organic Chemistry Laboratory Experiment. J. Chem. Educ. 2004, 81, 1181–1186. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, Y.-H.; Kong, X.-W.; Lai, Y.-S.; Ji, H.; Chen, Y.-P.; Peng, S.-X.; Park, K.H. Synthesis and Biological Evaluation of Nitric Oxide-Donating Thalidomide Analogues as Anticancer Agents: A Green Organic Chemistry Laboratory Experiment. J. Chem. Educ. 2009, 6, 466–474. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, P.; Wu, J.; Shang, Z. Metal-free C–N bond-forming reaction: Straightforward synthesis of anilines, through cleavage of aryl C–O bond and amide C–N bond. Tetrahedron Lett. 2013, 54, 3167–3170. [Google Scholar] [CrossRef]
- Maresca, A.; Carta, F.; Vullo, D.; Supuran, C.T. Dithiocarbamates strongly inhibit the β-class carbonic anhydrases from Mycobacterium tuberculosis: A new class of carbonic anhydrase inhibitors. Crystallographic and kinetic investigations. J. Enzyme Inhib. Med. Chem. 2013, 28, 407–411. [Google Scholar] [CrossRef]
- Carta, F.; Aggarwal, M.; Maresca, A.; Scozzafava, A.; McKenna, R.; Supuran, C.T. Dithiocarbamates: A new class of carbonic anhydrase inhibitors. Crystallographic and kinetic investigations. Chem. Comm. 2012, 48, 199–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekinci, D.; Kurbanoglu, N.I.; Salamcı, E.; Şentürk, M.; Supuran, C.T. Carbonic anhydrase inhibitors: Inhibition of human and bovine isoenzymes by benzenesulphonamides, cyclitols and phenolic compounds. J. Enzyme Inhib. Med. Chem. 2012, 27, 845–848. [Google Scholar] [CrossRef]
- Ekinci, D.; Karagoz, L.; Ekinci, D.; Senturk, M.; Supuran, C.T. Carbonic anhydrase inhibitors: In vitro inhibition of α isoforms (hCA I, hCA II, bCA III, hCA IV) by flavonoids: Inhibition of human and bovine isoenzymes by benzenesulphonamides, cyclitols and phenolic compounds. J. Enzyme Inhib. Med. Chem. 2013, 28, 283–288. [Google Scholar] [CrossRef]
- Alp, C.; Maresca, A.; Alp, N.A.; Gültekin, M.S.; Ekinci, D.; Scozzafava, A.; Supuran, C.T. Secondary/tertiary benzenesulfonamides with inhibitory action against the cytosolic human carbonic anhydrase isoforms I and II: Inhibition of human and bovine isoenzymes by benzenesulphonamides, cyclitols and phenolic compounds. J. Enzyme Inhib. Med. Chem. 2013, 28, 294–298. [Google Scholar] [CrossRef]
- Boztaş, M.; Çetinkaya, Y.; Topal, M.; Gülçin, İ.; Menzek, A.; Şahin, E.; Tanc, M.; Supuran, C.T. Synthesis and Carbonic Anhydrase Isoenzymes I, II, IX, and XII Inhibitory Effects of Dimethoxybromophenol Derivatives Incorporating Cyclopropane Moieties: Inhibition of human and bovine isoenzymes by benzenesulphonamides, cyclitols and phenolic compounds. J. Med. Chem. 2015, 58, 640–650. [Google Scholar] [CrossRef]
- Carta, F.; Vullo, D.; Maresca, A.; Scozzafava, A.; Supuran, C.T. Mono-/dihydroxybenzoic acid esters and phenol pyridinium derivatives as inhibitors of the mammalian carbonic anhydrase isoforms I, II, VII, IX, XII and XIV: Inhibition of human and bovine isoenzymes by benzenesulphonamides, cyclitols and phenolic compounds. J. Med. Chem. 2013, 21, 1564–1569. [Google Scholar] [CrossRef] [Green Version]
- M100–S22; Clinical and Laboratory Standards Institute Performance Standards for Antimicrobial Susceptibility Testing; The 8th Informational Supplement Document. CLSI: New York, NY, USA, 2012.
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to Sustain G2 Arrest After DNA Damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Alterio, V.; Hilvo, M.; Di Fiore, A.; Supuran, C.T.; Pan, P.; Parkkila, S.; Scaloni, A.; Pastorek, J.; Pastorekova, S.; Pedone, C.; et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc. Natl. Acad. Sci. USA 2009, 106, 16233–16238. [Google Scholar] [CrossRef] [Green Version]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aid. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozyreva, V.K.; Truong, C.-L.; Greninger, A.L.; Crandall, J.; Mukhopadhyay, R.; Chaturvedi, V.; Diekema, D.J. Validation and Implementation of Clinical Laboratory Improvements Act-Compliant Whole-Genome Sequencing in the Public Health Microbiology Laboratory. J. Clin. Microbiol. 2017, 55, 2502–2520. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, G.N.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963, 7, 95–99. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; MacKerell, A.D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 25, 2135–2145. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1998, 18, 1463–1472. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects: A next generation energy model for high resolution protein structure modeling. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comp.Aided Mol. Design 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comp.Aided Mol. Design 2010, 24, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Fisher, S.Z.; Aggarwal, M.; Kovalevsky, A.Y.; Silverman, D.N.; McKenna, R. Neutron Diffraction of Acetazolamide-Bound Human Carbonic Anhydrase II Reveals Atomic Details of Drug Binding: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Am. Chem. Soc. 2012, 134, 14726–14729. [Google Scholar] [CrossRef] [Green Version]
- de Bruyn Kops, C.; Stork, C.; Šícho, M.; Kochev, N.; Svozil, D.; Jeliazkova, N.; Kirchmair, J. GLORY: Generator of the Structures of Likely Cytochrome P450 Metabolites Based on Predicted Sites of Metabolism. Front. Chem. 2019, 7, 14726–14729. [Google Scholar] [CrossRef] [PubMed]
- Stork, C.; Embruch, G.; Šícho, M.; de Bruyn Kops, C.; Chen, Y.; Svozil, D.; Kirchmair, J. NERDD: A web portal providing access to in silico tools for drug discovery. Bioinformatics 2019, 7, 14726–14729. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-Y.; Dong, J.; Yang, Z.-J.; Yin, M.; Jiang, H.-L.; Lu, A.-P.; Chen, X.; Hou, T.-J.; Cao, D.-S. ChemFLuo: A web-server for structure analysis and identification of fluorescent compounds. Brief. Bioinformatics 2021, 22, 14726–14729. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Catalytic System | |||||
|---|---|---|---|---|---|---|
| H2SO4 a | Ce(III) Supp. b | H2SO4a /Ce(III) Supp. b | ||||
| Time (h) | %Yield | Time (h) | %Yield | Time (h) | %Yield | |
| A | 40 | 87 | 38 | 85 | 12 | 88 |
| B | 24 | 90 | 24 | 83 | 6 | 89 |
| C | 40 | 72 | 36 | 72 | 8 | 83 |
| D | 36 | 81 | 34 | 82 | 10 | 93 |
| Comp. | n | R1 | R2 or R3 | KI (nM) a | Selectivity Ratio | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| hCA I | hCA II | hCA VII | hCA IX | hCA XII | KI (hCA II)/KI (hCA VII) | KI (hCA II) /KI (hCA IX) | KI (hCA II) /KI (hCA XII) | ||||
| 1 | 0 | Cl | NH-CH2CH2CH2OH | 26.8 | 44.8 | 127.7 | 228.9 | NT | 0.35 | 0.19 | NT |
| 2 | 0 | Cl | NH-CH2CH2-C6H4(1,4)-4-SO2NH2 | 388.5 e | 41.8 e | NT | 28.0 e | NT | NT | 1.49 e | NT |
| 3 | 2 | NH-C6H4(1,4)-4-SO2NH2 | NH-CH2CH2-C6H4(1,4)-4-SO2NH2 | 8.5e | 4.8e | NT | 2.0e | NT | NT | 2.40e | NT |
| 4 | 0 | NH-CH2CH2CH2OH | 3-OH | 7523 | 3640 | NT | 27.5 | 65.8 | NT | 132.36 | 55.32 |
| 5 | 1 | Cl | NH-CH2CH2CH2OH | 3534 e | 57.4 e | 331.5 | 635.1 e | NT | 0.17 | 0.09 e | NT |
| 6 | 1 | Cl | NH-C6H4(1,4)-4-OH | 16.7e | 7.4e | NT | 0.4e | NT | NT | 18.50e | NT |
| 7 | 1 | Cl | NH-CH2-C6H4(1,4)-4-SO2NH2 | 731.3 e | 75.9 e | NT | 307.7 e | NT | NT | 0.25 e | NT |
| 8 | 1 | Cl | NH-CH2CH2-C6H4(1,4)-4-SO2NH2 | 391.7 e | 18.5e | NT | 190.0 e | NT | NT | 0.10 e | NT |
| 9 | 1 | NH-C6H4(1,4)-4-OH | NH-CH2CH(OH)CH2OH | 491.5 e | 66.9 e | NT | 32.8 e | NT | NT | 2.04 e | NT |
| 10 | 1 | NH-CH2-C6H4(1,4)-4-SO2NH2 | NH-CH2CH(OH)CH2OH | 76.1 e | 5.1e | NT | 1.0e | NT | NT | 5.10e | NT |
| 11 | 1 | NH-CH2-C6H4(1,4)-4-SO2NH2 | NH-CH2CH2-C6H4(1,4)-4-SO2NH2 | 313.5 e | 7.8e | NT | 2.4e | NT | NT | 3.25e | NT |
| 12 | 2 | NH-CH2-C6H4(1,4)-4-SO2NH2 | NH-CH2CH2-C6H4(1,4)-4-SO2NH2 | 62.3 e | 5.6e | NT | 1.6e | NT | NT | 3.50e | NT |
| 13 | 1 | NH-CH2CH2CH2OH | -H | 1960 | 277.4 | 95.7 | 4626 | NT | 2.90 | 0.06 | NT |
| 14 | 1 | NH-CH2CH2CH2OH | 2-OH | 5399 | 35.4 | 180.4 | 622.7 | NT | 0.20 | 0.06 | NT |
| 15 | 1 | NH-CH2CH2CH2OH | 3-OH | >10,000 | 3017 | 25.9 | >10,000 | NT | 116.48 | 0.30 | NT |
| 16 | 1 | NH-CH2CH2CH2OH | 4-OH | 799.9 | 36.0 | 201.0 | 160.9 | NT | 0.18 | 0.23 | NT |
| 17 | 1 | NH-CH2CH2CH2OH | 3-OH | 358.2 | 46.8 | NT | 22.2 | 492.9 | NT | 2.11 | 0.09 |
| 18 | 2 | Cl | NH-CH2CH2CH2OH | 52.0 e | 36.1 e | 75.7 | 231.2 e | NT | 0.48 | 0.16 e | NT |
| 19 | 2 | Cl | NH-CH2CH(OH)CH2OH | 9290 e | 78.0 e | 40.7 | 646.9 e | NT | 1.92 | 0.12 e | NT |
| 20 | 2 | Cl | NH-C6H4(1,4)-4-OH | 462.0 e | 65.5 e | NT | 192.0 e | NT | NT | 0.34 e | NT |
| 21 | 2 | NH-C6H4(1,4)-4-OH | -H | >10,000 | 3158 | 810.2 | >10,000 | NT | 3.90 | 0.32 | NT |
| 22 | 2 | NH-CH2CH2CH2OH | 2-OH | 7703 | 938.9 | 283.5 | >10,000 | NT | 3.32 | 0.09 | NT |
| 23 | 2 | NH-CH2CH(OH)CH2OH | 2-OH | >10,000 | 83.0 | 88.7 | >10,000 | NT | 0.94 | 0.01 | NT |
| 24 | 2 | NH-C6H4(1,4)-4-OH | 2-OH | 5013 | 63.8 | 280.8 | 227.4 | NT | 0.23 | 0.28 | NT |
| 25 | 2 | NH-CH2CH2CH2OH | 3-OH | 7487 | 4128 | 185.8 | >10,000 | NT | 25.99 | 0.41 | NT |
| 26 | 2 | NH-C6H4(1,4)-4-OH | 3-OH | 6627 | 1335 | 42.1 | >10,000 | NT | 31.71 | 0.13 | NT |
| 27 | 2 | NH-CH2CH2CH2OH | 4-OH | 8958 | 28.8 | 458.6 | >10,000 | NT | 0.06 | 0.00 | NT |
| 28 | 2 | NH-CH2CH(OH)CH2OH | 4-OH | 8223 | 184.4 | 594.2 | 8461 | NT | 0.31 | 0.02 | NT |
| 29 | 2 | NH-C6H4(1,4)-4-OH | 4-OH | 5806 | 730.4 | 88.6 | >10,000 | NT | 8.24 | 0.07 | NT |
| 30 | 2 | NH-CH2CH2CH2OH | -H | 5697 | 5133 | NT | 12.1 | 7.6 | NT | 424.21 | 675.39 |
| 31 | 2 | NH-CH2CH(OH)CH2OH | -H | 36.9 | 335.5 | NT | 248.3 | 4.4 | NT | 1.35 | 76.25 |
| 32 | 2 | NH-C6H4(1,4)-4-OH | -H | 90.6 | 7488 | NT | 2468 | 5.9 | NT | 3.03 | 1269.15 |
| 33 | 1 | NH-CH2CH2-C6H4(1,4)-4-SO2NH2 | -H | 48.0 | 57.9 | NT | 1967 | 6.2 | NT | 0.03 | 9.34 |
| 34 | 2 | NH-CH2CH(OH)CH2OH | 2-OH | 1672 | 5632 | NT | 2713 | 8.5 | NT | 2.08 | 662.59 |
| 35 | 2 | NH-C6H4(1,4)-4-OH | 2-OH | 3937 | 5470 | NT | 238.5 | 37.1 | NT | 22.94 | 147.44 |
| 36 | 1 | NH-CH2CH2-C6H4(1,4)-4-SO2NH2 | 2-OH | 447.5 | 82.0 | NT | 2722 | 8.3 | NT | 0.03 | 9.88 |
| 37 | 2 | NH-CH2CH2CH2OH | 3-OH | 675.3 | 73.5 | NT | 26.4 | 601.2 | NT | 2.78 | 0.12 |
| 38 | 2 | NH-CH2CH(OH)CH2OH | 3-OH | 6726 | 4443 | NT | 17.3 | 84.5 | NT | 256.82 | 52.58 |
| 39 | 2 | NH-C6H4(1,4)-4-OH | 3-OH | 742.5 | 154.1 | NT | 876.0 | 68.5 | NT | 0.17 | 2.25 |
| 40 | 1 | NH-CH2CH2-C6H4(1,4)-4-SO2NH2 | 3-OH | 48.8 | 18.9 | NT | 20.3 | 60.5 | NT | 0.93 | 0.31 |
| 41 | 2 | NH-CH2CH2-C6H4(1,4)-4-SO2NH2 | 3-OH | 608.2 | 41.4 | NT | 31.1 | 62.2 | NT | 1.33 | 0.67 |
| 42 | 2 | NH-CH2CH(OH)CH2OH | 4-OH | 43.5 | 96.3 | NT | 258.0 | 9.7 | NT | 0.37 | 9.93 |
| 43 | 2 | NH-C6H4(1,4)-4-OH | 4-OH | 4061 | 4837 | NT | 25.7 | 702.5 | NT | 188.21 | 6.88 |
| 44 | 1 | NH-CH2CH2-C6H4(1,4)-4-SO2NH2 | 4-OH | 858.9 | 252.0 | NT | 249.3 | 10.0 | NT | 1.01 | 25.2 |
| AAZ | - | - | - | 250.0 b | 12.1 b | 2.5 b | 25.8 b | 5.7 | 4.84 | 0.47 | 2.13 |
| BRZ | - | - | - | NT | 3.0 b | 2.8 c | 37.0 b | NT | 1.07 | 0.08 | NT |
| DCP | - | - | - | 1200.0 b | 38.0 b | 26.5 c | 50.0 b | 50.0 d | 1.44 | 0.76 | 0.76 |
| DZA | - | - | - | 50 000.0b | 9.0 b | 3.5 c | 52.0 b | NT | 2.57 | 0.17 | NT |
| EZA | - | - | - | 25.0 b | 8.0 b | 0.8 c | 34.0 b | 22.0 d | 10.26 | 0.23 | 0.36 |
| IND | - | - | - | 31.0 b | 15.0 b | NT | 24.0 b | 3.4 d | NT | 0.62 | 4.41 |
| MZA | - | - | - | 50.0 b | 14.0 b | 2.1 c | 27.0 b | 3.4 d | 6.67 | 0.52 | 4.12 |
| Comp. | n | R1 | R2 or R3 | MIC [µM] | |||
|---|---|---|---|---|---|---|---|
| EF | VRE 342 B | VRE 368 | VRE 725 B | ||||
| 1 | 0 | Cl | NH-CH2CH2CH2OH | >713 | >713 | >713 | >713 |
| 2 a | 0 | Cl | NH-CH2CH2-C6H4(1,4)-4-SO2NH2 | 132 | >528 | >528 | >528 |
| 3 a | 2 | NH-C6H4(1,4)-4-SO2NH2 | NH-CH2CH2-C6H4(1,4)-4-SO2NH2 | >395 | >395 | >395 | >395 |
| 4 | 0 | NH-CH2CH2CH2OH | 3-OH | >479 | >479 | >479 | >479 |
| 5 a | 1 | Cl | NH-CH2CH2CH2OH | 343 | 343 | 343 | 686 |
| 6 a | 1 | Cl | NH-C6H4(1,4)-4-OH | 157 | 157 | 314 | 157 |
| 7 a | 1 | Cl | NH-CH2-C6H4(1,4)-4-SO2NH2 | 16.49 | 65.98 | 32.99 | 131 |
| 8 a | 1 | Cl | NH-CH2CH2-C6H4(1,4)-4-SO2NH2 | 128 | 128 | 64.26 | >514 |
| 9 a | 1 | NH-C6H4(1,4)-4-OH | NH-CH2CH(OH)CH2OH | 34.67 | 34.67 | 69.34 | 34.67 |
| 10 a | 1 | NH-CH2-C6H4(1,4)-4-SO2NH2 | NH-CH2CH(OH)CH2OH | >475 | >475 | >475 | >475 |
| 11 a | 1 | NH-CH2-C6H4(1,4)-4-SO2NH2 | NH-CH2CH2-C6H4(1,4)-4-SO2NH2 | 197 | 98.80 | 197 | 98.80 |
| 12 a | 2 | NH-CH2-C6H4(1,4)-4-SO2NH2 | NH-CH2CH2-C6H4(1,4)-4-SO2NH2 | >387 | >387 | >387 | >387 |
| 13 b | 1 | NH-CH2CH2CH2OH | -H | 228 | 228 | 114 | 228 |
| 14 b | 1 | NH-CH2CH2CH2OH | 2-OH | 111 | 111 | 111 | >444 |
| 15 b | 1 | NH-CH2CH2CH2OH | 3-OH | 222 | 222 | 111 | 111 |
| 16 | 1 | NH-CH2CH2CH2OH | 4-OH | >444 | >444 | >444 | >444 |
| 17 b | 1 | NH-CH2CH2CH2OH | 3-OH | >467 | >467 | 467 | >467 |
| 18 a | 2 | Cl | NH-CH2CH2CH2OH | 330 | 330 | 661 | 661 |
| 19 a | 2 | Cl | NH-CH2CH(OH)CH2OH | >635 | >635 | >635 | >635 |
| 20 a | 2 | Cl | NH-C6H4(1,4)-4-OH | 38.01 | 76.03 | 38.01 | >608 |
| 21 | 2 | NH-C6H4(1,4)-4-OH | -H | 26.33 | 26.33 | 26.33 | 26.33 |
| 22 b | 2 | NH-CH2CH2CH2OH | 2-OH | 108 | 108 | 108 | 108 |
| 23 b | 2 | NH-CH2CH(OH)CH2OH | 2-OH | 52.83 | 52.83 | 52.83 | 105 |
| 24 b | 2 | NH-C6H4(1,4)-4-OH | 2-OH | 51.30 | 51.30 | 51.30 | 102 |
| 25 | 2 | NH-CH2CH2CH2OH | 3-OH | 27.13 | 27.13 | 27.13 | 108 |
| 26 | 2 | NH-C6H4(1,4)-4-OH | 3-OH | 51.30 | 51.30 | 25.65 | 51.30 |
| 27 | 2 | NH-CH2CH2CH2OH | 4-OH | >434 | >434 | >434 | 434 |
| 28 | 2 | NH-CH2CH(OH)CH2OH | 4-OH | >444 | >444 | >444 | >444 |
| 29 | 2 | NH-C6H4(1,4)-4-OH | 4-OH | 25.65 | 25.65 | 102 | 51.30 |
| 30 | 2 | NH-CH2CH2CH2OH | -H | >469 | >469 | >469 | >469 |
| 31 | 2 | NH-CH2CH(OH)CH2OH | -H | >455 | >455 | >455 | >455 |
| 32 | 2 | NH-C6H4(1,4)-4-OH | -H | 13.80 | 27.60 | 27.60 | 55.20 |
| 33 | 1 | NH-CH2CH2-C6H4(1,4)-4-SO2NH2 | -H | 97.44 | 194 | 389 | 194 |
| 34 | 2 | NH-CH2CH(OH)CH2OH | 2-OH | >443 | >443 | >443 | >443 |
| 35 b | 2 | NH-C6H4(1,4)-4-OH | 2-OH | >429 | >429 | >429 | >429 |
| 36 | 1 | NH-CH2CH2-C6H4(1,4)-4-SO2NH2 | 2-OH | >380 | >380 | >380 | >380 |
| 37 b | 2 | NH-CH2CH2CH2OH | 3-OH | >455 | >455 | >455 | >455 |
| 38 | 2 | NH-CH2CH(OH)CH2OH | 3-OH | >443 | >443 | >443 | >443 |
| 39 b | 2 | NH-C6H4(1,4)-4-OH | 3-OH | >429 | >429 | >429 | >429 |
| 40 | 1 | NH-CH2CH2-C6H4(1,4)-4-SO2NH2 | 3-OH | 190 | 190 | 380 | 190 |
| 41 | 2 | NH-CH2CH2-C6H4(1,4)-4-SO2NH2 | 3-OH | 372 | >372 | >372 | >372 |
| 42 | 2 | NH-CH2CH(OH)CH2OH | 4-OH | 55.39 | 110 | 221 | 221 |
| 43 b | 2 | NH-C6H4(1,4)-4-OH | 4-OH | 429 | >429 | >429 | >429 |
| 44 | 1 | NH-CH2CH2-C6H4(1,4)-4-SO2NH2 | 4-OH | 380 | 190 | >380 | 380 |
| AMP | – | – | – | 11.5 | 11.5 | 11.5 | 5.72 |
| VAN | – | – | – | – | 353 | 353 | 706 |
| Comp. | n | R1 | R2 or R3 | Tox IC50 [µM] |
|---|---|---|---|---|
| 7 a | 1 | Cl | NH-CH2-C6H4(1,4)-4-SO2NH2 | 6.19 ± 2.15 |
| 9 a | 1 | NH-C6H4(1,4)-4-OH | NH-CH2CH(OH)CH2OH | > 50 |
| 20 a | 2 | Cl | NH-C6H4(1,4)-4-OH | > 50 |
| 21 | 2 | NH-C6H4(1,4)-4-OH | -H | > 50 |
| 24 b | 2 | NH-C6H4(1,4)-4-OH | 2-OH | > 50 |
| 25 | 2 | NH-CH2CH2CH2OH | 3-OH | > 50 |
| 26 | 2 | NH-C6H4(1,4)-4-OH | 3-OH | > 50 |
| 29 | 2 | NH-C6H4(1,4)-4-OH | 4-OH | > 50 |
| 32 | 2 | NH-C6H4(1,4)-4-OH | -H | 6.51 ± 1.84 |
| DOX | 0.09 ± 1.12 |
| Ligand | KI (hCA IX) [nM] | Docking Score [kcal/mol] | E (MM/GBSA) [kcal/mol] |
|---|---|---|---|
| 4 | 27.5 | −8.612 | −55.08 |
| 17 | 22.2 | −8.499 | −67.42 |
| 30 | 12.1 | −7.124 | −69.19 |
| 37 | 26.4 | −7.627 | −75.03 |
| 38 | 17.3 | −7.664 | −90.82 |
| 40 | 20.3 | −7.068 | −59.25 |
| 41 | 31.1 | −7.088 | −65.38 |
| 43 | 25.7 | −6.958 | −78.89 |
| Ligand | MIC (E. faecalis) [µM] | Docking Score [kcal/mol] | IFD Score [kcal/mol] | E (MM/GBSA) [kcal/mol] |
|---|---|---|---|---|
| 7 | 16.49 | −7.324 | 3502.93 | −51.59 |
| 9 S-enantiomer | 34.67 | −7.930 | 3489.76 | −60.74 |
| 9 R-enantiomer | −7.539 | 3497.22 | −53.54 | |
| 21 | 26.33 | −10.013 | 3483.60 | −69.33 |
| 25 | 27.13 | −9.829 | 3492.28 | −57.21 |
| 32 | 13.80 | −9.126 | 3495.30 | −75.35 |
| Ligand | QPlogPoct/w | QPlogS | Oral Absorp.% | QPlogHERG | QPlogBB |
|---|---|---|---|---|---|
| 7 | −0.1 | −4.3 | 14 | −6.6 | −3.7 |
| 9 S-enantiomer | −0.5 | −3.4 | 11 | −6.8 | −4.2 |
| 9 R-enantiomer | −0.7 | −3.4 | 7 | −6.9 | −4.5 |
| 21 | 3.5 | −7.3 | 27 | −9.1 | −4.5 |
| 25 | 2.2 | −6.0 | 17 | −8.2 | −4.8 |
| 26 | 2.5 | −6.5 | 11 | −8.7 | −5.2 |
| 32 | 4.5 | −7.8 | 58 | −9.4 | −3.5 |
| Ligand | Probability-G | Probability-B |
|---|---|---|
| 7 | 0.51267 | 0.0162054 |
| 9 | 0.249959 | 0.00907222 |
| 21 | 0.73961 | 0.02375 |
| 25 | 0.641677 | 0.0461065 |
| 26 | 0.688407 | 0.0478596 |
| 32 | 0.649607 | 0.0226621 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Havránková, E.; Garaj, V.; Mascaretti, Š.; Angeli, A.; Soldánová, Z.; Kemka, M.; Motyčka, J.; Brázdová, M.; Csöllei, J.; Jampílek, J.; et al. Novel 1,3,5-Triazinyl Aminobenzenesulfonamides Incorporating Aminoalcohol, Aminochalcone and Aminostilbene Structural Motifs as Potent Anti-VRE Agents, and Carbonic Anhydrases I, II, VII, IX, and XII Inhibitors. Int. J. Mol. Sci. 2022, 23, 231. https://doi.org/10.3390/ijms23010231
Havránková E, Garaj V, Mascaretti Š, Angeli A, Soldánová Z, Kemka M, Motyčka J, Brázdová M, Csöllei J, Jampílek J, et al. Novel 1,3,5-Triazinyl Aminobenzenesulfonamides Incorporating Aminoalcohol, Aminochalcone and Aminostilbene Structural Motifs as Potent Anti-VRE Agents, and Carbonic Anhydrases I, II, VII, IX, and XII Inhibitors. International Journal of Molecular Sciences. 2022; 23(1):231. https://doi.org/10.3390/ijms23010231
Chicago/Turabian StyleHavránková, Eva, Vladimír Garaj, Šárka Mascaretti, Andrea Angeli, Zuzana Soldánová, Miroslav Kemka, Jozef Motyčka, Marie Brázdová, Jozef Csöllei, Josef Jampílek, and et al. 2022. "Novel 1,3,5-Triazinyl Aminobenzenesulfonamides Incorporating Aminoalcohol, Aminochalcone and Aminostilbene Structural Motifs as Potent Anti-VRE Agents, and Carbonic Anhydrases I, II, VII, IX, and XII Inhibitors" International Journal of Molecular Sciences 23, no. 1: 231. https://doi.org/10.3390/ijms23010231