
(20S) Ginsenoside Rh2 Exerts Its Anti-Tumor Effect by Disrupting the HSP90A-Cdc37 System in Human Liver Cancer Cells


Abstract
:1. Introduction
2. Results
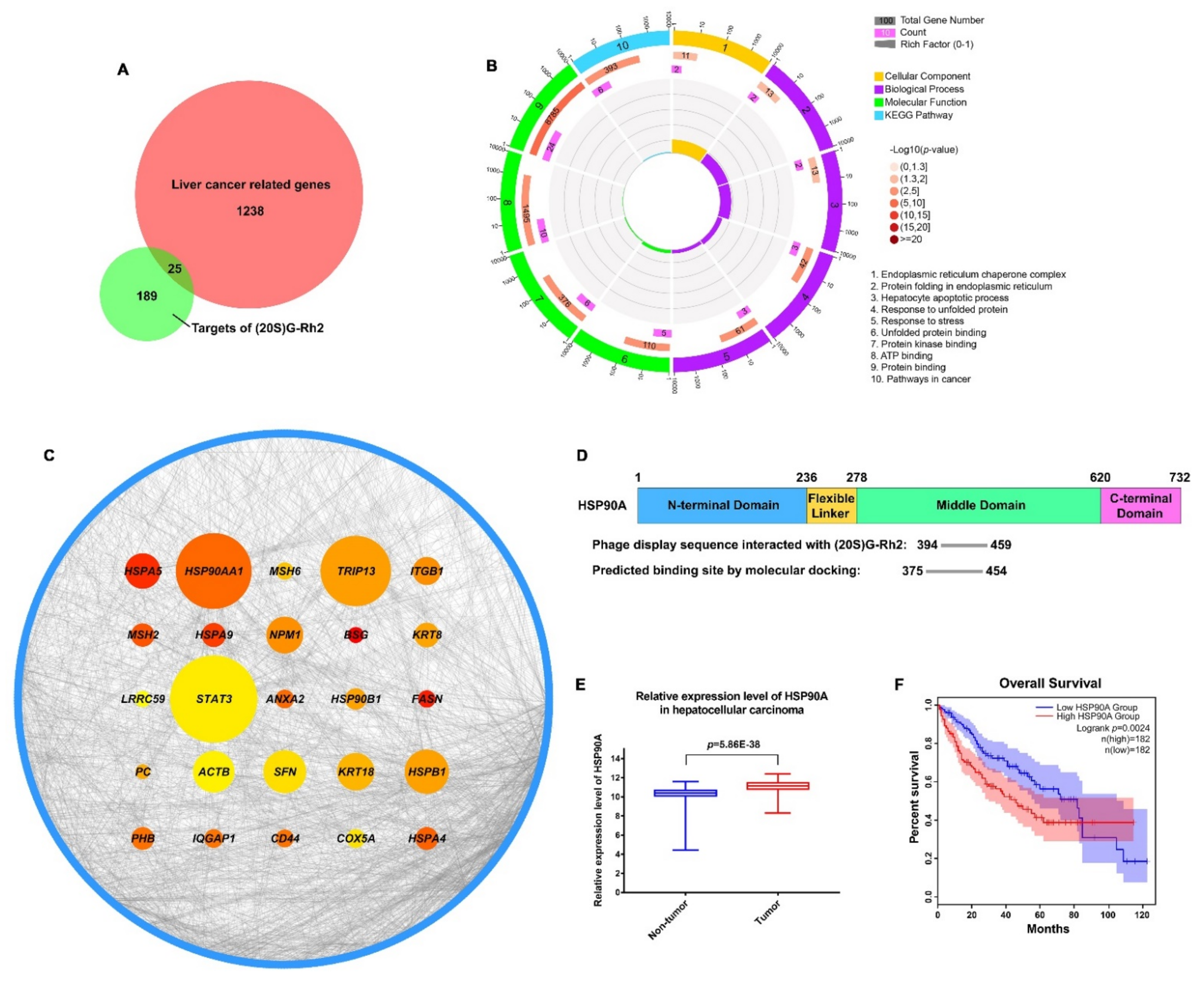
2.1. HSP90AA1 May Serve as a Critical Liver Cancer-Related Gene Targeted by (20S) G-Rh2
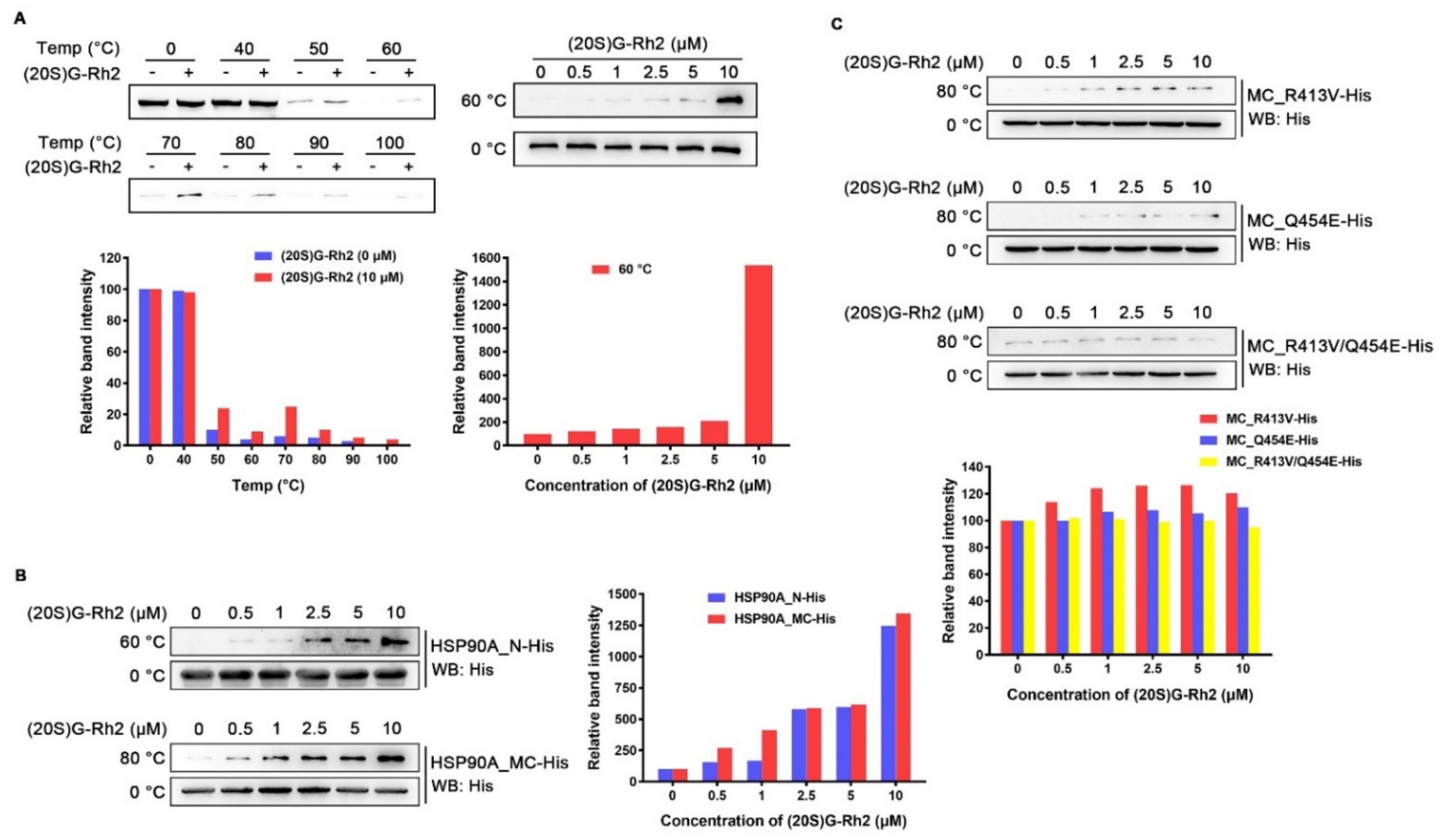
2.2. Binding Modes of HSP90A and (20S) G-Rh2 Were Predicted by Molecular Docking
2.3. (20S) G-Rh2 Interacted with HSP90A In Vitro
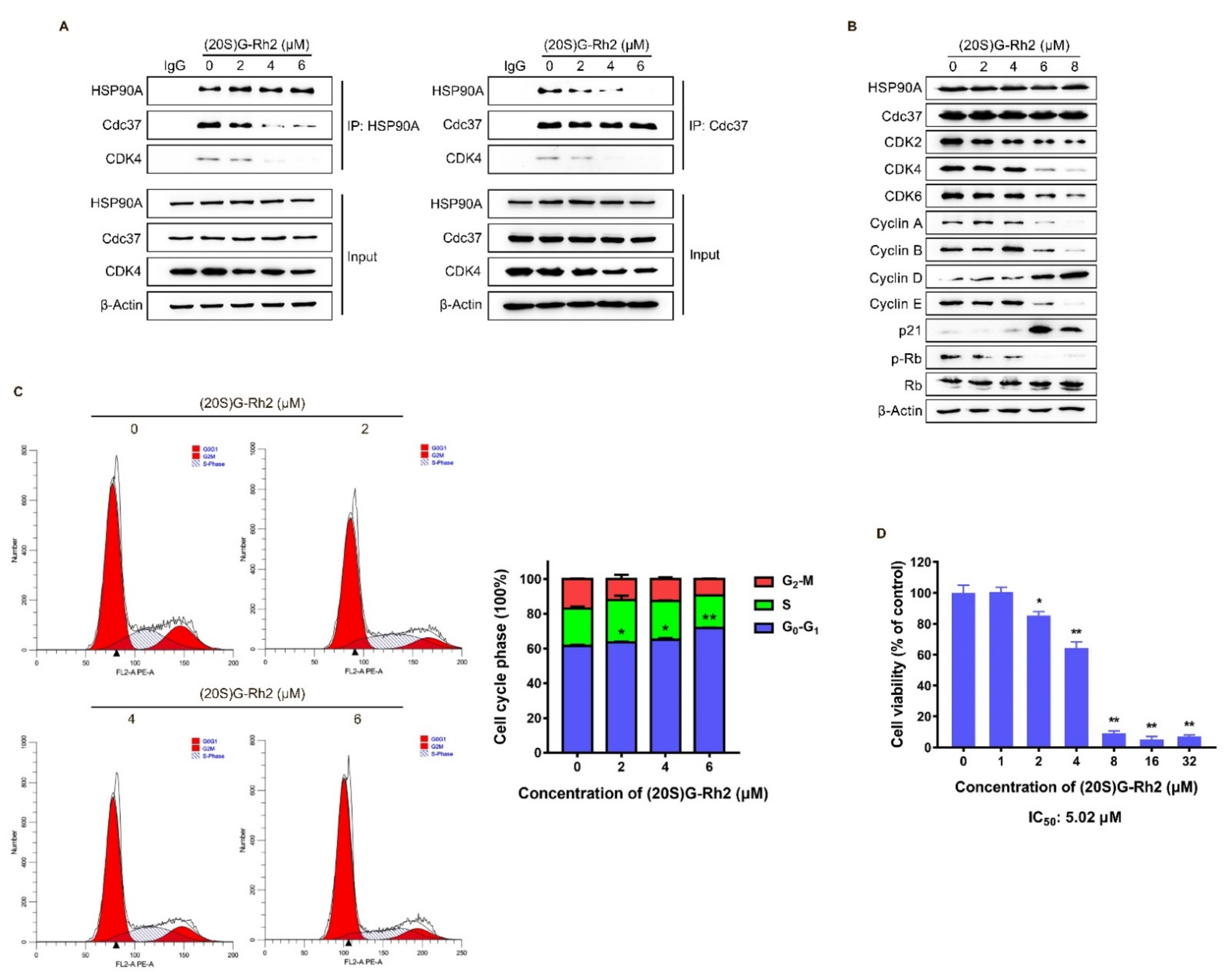
2.4. (20S) G-Rh2 Induced CDKs Degradation and Cell Cycle Arrest by Disrupting the Interaction between HSP90A and Cdc37
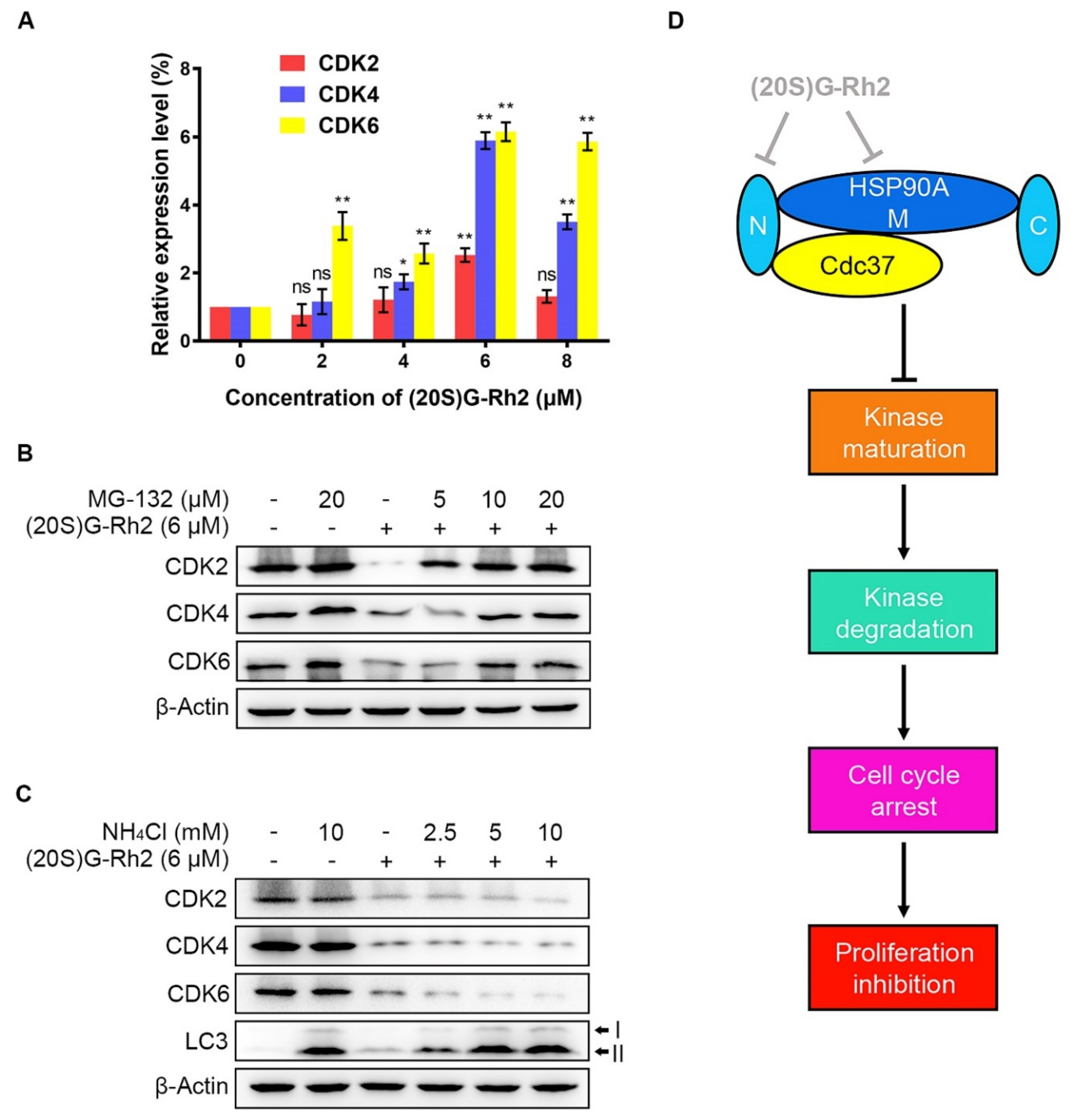
2.5. (20S) G-Rh2 Inhibited CDKs Maturation and Induced Proteasome Degradation of CDKs
3. Discussion
4. Materials and Methods
4.1. Reagents and Plasmids
4.2. Cell Lines and Culture
4.3. Protein Mass Spectrometry Analysis
4.4. Phage Display Screening
4.5. Network Topology Analysis
4.6. Gene Ontology and Pathway Enrichment Analysis
4.7. Molecular Docking Analysis
4.8. Prokaryotic Expression of the Truncated HSP90A In Vitro
4.9. Thermal Shift Assay
4.10. Co-Immunoprecipitation
4.11. Cell Cycle Analysis
4.12. Cell Viability Assay
4.13. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Swords, R.; Carew, J.S.; Nawrocki, S.T.; Bhalla, K.; Giles, F.J. Targeting HSP90 for cancer therapy. Br. J. Cancer 2009, 100, 1523–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, S.; Buchner, J. Molecular chaperones—Cellular machines for protein folding. Angew. Chem. Int. Edit. 2002, 41, 1098–1113. [Google Scholar] [CrossRef]
- Chen, B.; Zhong, D.B.; Monteiro, A. Comparative genomics and evolution of the HSP90 family of genes across all kingdoms of organisms. BMC Genom. 2006, 7, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Liu, J.; Wang, X.; Cheng, X.; Wang, Y.; Wu, N. Essential role of the first intron in the transcription of hsp90beta gene. FEBS Lett. 1997, 413, 92–98. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.L.; Yu, J.; Cheng, X.K.; Ding, L.; Heng, F.Y.; Wu, N.H.; Shen, Y.F. Regulation of human hsp90 alpha gene expression. FEBS Lett. 1999, 444, 130–135. [Google Scholar] [CrossRef] [Green Version]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Backe, S.J.; Sager, R.A.; Woodford, M.R.; Makedon, A.M.; Mollapour, M. Post-translational modifications of Hsp90 and translating the chaperone code. J. Biol. Chem. 2020, 295, 11099–11117. [Google Scholar] [CrossRef]
- Nitika; Porter, C.M.; Truman, A.W.; Truttmann, M.C. Post-translational modifications of Hsp70 family proteins: Expanding the chaperone code. J. Biol. Chem. 2020, 295, 10689–10708. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Q.Y.; You, Q.D. Targeting the HSP90-CDC37-kinase chaperone cycle: A promising therapeutic strategy for cancer. Med. Res. Rev. 2021, 27. [Google Scholar] [CrossRef]
- Calderwood, S.K. Cdc37 as a co-chaperone to Hsp90. Subcell. Biochem. 2015, 78, 103112. [Google Scholar] [CrossRef]
- Gray, P.J.; Prince, T.; Cheng, J.; Stevenson, M.A.; Calderwood, S.K. Targeting the oncogene and kinome chaperone CDC37. Nat. Rev. Cancer 2008, 8, 491–495. [Google Scholar] [CrossRef] [Green Version]
- Verba, K.A.; Wang, R.Y.R.; Arakawa, A.; Liu, Y.X.; Shirouzu, M.; Yokoyama, S.; Agard, D.A. STRUCTURAL BIOLOGY Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 2016, 352, 1542–1547. [Google Scholar] [CrossRef] [Green Version]
- Bagatell, R.; Whitesell, L. Altered Hsp90 function in cancer: A unique therapeutic opportunity. Mol. Cancer Ther. 2004, 3, 1021–1030. [Google Scholar]
- Taha, E.A.; Ono, K.; Eguchi, T. Roles of Extracellular HSPs as Biomarkers in Immune Surveillance and Immune Evasion. Int. J. Mol. Sci. 2019, 20, 4588. [Google Scholar] [CrossRef] [Green Version]
- Calderwood, S.K.; Gong, J.L. Heat Shock Proteins Promote Cancer: It’s a Protection Racket. Trends Biochem. Sci. 2016, 41, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.X.; Xiao, H.Y.; Cao, L. Recent advances in heat shock proteins in cancer diagnosis, prognosis, metabolism and treatment. Biomed. Pharmacother. 2021, 142, 14. [Google Scholar] [CrossRef]
- Husnjak, K.; Dikic, I. Ubiquitin-Binding Proteins: Decoders of Ubiquitin-Mediated Cellular Functions. Annu. Rev. Biochem. 2012, 81, 291–322. [Google Scholar] [CrossRef]
- Kirisako, T.; Kamei, K.; Murata, S.; Kato, M.; Fukumoto, H.; Kanie, M.; Sano, S.; Tokunaga, F.; Tanaka, K.; Iwai, K. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006, 25, 4877–4887. [Google Scholar] [CrossRef]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Pedro, J.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Hurley, J.H.; Young, L.N. Mechanisms of Autophagy Initiation. Annu. Rev. Biochem. 2017, 86, 225–244. [Google Scholar] [CrossRef]
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef]
- Anwanwan, D.; Singh, S.K.; Singh, S.; Saikam, V.; Singh, R. Challenges in liver cancer and possible treatment approaches. Biochim. Biophys. Acta-Rev. Cancer 2020, 1873, 7. [Google Scholar] [CrossRef]
- El-Serag, H.B. CURRENT CONCEPTS Hepatocellular Carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef]
- Chung, K.S.; Cho, S.H.; Shin, J.S.; Kim, D.H.; Choi, J.H.; Choi, S.Y.; Rhee, Y.K.; Hong, H.D.; Lee, K.T. Ginsenoside Rh2 induces cell cycle arrest and differentiation in human leukemia cells by upregulating TGF-beta expression. Carcinogenesis 2013, 34, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Li, B.H.; Zhao, J.O.; Wang, C.Z.; Searle, J.; He, T.C.; Yuan, C.S.; Du, W. Ginsenoside Rh2 induces apoptosis and paraptosis-like cell death in colorectal cancer cells through activation of p53. Cancer Lett. 2011, 301, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Chen, C.; Li, Z.M.; Yang, Y.; Xing, C.Q.; Li, Y.; Jin, Y.H. Specific Interaction with Human Serum Albumin Reduces Ginsenoside Cytotoxicity in Human Umbilical Vein Endothelial Cells. Front. Pharmacol. 2020, 11, 7. [Google Scholar] [CrossRef]
- Lee, K.Y.; Park, J.A.; Chung, E.; Lee, Y.H.; Kim, S.I.; Lee, S.K. Ginsenoside-Rh2 blocks the cell cycle of SK-HEP-1 cells at the G1/S boundary by selectively inducing the protein expression of p27kip1. Cancer Lett. 1996, 110, 193–200. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef] [PubMed]
- Pinero, J.; Queralt-Rosinach, N.; Bravo, A.; Deu-Pons, J.; Bauer-Mehren, A.; Baron, M.; Sanz, F.; Furlong, L.I. DisGeNET: A discovery platform for the dynamical exploration of human diseases and their genes. Database 2015, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for annotation, visualization, and integrated discovery. Genome. Biol. 2003, 4, 11. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.J.; Li, Y.; Song, Z.G.; Zhu, H.Y.; Jin, Y.H. The interaction of serum albumin with ginsenoside Rh2 resulted in the downregulation of ginsenoside Rh2 cytotoxicity. J. Ginseng. Res. 2017, 41, 330–338. [Google Scholar] [CrossRef]
- Wang, Y.S.; Lin, Y.J.; Li, H.; Li, Y.; Song, Z.G.; Jin, Y.H. The identification of molecular target of (20S) ginsenoside Rh2 for its anti-cancer activity. Sci. Rep. 2017, 7, 12. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucl. Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nature Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Tang, Z.F.; Kang, B.X.; Li, C.W.; Chen, T.X.; Zhang, Z.M. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucl. Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Sun, L.; Xu, C.; Yu, F.; Zhou, H.; Zhao, Y.; Zhang, J.; Cai, J.; Mao, C.; Tang, L.; et al. Structure insights into mechanisms of ATP hydrolysis and the activation of human heat-shock protein 90. Acta Biochim. Biophys. Sin. 2012, 44, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Roe, S.M.; Ali, M.M.U.; Meyer, P.; Vaughan, C.K.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. The mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50(cdc37). Cell 2004, 116, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Pearl, L.H. Hsp90 and Cdc37—A chaperone cancer conspiracy. Curr. Opin. Genet. Dev. 2005, 15, 55–61. [Google Scholar] [CrossRef]
- Hallett, S.T.; Pastok, M.W.; Morgan, R.M.L.; Wittner, A.; Blundell, K.L.I.M.; Felletar, I.; Wedge, S.R.; Prodromou, C.; Noble, M.E.M.; Pearl, L.H.; et al. Differential Regulation of G1 CDK Complexes by the Hsp90-Cdc37 Chaperone System. Cell Rep. 2017, 21, 1386–1398. [Google Scholar] [CrossRef]
- Prince, T.; Sun, L.; Matts, R.L. Cdk2: A genuine protein kinase client of Hsp90 and Cdc37. Biochemistry 2005, 44, 15287–15295. [Google Scholar] [CrossRef]
- Oh, J.I.; Chun, K.H.; Joo, S.H.; Oh, Y.T.; Lee, S.K. Caspase-3-dependent protein kinase C delta activity is required for the progression of Ginsenoside-Rh2-induced apoptosis in SK-HEP-1 cells. Cancer Lett. 2005, 230, 228–238. [Google Scholar] [CrossRef]
- Hartl, F.U.; Hayer-Hartl, M. Protein folding—Molecular chaperones in the cytosol: From nascent chain to folded protein. Science 2002, 295, 1852–1858. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, R.I. Regulation of the heat shock transcriptional response: Cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998, 12, 3788–3796. [Google Scholar] [CrossRef] [Green Version]
- Parsell, D.A.; Lindquist, S. The function of heat-shock proteins in stress tolerance: Degradation and reactivation of damaged proteins. Annu. Rev. Genet. 1993, 27, 437–496. [Google Scholar] [CrossRef]
- Hamilton, E.; Infante, J.R. Targeting CDK4/6 in patients with cancer. Cancer Treat. Rev. 2016, 45, 129–138. [Google Scholar] [CrossRef] [Green Version]
- VanArsdale, T.; Boshoff, C.; Arndt, K.T.; Abraham, R.T. Molecular Pathways: Targeting the Cyclin D-CDK4/6 Axis for Cancer Treatment. Clin. Cancer Res. 2015, 21, 2905–2910. [Google Scholar] [CrossRef] [Green Version]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef]
- Schafer, K.A. The cell cycle: A review. Vet. Pathol. 1998, 35, 461–478. [Google Scholar] [CrossRef]
- Ji, C.H.; Kwon, Y.T. Crosstalk and Interplay between the Ubiquitin-Proteasome System and Autophagy. Mol. Cells 2017, 40, 441–449. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Banerji, U.; Tavana, B.; George, G.C.; Aaron, J.; Kurzrock, R. Targeting the molecular chaperone heat shock protein 90 (HSP90): Lessons learned and future directions. Cancer Treat. Rev. 2013, 39, 375–387. [Google Scholar] [CrossRef]
- Neckers, L.; Workman, P. Hsp90 Molecular Chaperone Inhibitors: Are We There Yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef] [Green Version]
- Doi, T.; Kurokawa, Y.; Sawaki, A.; Komatsu, Y.; Ozaka, M.; Takahashi, T.; Naito, Y.; Ohkubo, S.; Nishida, T. Efficacy and safety of TAS-116, an oral inhibitor of heat shock protein 90, in patients with metastatic or unresectable gastrointestinal stromal tumour refractory to imatinib, sunitinib and regorafenib: A phase II, single-arm trial. Eur. J. Cancer 2019, 121, 29–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, R.; Tummalapalli, S.R.; Rotella, D.P. Progress in the Discovery and Development of Heat Shock Protein 90 (Hsp90) Inhibitors. J. Med. Chem. 2014, 57, 8718–8728. [Google Scholar] [CrossRef]
- Biamonte, M.A.; Van de Water, R.; Arndt, J.W.; Scannevin, R.H.; Perret, D.; Lee, W.C. Heat Shock Protein 90: Inhibitors in Clinical Trials. J. Med. Chem. 2010, 53, 3–17. [Google Scholar] [CrossRef]
- Chen, C.; Lv, Q.; Li, Y.; Jin, Y.H. The Anti-Tumor Effect and Underlying Apoptotic Mechanism of Ginsenoside Rk1 and Rg5 in Human Liver Cancer Cells. Molecules 2021, 26, 3926. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Result | Estimated Free Energy of Binding (kcal/mol) | Estimated Inhibition Constant, Ki (298.15 K) |
|---|---|---|
| 4BQG:(20S) G-Rh2 result 1 | −7.25 | 4.89 μM |
| 4BQG:(20S) G-Rh2 result 2 | −5.01 | 212.28 μM |
| 3Q6M:(20S) G-Rh2 | −6.32 | 23.18 μM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Wang, Y.-S.; Zhang, E.-T.; Li, G.-A.; Liu, W.-Y.; Li, Y.; Jin, Y.-H. (20S) Ginsenoside Rh2 Exerts Its Anti-Tumor Effect by Disrupting the HSP90A-Cdc37 System in Human Liver Cancer Cells. Int. J. Mol. Sci. 2021, 22, 13170. https://doi.org/10.3390/ijms222313170
Chen C, Wang Y-S, Zhang E-T, Li G-A, Liu W-Y, Li Y, Jin Y-H. (20S) Ginsenoside Rh2 Exerts Its Anti-Tumor Effect by Disrupting the HSP90A-Cdc37 System in Human Liver Cancer Cells. International Journal of Molecular Sciences. 2021; 22(23):13170. https://doi.org/10.3390/ijms222313170
Chicago/Turabian StyleChen, Chen, Yu-Shi Wang, En-Ting Zhang, Gang-Ao Li, Wen-Yuan Liu, Yang Li, and Ying-Hua Jin. 2021. "(20S) Ginsenoside Rh2 Exerts Its Anti-Tumor Effect by Disrupting the HSP90A-Cdc37 System in Human Liver Cancer Cells" International Journal of Molecular Sciences 22, no. 23: 13170. https://doi.org/10.3390/ijms222313170