1. Introduction
Sphingosine-1-phosphate (S1P) is a bioactive sphingolipid, which can be generated by activation of sphingosine kinases with various stimuli, including growth factors, hormones, bacterial stimuli, and cytokines [
1,
2]. Because S1P can be degraded by S1P lyase or dephosphorylated by S1P phosphatases in the tissues, the constitutive S1P level in tissues is very low (10–30 nM) [
3]. In contrast, the S1P level in the blood is very high (150–1000 nM) because platelets lack S1P lyase and erythrocytes lack both S1P lyase and S1P phosphatases [
3,
4]. This sharp gradient between the S1P level in the blood and the S1P level in the tissues controls the migration of immune cells from blood to peripheral tissues, subsequently influencing the inflammatory response in the tissues [
3,
4].
S1P plays multifaceted roles in bone homeostasis. First, S1P controls monocytes (osteoclast precursors) migration from the blood to bone tissues, subsequently affecting osteoclastogenesis [
5,
6]. Second, S1P promotes osteoblast chemotaxis and survival, subsequently increasing RANKL production and affecting osteoclastogenesis [
7]. Higher circulating S1P levels were associated with lower bone mineral density and higher bone resorption markers in postmenopausal women [
8]. Third, under physiological condition, S1P can also promote osteoblast proliferation. Deficiency in S1P lyase in mice or pharmacological inhibition of S1P lyase led to an increased in the S1P level, and markedly enhanced bone mass and strength in animals [
9].
S1P binds to five G protein-coupled S1P receptors (S1PR1–5). S1PR1 is coupled to G
i; S1PR2 and S1PR3 are coupled to G
i, G
q, and G
12/13 proteins; whereas S1PR4 and S1PR5 are coupled chiefly to G
i and G
12/13 in response to S1P [
1]. S1PR2 regulates various cellular signaling pathways, including adenylate cyclase, phospholipase C (PLC), phosphoinositide-3 kinase (PI3K), nuclear kappa-B (NF-κB), extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), p38 mitogen-activated kinase (MAPK), and small G proteins Rac and Rho [
1,
10,
11,
12]. Previously, there were conflicting results regarding how S1PR2 regulates osteogenesis. Ishii et al. [
13] demonstrated that a strain of S1PR2-deficient mice increased bone volume, trabecular thickness, and trabecular density compared with wild-type mice. However, Higashi et al. [
14] showed that a high concentration of S1P (2 μM) enhanced RUNX2 expression by promoting S1PR2/RhoA/ROCK/Smad1/5/8 signaling. Inhibition of S1PR2 by a S1PR2 antagonist, JTE013, reduced RUNX2 induced by S1P [
14]. They also showed that a S1PR2 agonist, CYM5520, enhanced trabecular bone volume in animals [
14]. Another study [
15] demonstrated that treatment with a low dose of JTE013 (1 μM) increased the proliferation of murine BMSCs by enhancing p-ERK expression. However, treatment with JTE013 (1 μM) reduced osteopontin expression in murine BMSCs [
15].
Additionally, previous studies demonstrated that S1PR2 not only controls S1P signaling, but also regulates many other signaling pathways induced by other factors [
16,
17,
18,
19]. Our studies showed that treatment with a S1PR2 shRNA, or the specific S1PR2 antagonist, JTE013, suppressed PI3K, MAPKs, and NF-κB protein kinases induced by an oral bacterial pathogen
Aggregatibacter actinomycetemcomitans, subsequently reduced inflammatory cytokines (IL-1β, IL-6, and TNF-α) levels in murine bone marrow-derived monocytes and macrophages (BMMs) compared with controls [
20,
21]. Additionally, treatment with the S1PR2 shRNA or JTE013 decreased podosome components (cell adhesion structural units, including PI3K, Src, Pyk2, integrin β3, F-actin, and paxillin levels) induced by RANKL, resulting in the inhibition of osteoclastogenesis induced by RANKL compared with the controls [
21]. In contrast, S1P (<1 μM) could not induce a significant inflammatory cytokine production in BMMs [
22], and treatment with S1P (1 to 1000 nM) in a single culture of murine BMMs did not affect RANKL-induced osteoclast differentiation [
7]. In animal studies, treatment with JTE013 in mice alleviated periodontal inflammatory bone loss induced by ligature placement [
23] and attenuated osteoporosis induced by RANKL [
13]. Moreover, in a streptozotocin-induced type I diabetes animal model, treatment with JTE013 attenuated streptozotocin-induced apoptosis of pancreatic beta cells and decreased the incidence of diabetes [
17]. Additionally, in a high-fat die-induced type II diabetes animal model, treatment with JTE013 improved glucose tolerance and increased insulin sensitivity in mice [
18]. These studies demonstrated that S1PR2 modulates many cell signaling pathways that are induced by many factors.
Bone is generated by osteoblasts, which are derived from mesenchymal stem cells. Mesenchymal stem cells undergo cell proliferation, synthesize bone matrix proteins, and deposit minerals on bone matrix, forming bone tissues. Osteoblasts synthesize very dense and cross-linked collagen, osteogenic enzymes, including alkaline phosphatase (ALP), and proteins, including Wnts, bone morphogenetic proteins (BMPs), osteocalcin (OCN), and osterix (OSX). Additionally, osteoblasts produce calcium and phosphate-based minerals, which are deposited into the organic matrix, forming mineralized bone tissues.
Osteogenesis is regulated by various signaling pathways, including the Wnt and BMPs signaling pathways [
24,
25,
26,
27,
28]. The Wnt signaling pathways can be classified as Wnt canonical and Wnt non-canonical pathways. The Wnt canonical pathway is mediated by β-catenin. In the absence of Wnt, cytoplasmic β-catenin is constantly degraded by the Axin complex, which is composed of the scaffolding protein Axin, the tumor suppressor adenomatous polyposis coli gene product (APC), casein kinase 1 (CK1), and glycogen synthase kinase 3 (GSK3). CK1 and GSK3 sequentially phosphorylate the amino terminal region of β-catenin, resulting in β-catenin recognition by β-Trcp, an E3 ubiquitin ligase subunit, and subsequent β-catenin ubiquitination and proteasomal degradation [
29]. In the presence of Wnt, Wnt ligands bind to a complex of the seven-pass transmembrane Frizzled (Fz) receptor and low-density lipoprotein receptor–related protein (LPR) 5/6. The formation of the Wnt-Fz-LPR5/6 complex, together with the recruitment of scaffolding protein Disheveled (Dvl), results in LPR5/6 phosphorylation and the recruitment of the Axin complex to the receptors [
29]. These events inhibit the degradation of β-catenin in the cytoplasm, resulting in accumulation of β-catenin in the cytoplasm and followed by β-catenin nuclear translocation [
25,
30]. The Wnt non-canonical pathways are classified as the Wnt/planar cell polarity (PCP) pathway and the Wnt/Ca
2+ pathway. In the Wnt/PCP pathway, Wnt ligands bind to Fz receptors that activate small GTPase Rac1, RhoA, and downstream JNK, which control the rearrangements in the cytoskeleton and gene expression [
25,
30]. In the Wnt/Ca
2+ pathway, Wnt ligands bind to Fz receptors, which activate heterotrimeric G proteins and PLC [
31,
32]. Activation of PLC triggers the generation of inositol 1,4,5-triphosphate (IP3) and 1,2-diacylglycerol (DAG) [
31,
32]. IP3 diffuses through the cytosol and interacts with the calcium channels on the membrane of endoplasmic reticulum (ER), resulting in the release of Ca
2+ [
31,
32]. Ca
2+ subsequently activates the calcium/calmodulin-activated protein kinase (CaMK)II [
31,
32]. Ca
2+ and DAG can further activate protein kinase C (PKC) [
31,
32].
The BMPs signaling involves BMPs dimers that bind to BMP type II receptors, which heterodimerize with type I receptors and subsequently activate the receptor-regulated (R)-Smads (Smad 1, 5, and 8). Phosphorylation of Smad 1, 5, and 8 heterodimers form a complex with the only common mediator, (Co)-Smad (Smad4). Following nuclear translocation of the R-Smad/Co-Smad complex, the complex activates multiple transcriptional factors, including RUNX2 [
27,
28,
33]. BMPs can also activate MAPKs, which in turn promote RUNX2 expression [
33,
34].
Additionally, osteoblasts possess matrix vesicles (MVs), which are extracellular membrane-bounded microparticles, composed of cytoskeletal and surface proteins (including actin and integrins), as well as proteins and enzymes involved in synthesis and transport of Ca
2+ and PO4
3−, including tissue non-specific alkaline phosphatase (TNAP), phosphoethanolamine/phosphocholine phosphatase (PHOSPHO-1), annexins, and phospholipases [
35,
36,
37,
38,
39]. MVs also possess phospholipids, clathrin heavy chain, syntaxin, and G proteins [
38,
39]. The major function of MVs is the initiation of mineralization. Previous studies revealed that apatite initially is formed within MVs. Then, apatite platelets propagate and are inserted into collagen fibrils, leading to matrix mineralization [
35,
36,
37,
38,
39]. The biosynthesis of MVs is involved with the endosomal pathway via exocytosis [
38], and the actin network is extensively involved during MVs formation [
40,
41].
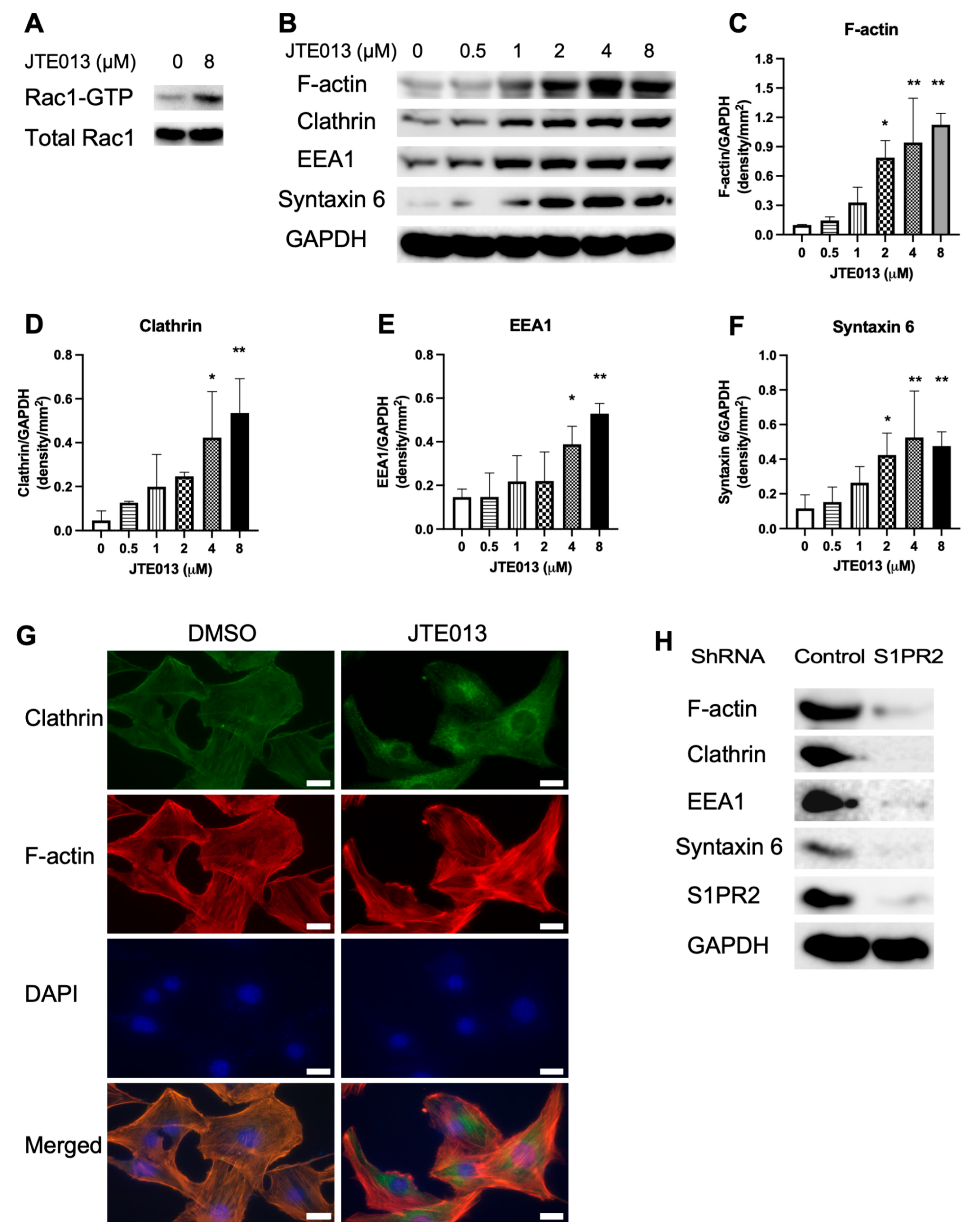
Vesicle trafficking is involved in both endocytosis and exocytosis. Endocytosis is coupled with exocytosis because endocytosis maintains exocytosis via recycling vesicles to prevent vesicle exhaustion [
42]. Vesicle trafficking is regulated by G proteins, including Rac [
43,
44]. Rac activates the Wiskott–Aldrich syndrome protein family verprolin-homologous protein (WAVE) [
43,
44]. Activation of WAVE leads to the formation of the actin-related protein 2/3 (Arp2/3) complex, subsequently resulting in polymerization of actin, which polymerizes globular (G) actin monomers to form filament (F)-actin [
43,
44]. Polymerization of actin subsequently promotes vesicle trafficking [
43,
44]. Previous studies have demonstrated that S1PR2 inhibits Rac activity [
11,
12]; also, S1PR2 deficiency in murine bone marrow-derived macrophages enhanced Rac1-GTP and F-actin stimulated by
Escherichia coli (
E. coli), leading to increased bacterial phagocytosis [
19]. Treatment with the S1PR2 antagonist, JTE013, also promoted bacterial clearance and improved survival in mice challenged with
E. coli [
19]. In this study, we treated murine bone marrow-derived stromal cells (BMSCs) with control vehicle DMSO, JTE013 (0.5 to 8 μM), a control shRNA, or a S1PR2 shRNA to determine the effects of JTE013 or the S1PR2 shRNA on osteogenesis.
3. Discussion
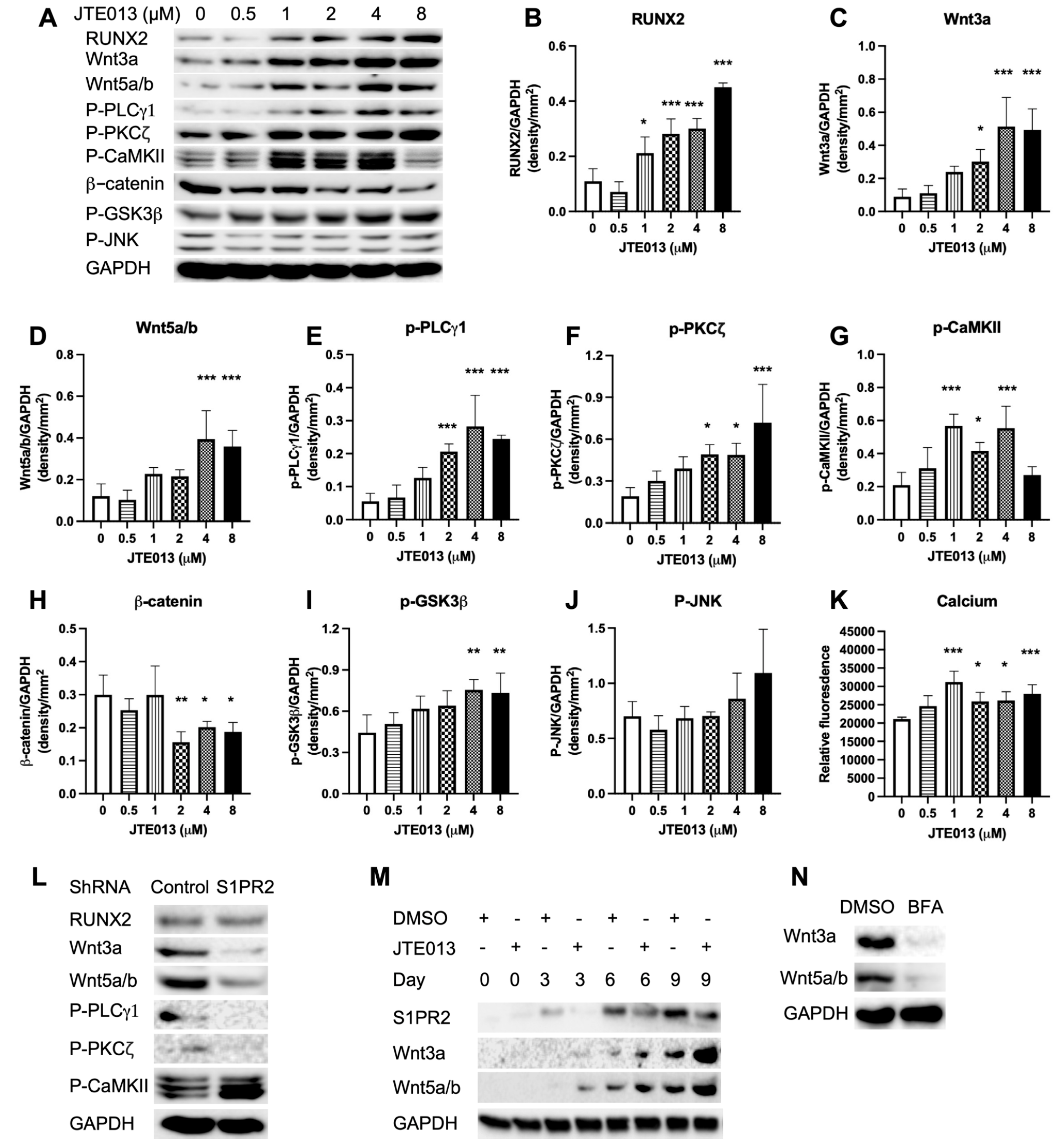
In this in vitro study, we demonstrated that treatment with a S1PR2 specific antagonist (JTE013) or a S1PR2 shRNA increased ALP and ARS staining, and enhanced osteogenic gene (ALPL, RUNX2, OCN, OSX) expressions in murine BMSCs cultured in osteogenic media. Protein analysis revealed that treatment with JTE013 increased vesicle trafficking-associated proteins (F-actin, EEA1, clathrin, syntaxin 6) and enhanced Wnt/Ca
2+ signaling-associated proteins (Wnt3a, Wnt5a/b, p-PLCγ1, p-CaMKII, and p-PKCζ). Additionally, low doses of JTE013 (1 to 2 μM) increased BMPs/Smad signaling-associated proteins (BMP2, BMP7, BMPR1A, BMPRII, and p-Smad
1/5/9). In contrast, treatment with the S1PR2 shRNA inhibited vesicle trafficking-associated proteins (F-actin, EEA1, clathrin, syntaxin 6), reduced Wnt3a, Wnt5a/b, p-PLCγ1, and p-PKCζ protein levels, but increased the p-CaMKII protein level compared with the control shRNA treatment. Treatment with the S1PR2 shRNA also suppressed BMP2, BMP7, and p-Smad
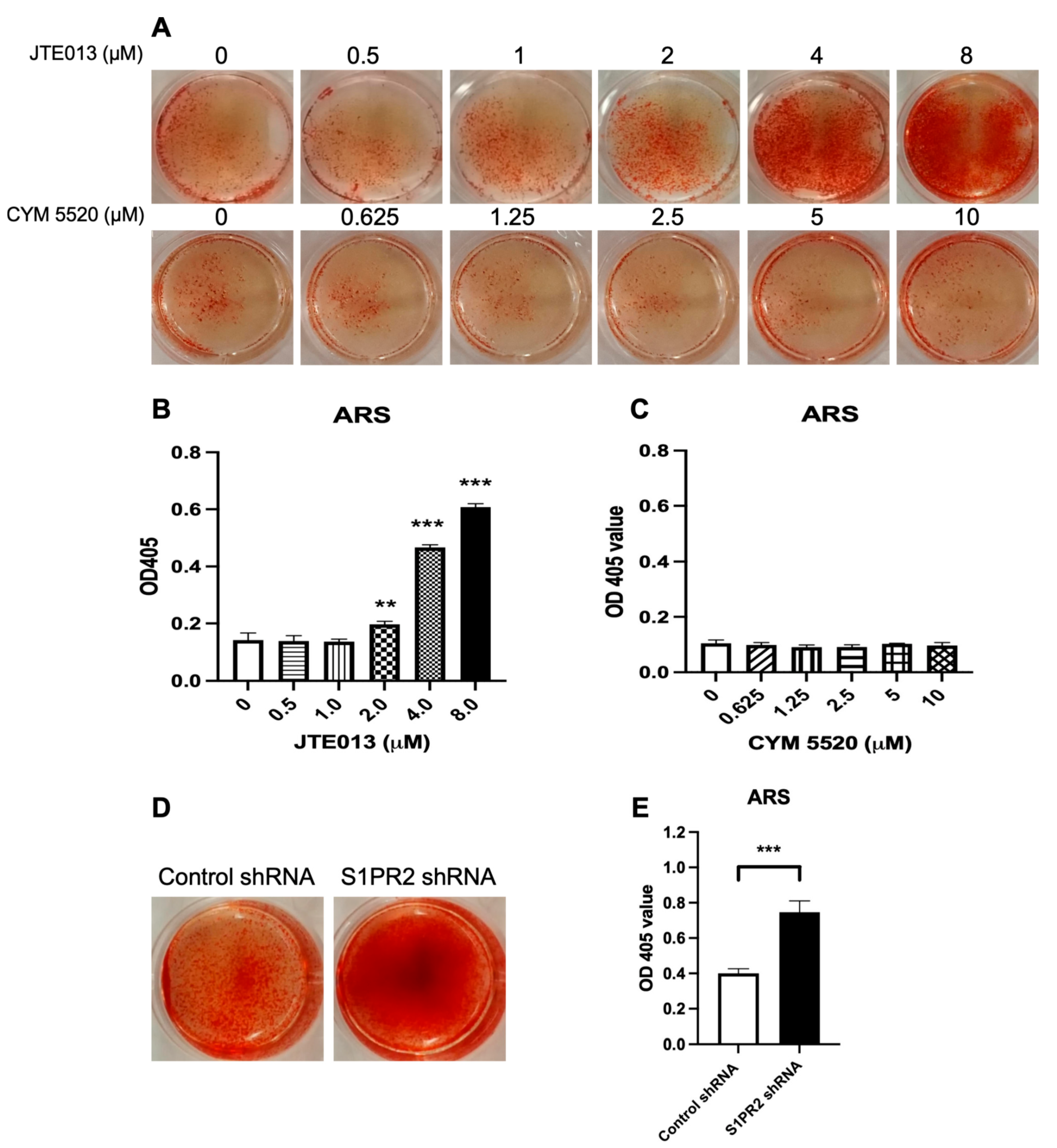
1/5/9 protein expressions compared with the control shRNA treatment. These data support that the S1PR2 shRNA acted on different cell signaling pathways in murine BMSCs. The activation of p-CaMKII was independent of Wnt protein expressions. We observed an increase of ARS (mineralization staining) in murine BMSCs treated with the S1PR2 shRNA (
Figure 3D–E), which might be associated with the activation of p-CaMKII in BMSCs treated with the S1PR2 shRNA. Future studies are needed to determine how the S1PR2 shRNA regulates cell signaling pathways affecting osteogenesis.
It is well-known that vesicle trafficking plays an important role in bacterial endocytosis/phagocytosis [
48] and synaptic vesicle release in neurological diseases [
49]. However, little is known about the role of vesicle trafficking in osteogenesis. We are the first to emphasize that vesicle trafficking is an important signaling pathway in osteogenesis. In this in vitro experiment, vesicle trafficking was involved in the uptake of proteins and minerals from cell culture media, the synthesis and transport of the intracellular vesicle pool via endosomes and the Golgi network, and the exocytosis of proteins and matrix vesicles on the plasma membrane. Endosomes function to sort cargo proteins, lipids, and minerals [
50]. Endosomal proteins and lipids can either be delivered to the cell surface via the recycling of the endosome pathway, or they can be transported to the Golgi network before reaching the lysosomes for degradation. The increased expressions of EEA1 and clathrin suggest that JTE013 promoted endocytosis in BMSCs cultured in osteogenic media. Additionally, clathrin is involved in exocytosis, as clathrin depletion blocked exocytosis [
51]. Syntaxin 6, a SNARE (soluble N-ethylmaleimide sensitive factor activating protein receptor) protein, plays an essential role in vesicle fusion to transport cargos from endosomes to Golgi network as well as vesicle fusion with the plasma membrane to release intracellular vesicles. We observed a dose-dependent increase of ARS staining, a symbol of mineralization, in BMSCs treated with JTE013, which might be associated with the enhanced generation of matrix vesicles inside of BMSCs and increased exocytosis of matrix vesicles on the plasma membrane in JTE013-treated BMSCs.
Rac acts on multiple roles in vesicle trafficking. First, Rac can induce actin polymerization, which drives vesicle trafficking [
43]. Second, Rac can directly activate PLC, leading to the generation of inositol triphosphate (IP3), which increases calcium release [
52,
53]. Calcium, in turn, plays an essential role in exocytosis. A majority of the secretory granules accumulated in the cytosol, creating a large, undocked vesicle pool, known as the reserve pool. Exocytosis undergoes a series of events: recruitment of vesicles to the plasma membrane, specific tethering at the plasma membrane, priming, and then triggering membrane fusion [
54]. Calcium functions both in priming and vesicle fusion. The first priming step requires a moderate increase in Ca
2+, as well as the presence of adenosine triphosphate magnesium, whereas a rapid substantial elevation of Ca
2+ is required for vesicle fusion [
54]. In this study, we observed an increased p-PLCγ1 protein expression and Ca
2+ release, which might be associated with the increased Rac1-GTP in JTE013-treated BMSCs. Additionally, calcium influx triggers endocytosis because reducing or buffering calcium influx abolished endocytosis and reduced exocytosis [
42]. The enhanced ARS staining in JTE013-treated BMSCs supports that more matrix vesicles were deposited on the plasma membrane of BMSCs treated with JTE013.
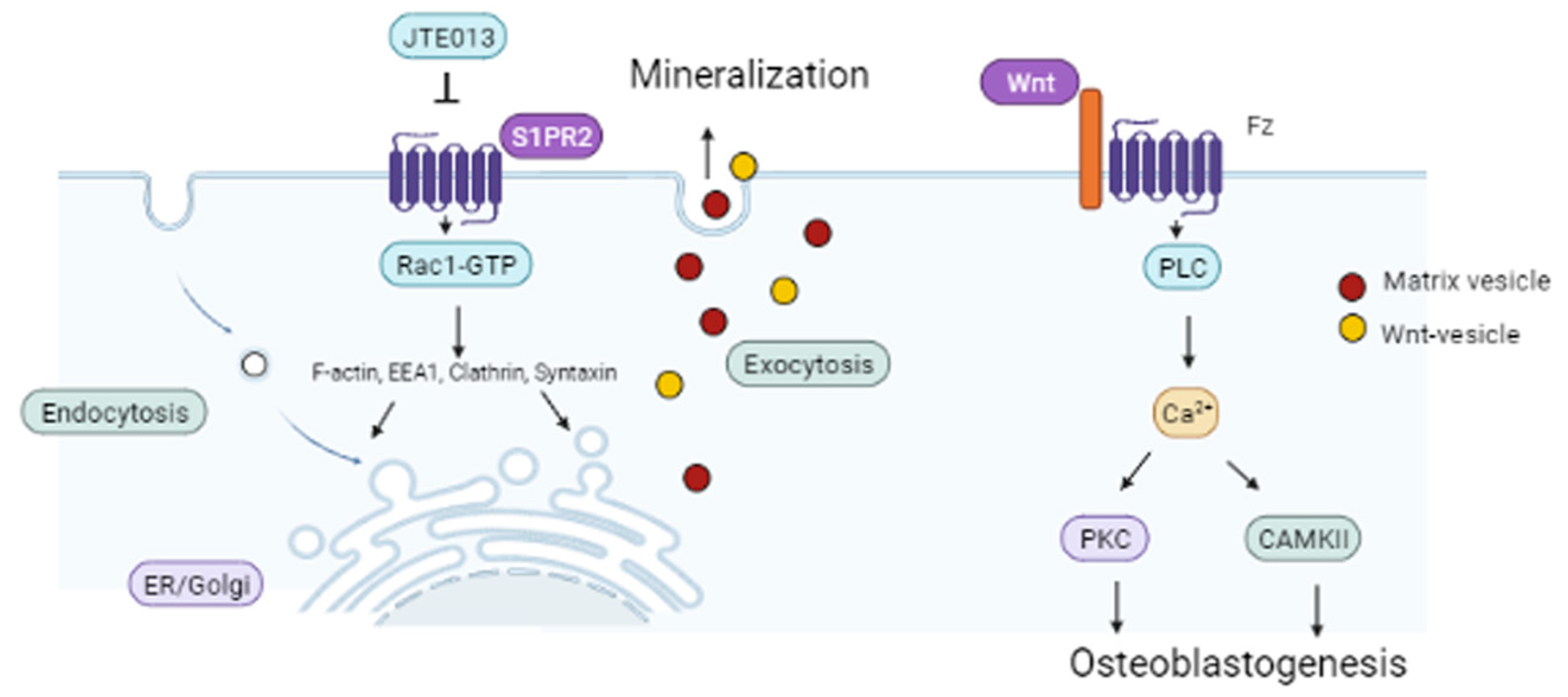
Vesicle trafficking not only plays an important role in mineralization, but also is involved in protein synthesis and secretion, including with Wnts. Following synthesis, Wnt proteins are processed in the ER, where they undergo post-translational modification (an ER-localized enzyme catalyzes the transfer of palmitoleic acid onto Wnts) [
55]. Wnt chaperone Wntless (Wls) binds to Wnts through its palmitoleic acid moiety to transport Wnts from the ER via the Golgi to the plasma membrane [
55]. Following the delivery of Wnts to the plasma membrane, Wls undergoes endocytosis and is recycled back to the Wnt-producing cells, where it binds to newly synthesized Wnt proteins and traffics them to the plasma membrane [
55]. Yamamoto et al. [
56] demonstrated that the secretion of Wnt3a is attenuated by the depletion of clathrin, which suggests that endocytosis is involved in Wnt3a secretion. In this study, we observed a dose-dependent increase of Wnt3a and Wnt5a/b protein expressions in BMSCs treated with JTE013, which might be associated with increased endocytosis of Wls, as well as increased synthesis and export of Wnts in JTE013-treated BMSCs (
Figure 8). In this study, BMSCs treated with the S1PR2 shRNA suppressed vesicle trafficking-associated protein levels (
Figure 5H), and attenuated Wnt3a and Wnt5a/b protein levels (
Figure 6L) compared with BMSCs treated with the control shRNA. Additionally, inhibition of vehicle trafficking by brefeldin A also suppressed Wnt3a and Wnt5a/b protein levels in murine BMSCs cultured in osteogenic media (
Figure 6N). Our study results are congruent with those reported by Yamamoto et al. [
56], supporting that Wnt protein expressions are dependent on vesicle trafficking.
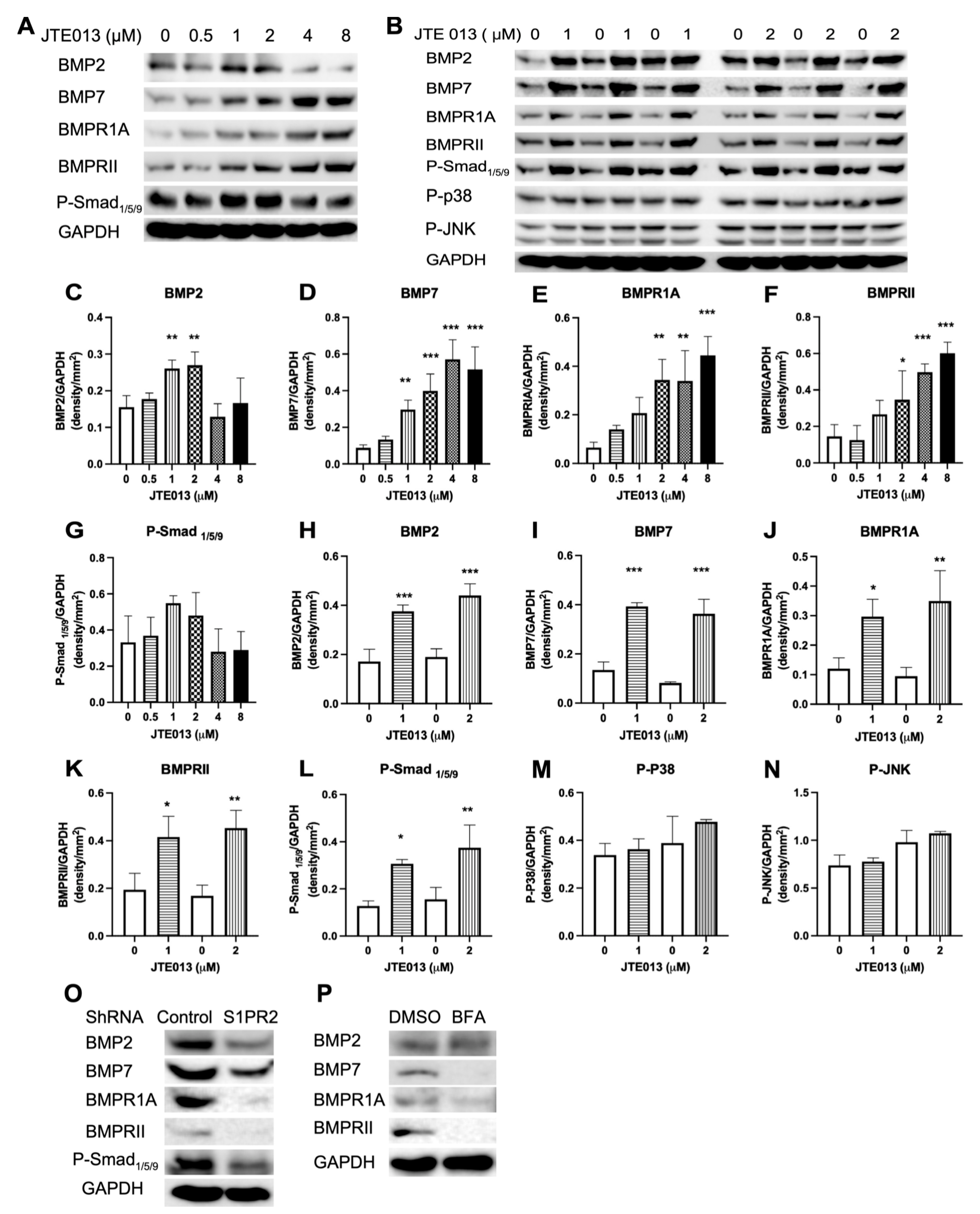
In addition to Wls, BMP receptors also undergo endocytosis, subsequently influencing BMP signaling [
57,
58,
59]. It has been demonstrated that the endocytosis of BMPRs is via clathrin-coated vesicles, cholesterol-rich lipid rafts, and caveolin-rich domains [
57,
58,
59]. Increased vesicle trafficking allows for the recycling of BMPRs to the plasma membrane, which may support signal renewal [
57]. Additionally, the endocytosis of BMPRs enables BMPRs to interact with other proteins that mediate the propagation of signaling to different intracellular locations [
57,
59]. Hartung et al. [
58] used chemicals to deplete cholesterol or disrupt clathrin-mediated endocytosis, which did not affect the initial p-Smad expression. Instead, these chemicals reduced BMP2-induced ALP expression, which suggested that the continuation of Smad signaling requires BMPRs endocytosis [
58]. In this study, we observed a dose-dependent increase of BMP7, BMPR1A, and BMPRII in JTE013-treated BMSCs (
Figure 7A,D–F). BMSCs treated with the S1PR2 shRNA inhibited vesicle trafficking-associated proteins (
Figure 5H) and reduced BMP7, BMPR1A, and BMPRII protein expressions in BMSCs compared with BMSCs treated with the control shRNA (
Figure 7O). Additionally, inhibition of protein transport from the ER to Golgi by brefeldin A suppressed BMP, BMPR1A, and BMPRII protein expressions in BMSCs compared with the control (
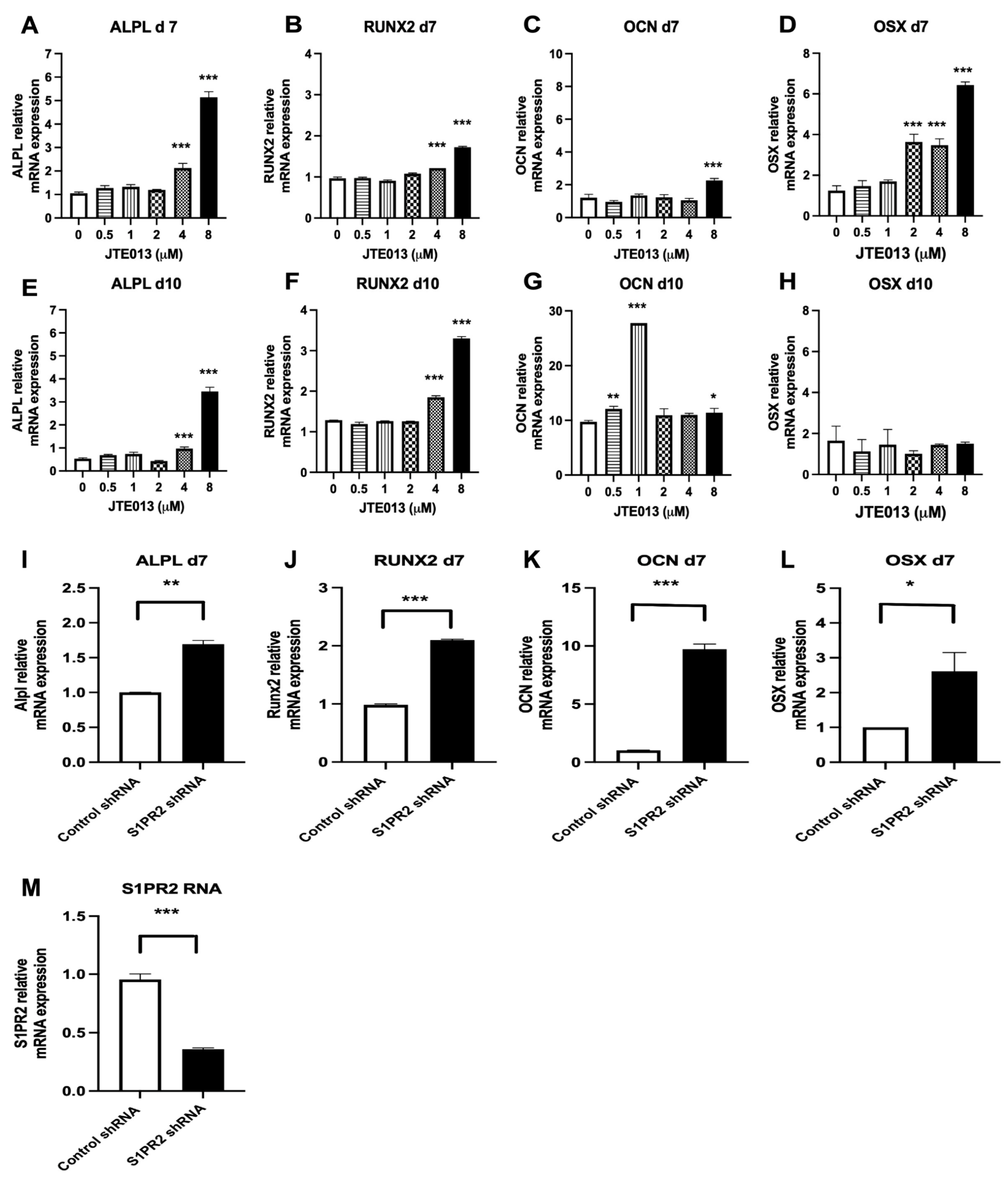
Figure 7P). These data suggest that the synthesis and transport of BMP7, BMR1A, and BMPRII might be involved in the ER–Golgi signaling pathway. We observed an increase of OCN mRNA levels on day 10 in BMSCs treated with JTE013 (1μM) (
Figure 4G), which might be associated with enhanced BMPs/Smad signaling in these cells (
Figure 7B,H–L).
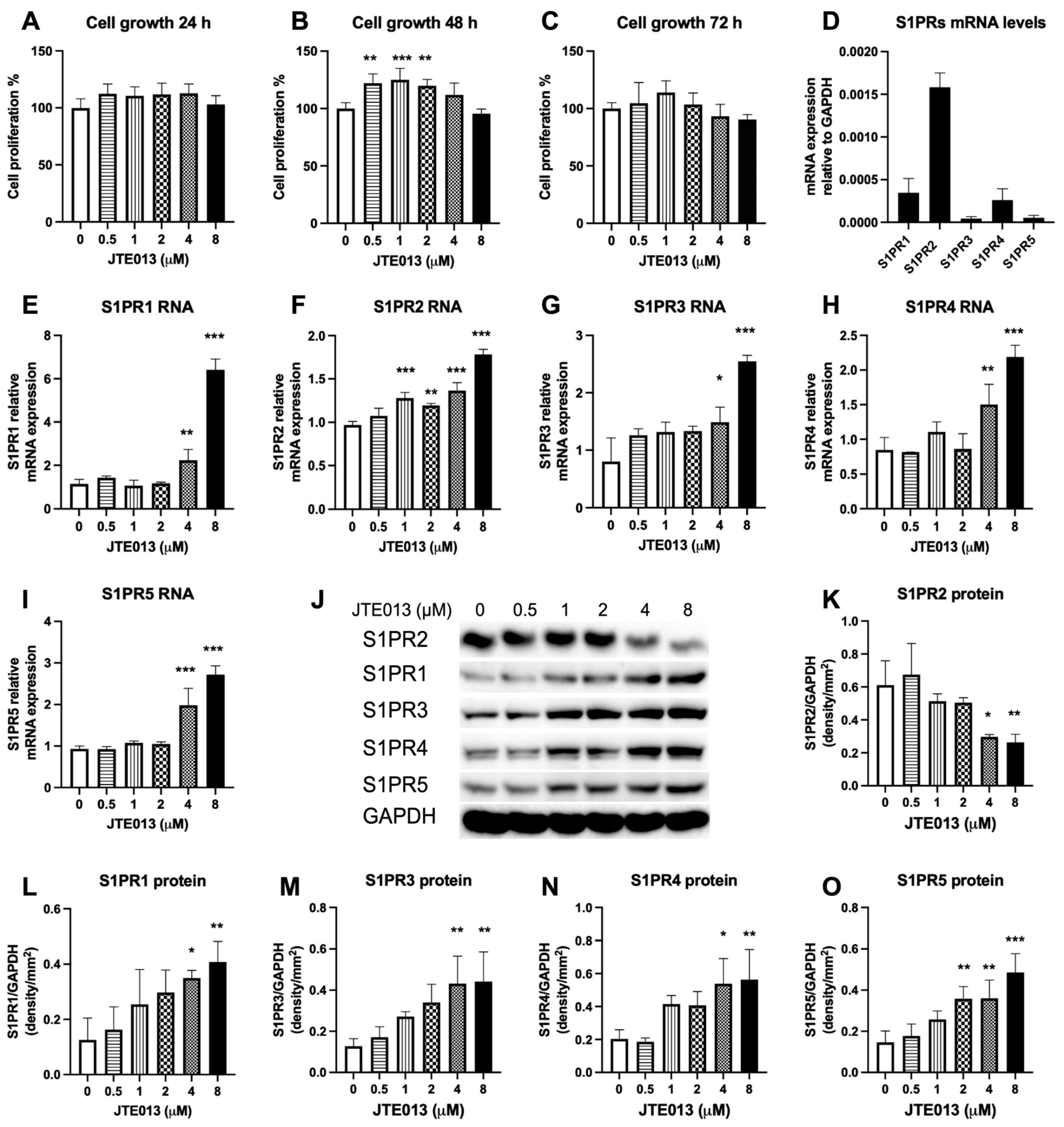
Because S1PR2 couples with multiple G
i, G
q, and G
12/13 proteins, with S1PR1 co-coupled to G
i, S1PR3 co-coupled with G
i, G
q, and G
12/13 proteins, and S1PR4 and S1PR5 co-coupled to G
i and G
12/13 [
1], other S1PRs (S1PR1, S1PR3, S1PR4, and S1PR5) might have overlapping and compensatory roles. In this study, we observed elevated expressions of both mRNA and protein levels of S1PR1, S1PR3, S1PR4, and S1PR5 in murine BMSCs treated with a high dose of JTE013 (4 or 8 μM). This might be caused by the stimulation of compensatory increase of these S1PRs by JTE013 or due to increased biosynthesis of these S1PRs via the ER–Golgi signaling pathway. A previous study [
60] indicated that JTE013 was both an S1PR2 and S1PR4 antagonist because JTE013 (10 μM) was reported to inhibit S1P-induced p-ERK expression in MAD-MB-453 breast cancer cells, and a S1PR4 siRNA also reduced the S1P-induced p-ERK, while a S1PR2 siRNA failed to reduce the S1P-induced p-ERK expression in MDA-MB-453 breast cancer cells [
60]. However, the study did not reveal S1PR2, S1PR4 mRNA, or protein levels in MDA-MB-453 breast cancer cells treated with JTE013. In our previous study [
20], knockdown of S1PR2 by a S1PR2 shRNA in murine BMMs reduced p-ERK protein levels at 2 and 4 h that were induced by an oral bacterial pathogen (
Aggregatibacter actinomycetemcomitans), which demonstrated that S1PR2 controls p-ERK expression. S1PR4 might have overlapping role in controlling p-ERK expression. In this study, JTE013 (4 to 8 μM) only reduced S1PR2 protein expression without attenuation of S1PR2 mRNA levels, which suggested that JTE013 possibly targets S1PR2 protein for degradation. Although both JTE013 (4 or 8 μM) and the S1PR2 shRNA reduced S1PR2 protein level in murine BMSCs, JTE013 promoted vesicle trafficking, while the S1PR2 shRNA inhibited vesicle trafficking. The increased vesicle trafficking in JTE013-treated cells might be caused by some off-target effects, including the effects caused by enhancing S1PR1, S1PR3, S1PR4, and S1PR5 mRNA and related protein levels in murine BMSCs (
Figure 1E–O).
Because S1PR2 couples with multiple G
i, G
q, and G
12/13 proteins, the inhibition of S1PR2 by JTE013 influences multiple signaling pathways. These signaling pathways could also cross talk to each other, thus affecting osteogenesis. Previous studies have demonstrated that the R-Smad linker region contains conserved MAPKs and GSK3 phosphorylation sites, which are crucial targets for regulating the stability of activated R-Smads [
59,
61]. Phosphorylation of the Smad1 linker by MAPKs and GSKs leads to Smad1 protein ubiquitination via Smurf1, a member of the E3 ubiquitin ligase family, resulting in Smad1 protein degradation [
59,
61]. GSK3 can also phosphorylate Smad4, leading to Smad4 ubiquitination and degradation [
62]. Additionally, CaMKII phosphorylates Smad 2 and Smad4, and to a lesser extent, Smad3 [
63]. PKC phosphorylates Smad 3 and Smad 2 [
63]. In higher doses of JTE013, we observed an increase of p-GSK3β (
Figure 6A,H), but lower activation of p-Smad
1/5/9 (
Figure 7A,G), which might be associated with increased phosphorylation of the link region of Smad
1/5/9 by GSK3 or by other protein kinases leading to Smad
1/5/9 ubiquitination and degradation. A previous study [
64] also demonstrated that PKC phosphorylated N-terminal serine residues of β-catenin and promoted β-catenin degradation. In BMSCs treated with JTE013, we observed a dose-dependent increase of p-PKCζ and p-GSK3β (
Figure 6A,F,I), which might have contributed to the degradation of β-catenin (
Figure 6A,H) in this study. In BMSCs treated with JTE013 (8 μM), the p-CaMKII was not activated (
Figure 6A,G). This might be associated with cross talk with other signaling pathways that affect CaMKII activation. Future studies are needed to determine which signaling pathways cross talk with Wnt/Ca
2+, affecting CaMKII activation.
In this study, the results conflict with those from a previous study [
14]. Previously, Higashi et al. [
14] showed that treatment with JTE013 (10 μM) in either mouse osteoblast-like MC3T3-E1 cells or primary osteoblasts inhibited RUNX2 induced by S1P (2 μM). However, the constitution levels of S1P in most tissues are very low (10–30 nM) because S1P can be degraded either by S1P lyase or dephosphorylated by S1P phosphatase [
2,
4]. Therefore, using S1P (2 μM) to stimulate osteoblasts is an artificial effect. In this study, we only cultured BMSCs in osteogenic media without the addition of S1P. Second, when Higashi et al. [
14] determined the RUNX2 protein level, they only cultured osteoblast cells in serum-free media for 24 h and then cultured cells in cell growth media with S1P (2 μM) for 24 h. In our study, we harvested protein from BMSCs cultured in osteogenic media with DMSO or JTE013 for 9 days. Osteogenesis from BMSCs is a long-term process. In the early stages (before day 3), osteogenic proteins (BMPs, RUNX2, Wnts, and β-catenin) were undetectable in BMSCs cultured in osteogenic media (data not shown). We observed a reduction of protein concentration in BMSCs treated with a high dose of JTE013 (4 or 8 μM) on day 9 (data not shown), which suggested that the high dose of JTE013 inhibited cell growth. However, because JTE013 enhanced Wnt protein expressions in BMSCs (
Figure 6A,C,D), and because Wnts determine the fate of stem cell differentiation [
26], we observed increased RUNX2 protein expression in JTE013-treated BMSCs (
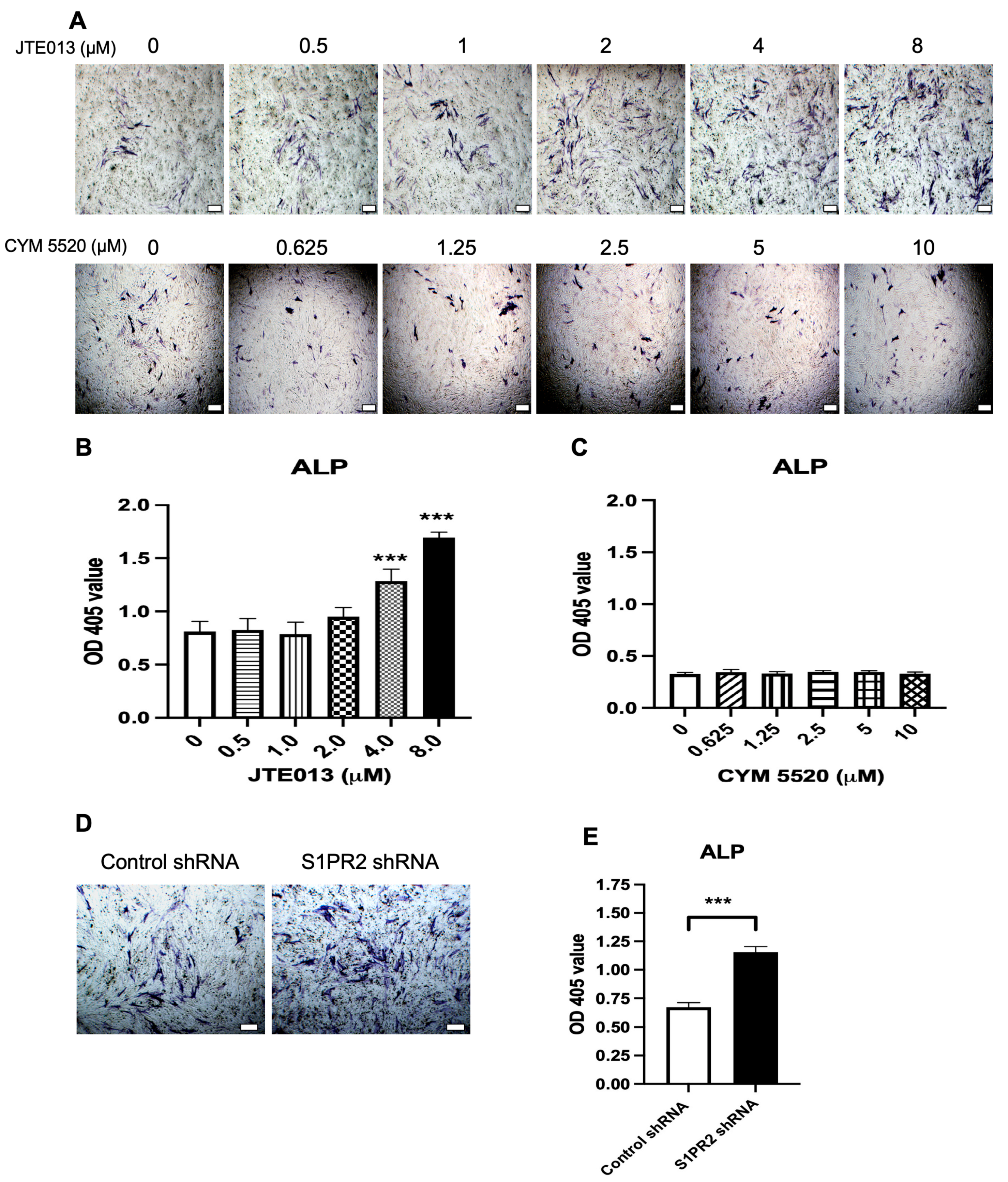
Figure 7A,B). Our ALP and ARS staining (
Figure 2 and
Figure 3) and our osteogenic genes’ (ALPL, RUNX2, OCN, OSX) expressions in BMSCs (
Figure 4) also supported that JTE013 promoted the differentiation of osteoblasts. Higashi et al. [
14] also demonstrated that C57BL/6N mice treated with a S1PR2 agonist, CYM5520, had enhanced bone volume in the tibia of mice. However, our results showed that CYM5520 had no effect on ALP staining and ARS staining in BMSCs cultured in osteogenic media (
Figure 2 and
Figure 3). Future studies are needed to determine how CYM5520 affects osteogenesis. This study results were consistent with Price et al.’s study [
15], which showed that a low dose of JTE013 (1 μM) promoted the growth of murine BMSCs (
Figure 1B). Price et al. [
15] did not determine the effect of a high dose of JTE013 on osteogenesis. This study shows that only high doses of JTE013 (above 4 μM) significantly reduced S1PR2, while low doses of JTE013 (0.5 to 2 μM) failed to attenuate the S1PR2 protein level (
Figure 1J–K).
Our understanding of the signaling pathways that regulate osteogenesis is evolving rapidly. Herein, we demonstrated that BMSCs treated with the S1PR2 antagonist, JTE013, enhanced osteogenesis, which is associated with promoting vesicle trafficking, Wnt/Ca2+, and BMPs/Smad signaling. We emphasize that vesicle trafficking plays an essential role in osteogenesis by increasing the intracellular biosynthesis of osteogenic proteins and mineral vesicles, and by exporting Wnts and matrix vesicles on the plasma membrane, thus promoting osteogenesis and bone matrix mineralization.
4. Materials and Methods
4.1. Cells and Reagents
Primary murine bone marrow stromal cells (BMSCs) were obtained from Dr. James Cray’s laboratory (the Ohio State University, Columbus, OH, USA). These BMSCs were isolated from femurs of several male adult CD-1 mice by Stem Cell Core, Augusta University (Augusta, Georgia, USA). The isolation of BMSCs was conducted using a protocol described previously [
65]. Briefly, bone marrow cells were cultured in complete isolation media (RPMI-1640 culture media with 10% FBS and 100 U/mL penicillin and streptomycin) for 3 h. Then, the media containing non-adherent cells were removed. The adherence cells were washed with PBS and were cultured in the complete isolation media for 3–4 weeks with the media changed every 3–4 days. At 70–80% confluence, the cells were lifted by incubation with trypsin/EDTA. Then, the cells were selected for being positive to stem cell antigen 1 (Sca-1), but negative for anti-mouse CD11b, CD45R/B22O, and Pan DC monoclonal antibodies by using antibody-conjugated magnetic nanoparticle beads. These procedures removed monocytes, granulocytes, macrophages, dendritic cells, natural killer cells, B lymphocytes, T lymphocytes, and macrophage progenitors from BMSCs. BMSCs were grown in high glucose Dulbecco’s Modified Eagle Medium (DMEM), supplemented with 10% fetal bovine serum (FBS, USDA approved source, Fisher Scientific, Suwanee, GA, USA) and 100 U/mL penicillin and streptomycin. A passage number of 9 murine BMSCs were used in this study. JTE013 and CYM5520 were purchased from Cayman Chemical (Ann Arbor, MI, USA), dissolved in DMSO, and diluted using cell culture media. Dexamethasone, ascorbic acid, β-glycerophosphate, polybrene, alizarin red S, and a leukocyte alkaline phosphatase kit were purchased from Sigma Aldrich (St. Louis, MO, USA). DMSO, 2-amino-2-methyl-1-propanol (AMP), p-nitrophenol, puromycin, brefeldin A, p-nitrophenyl phosphate, acetic acid, ammonium hydroxide, DMEM, penicillin, and streptomycin were purchased from Fisher Scientific.
4.2. Osteogenic Induction
The osteogenic medium was high glucose DMEM, supplemented with 10% FBS, 10 mM β-glycerophosphate, 0.1 μM dexamethasone, 0.5 mM ascorbic acid, and 100 U/mL penicillin and streptomycin. First, 5 × 104 BMSCs were placed in each well of 24-well plates (for ALP and ARS assay, protein quantification, and RNA extraction) or 4 × 105 BMSCs were placed in 60 mm dishes (for Western blot assay) and cultured in high glucose DMEM, supplemented with 10% FBS and 100 U/mL penicillin and streptomycin for 24 h. The next day, the cell culture media were replaced with osteogenic media. The osteogenic medium was changed every 2 days. Because osteoblastogenesis is a long-term process, murine BMSCs were cultured in osteogenic media with the presence of either JTE013, DMSO, a S1PR2 shRNA, or a control shRNA from 30 min to 21 days.
4.3. Cell Proliferation Assay
For the cell proliferation assay, 1 × 104 BMSCs were placed in each well of 96-well plates in high glucose DMEM media supplemented with 10% FBS and 100 U/mL penicillin and streptomycin. The second day, the cell culture media were changed with osteogenic medium containing either DMSO or JTE013 (0.5 to 8 μM) for 24 to 72 h. Cell growth was quantified by a CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA).
4.4. ALP Staining and Quantification
BMSCs were fixed with 10% buffered formalin (Fisher Scientific) for 1 min at room temperature (RT). After washing with deionized water once, the cells were stained in alkaline-dye solution (prepared from a leukocyte alkaline phosphatase kit according to Sigma Aldrich’s instructions) at RT for 30 min. After removing the dye solution, the BMSCs were washed with deionized water twice and photographed. The ALP quantification was performed according to a previous study [
66]. Briefly, BMSCs were lysed in 200 μL lysis buffer (containing 25 mM Tris-HCL with 0.5% Triton X-100) at 4 °C for 2 h. After scraping and collecting the lysate in 1.5 mL tubes, the cells were vortexed for 1 min and centrifuged at 2500×
g for 15 min at 4 °C. Then, 50 μL of supernatant derived from each sample was transferred to a new well of the 96-well plate, mixed with 50 μL of 50 mM p-nitrophenyl phosphate in AMP buffer, and incubated at 37 °C in an incubator for 30 min. A 10 times serial dilution of p-nitrophenol (stock 5000 μM) diluted in AMP buffer was used as standard curve. The amount of released p-nitrophenol was measured at an absorbance of 405 by a VersaMax microplate reader (Molecular Devices, San Jose, CA, USA). The OD405 value was calibrated by protein concentration in cell protein lysates.
4.5. ARS Staining and Quantification
BMSCs were fixed with 10% buffered formalin for 20 min at RT. After washing with deionized water once, the cells were stained in 40 mM ARS solution (pH 4.2) for 20 min. After aspirating the staining solution, the cells were washed with deionized water three times and photographed. The ARS quantification was performed as reported previously [
67]. Briefly, BMSCs were incubated with 300 μL of 10% acetic acid at RT for 30 min with shaking. After scraping and collecting the cell lysate in a 1.5 mL tube, the cells were vortexed for 30 s. The slurry was overlaid with 250 μL mineral oil, heated at 85 °C for 10 min, and transferred to ice for 5 min. Then the slurry was centrifuged at 20,000×
g for 15 min. Next, 250 μL of supernatant was transferred to a new 1.5 mL tube and mixed with 100 μL of 10% ammonium hydroxide to neutralize the acid. After adjusting the pH value of mixture to 4.2, aliquots of 100 μL of the mixture were loaded in triplicate in a 96-well plate. A 10 times serial dilution of ARS solution was used as standard curve. The absorbance 405 was detected by the VersaMax microplate reader and calibrated by the protein concentration in cell protein lysates.
4.6. A S1PR2 shRNA or a Control shRNA Lentiviral Vector Treatment
The S1PR2 shRNA and control shRNA lentiviral vectors were constructed and purified as previously described [
20]. 4 × 10
5 murine BMSCs were infected with either the S1PR2 shRNA lentiviral vector or the control shRNA lentiviral vector (multiplicity of infection 25) with the presence of polybrene (10 μg/mL) in high glucose DMEM media with 10% FBS and 100 U/mL penicillin and streptomycin. Then, 24 h after the lentiviral infection, the media were changed with high glucose DMEM media with 10% FBS, 100 U/mL penicillin and streptomycin, and puromycin (2 μg/mL) for 1 day. Then, the media were changed with osteogenic media with puromycin (2 μg/mL) for another 2 days and later changed with osteogenic media with puromycin (1 μg/mL) for two weeks. The osteogenic media were changed every two days.
4.7. RNA Extraction and Real-Time Polymerase Chain Reaction (PCR)
Total RNA was isolated from cells using TRIZOL (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions. Complementary DNA was synthesized by a TaqMan reverse transcription kit (Life Technologies) using random hexamers and total RNA (1 μg). Each reverse transcription cDNA sample (20 μL) was diluted 5 times by ultra-pure distilled water and 5 μL of diluted cDNA was used for each real-time PCR reaction. Real-time PCR was performed using a StepOnePlus Real-Time PCR System (Life Technologies) and TaqMan gene expression master mix (Life Technologies). PCR conditions for this study were as follows: 50 °C for 2 min, 95 °C for 10 min, 40 cycles of 95 °C for 15 s, and 60 °C for 1 min. The following amplicon primers were obtained from Life Technologies: S1PR1 (Mm02619656_s1), S1PR2 (ARFVPA4), S1PR3 (Mm02620181_s1), S1PR4 (Mm00468695_s1), S1PR5 (mM02620565_s1), ALPL (Mm00475834_m1), RUNX2 (Mm00501584_m1), OCN (Mm03413826_mH), OSX (Mm04209856_m1), and GAPDH (Mm99999915_g1). The amplicon concentration was determined using threshold cycle values compared with standard curves for each primer. Sample mRNA levels were normalized to an endogenous control GAPDH expression and were expressed as fold changes as compared with control groups.
4.8. Protein Isolation and Western Blot Analysis
Proteins were extracted by RIPA cell lysis buffer (Cell signaling Technology, Danvers, MA, USA). The protein concentration was determined by a DCTM protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA). Rac1-GTP was analyzed by a Rac1 pull down activation assay biochem kit (Cytoskeleton, Inc., Denver, CO, USA). For the analysis, 25 μg of protein was loaded on 10% Tris-HCl gels and electro-transferred to nitrocellulose membranes. Membranes were blocked with milk for 1 h at RT and incubated with the primary antibody overnight at 4 °C. The antibodies of the clathrin heavy chain, early endosome antigen 1 (EEA1), syntaxin 6, Runx2, Wnt3a, Wnt5a/b, p-PLCγ1, p-PKCζ, p-CaMKII, β-catenin, p-GSK3β, p-JNK, p-p38, p-Smad 1/5/9, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies of F-actin, BMP2, and BMPR1A were obtained from Abcam (Cambridge, MA, USA). Antibodies of BMP7 and BMPRII were purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). The S1PR1 antibody (PA1-1040), S1PR3 antibody (PA5-23225), S1PR4 antibody (PA5-102054), and S1PR5 antibody (PA5-98877) were all purchased from Thermo Fisher Scientific (Waltham, MA, USA). The S1PR2 antibody (SAB4503614) was obtained from Sigma Aldrich. All primary antibodies were incubated at 1:500 to 1:1000 dilution overnight at 4 °C. After washing, the nitrocellulose membranes were incubated at RT for 1.5 h with horseradish peroxidase-conjugated secondary antibodies (1:1000, Cell Signaling Technology) and developed using SuperSignal West Pico Chemiluminescent Substrate (Life Technologies, Carlsbad, CA, USA). Digital images were recorded by a G-BOX chemiluminescence imaging system (Syngene, Frederick, MD, USA). Protein densitometry was analyzed by the GeneTools software (Syngene) and normalized by GAPDH expression.
4.9. Immunofluorescence Staining
2.5 × 104 BMSCs were placed on a coverslip in 35 mm dishes in high glucose DMEM media with 10% FBS. After 4 h, BMSCs were starved by being treated with high glucose DMEM media with 1% FBS overnight. The second day, the cell culture media were changed to osteogenic media with DMSO or JTE013 (8 μM) for 30 min. BMSCs were fixed by 4% paraformaldehyde for 10 min at RT. After three washes with PBS, the cells were permeabilized by treatment with 0.1% Triton X-100 in PBS for 10 min at RT. After another three washes with PBS, the BMSCs were blocked with 5% goat serum for 1 h at RT. Following another three washes with PBS, the BMSCs were incubated with anti-clathrin heavy chain (1:100, Cell Signaling Technology) with 1% bovine serum albumin (BSA) overnight at 4 °C. Cells were washed again with PBS, and incubated with an Alexa fluor 488-labeled goat anti-rabbit secondary antibody (1:400, Fisher Scientific, Suwanee, GA, USA) with 1% BSA for 1 h at RT. After washing with PBS, BMSCs were incubated with rhodamine phalloidin (1:140, Cytoskeleton, Inc., Denver, CO, USA) with 1% BSA for 1 h at RT. Cells were washed again with PBS and mounted on a slide with Prolong Gold anti-fade reagent with DAPI (Fisher Scientific) overnight at RT. Digital images were recorded by a Zeiss Axio Imager A1 fluorescent microscope.
4.10. Calcium Quantification
The calcium content in BMSCs was measured by a Fluo-4 NW Calcium Assay kit (Thermo Fisher Scientific, Waltham, MA, USA). The fluorescence signal was recorded by a FLUOstar OPTIMA microplate reader (BMG LABTECH Inc., Cary, NC, USA).
4.11. Statistical Analysis
Data were analyzed by one-way ANOVA with Dunnett’s multiple comparisons test. All statistical tests were performed using GraphPad Prism software (GraphPad Software Inc., La Jolla CA, USA). Values are expressed as means ± standard error of the means (SEM) of three independent experiments. A p value of 0.05 or less was considered significant.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







