
Reactive Oxygen Species Mediate 6c-Induced Mitochondrial and Lysosomal Dysfunction, Autophagic Cell Death, and DNA Damage in Hepatocellular Carcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
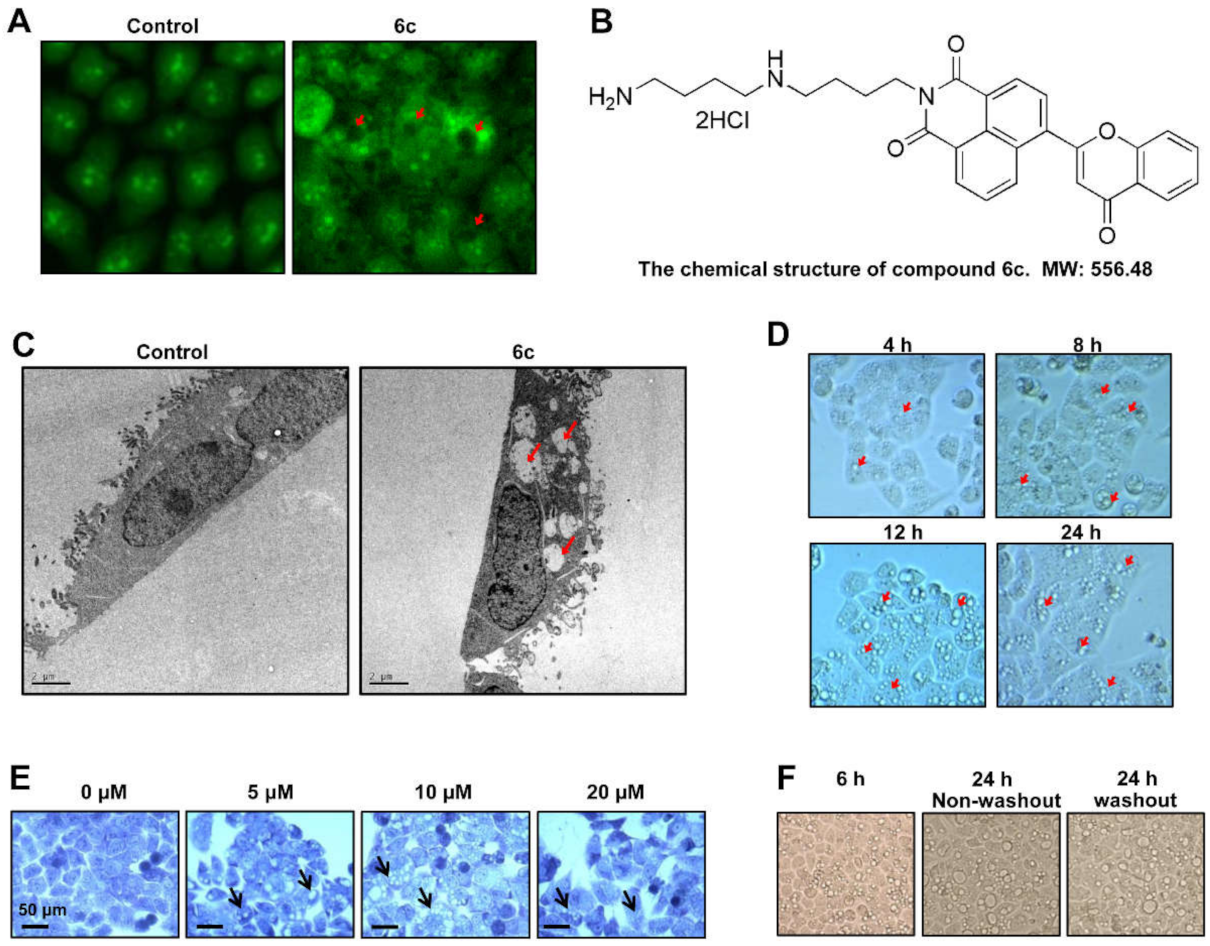
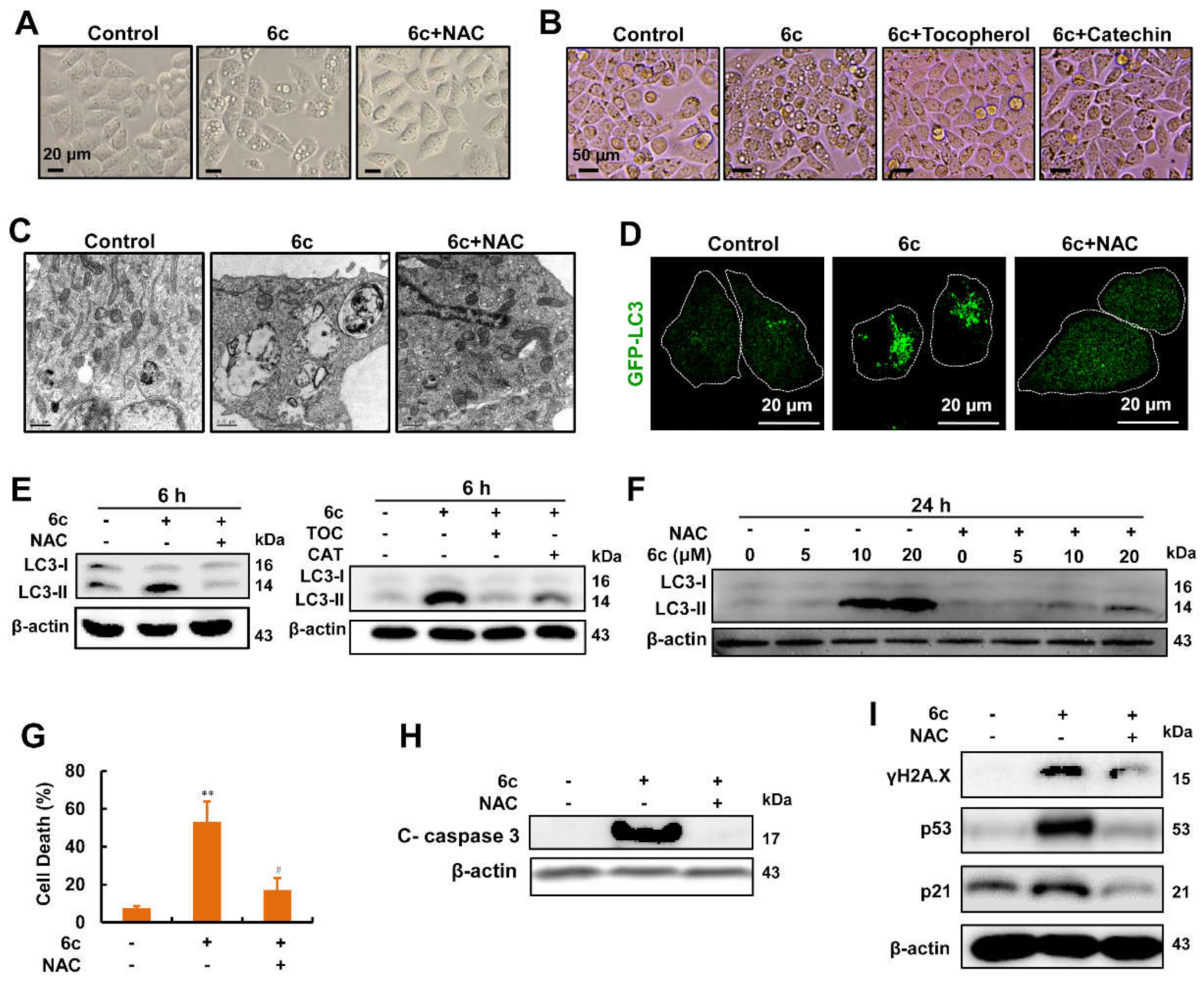
2.1. Compound 6c Induced Extensive Cytoplasmic Vacuolations
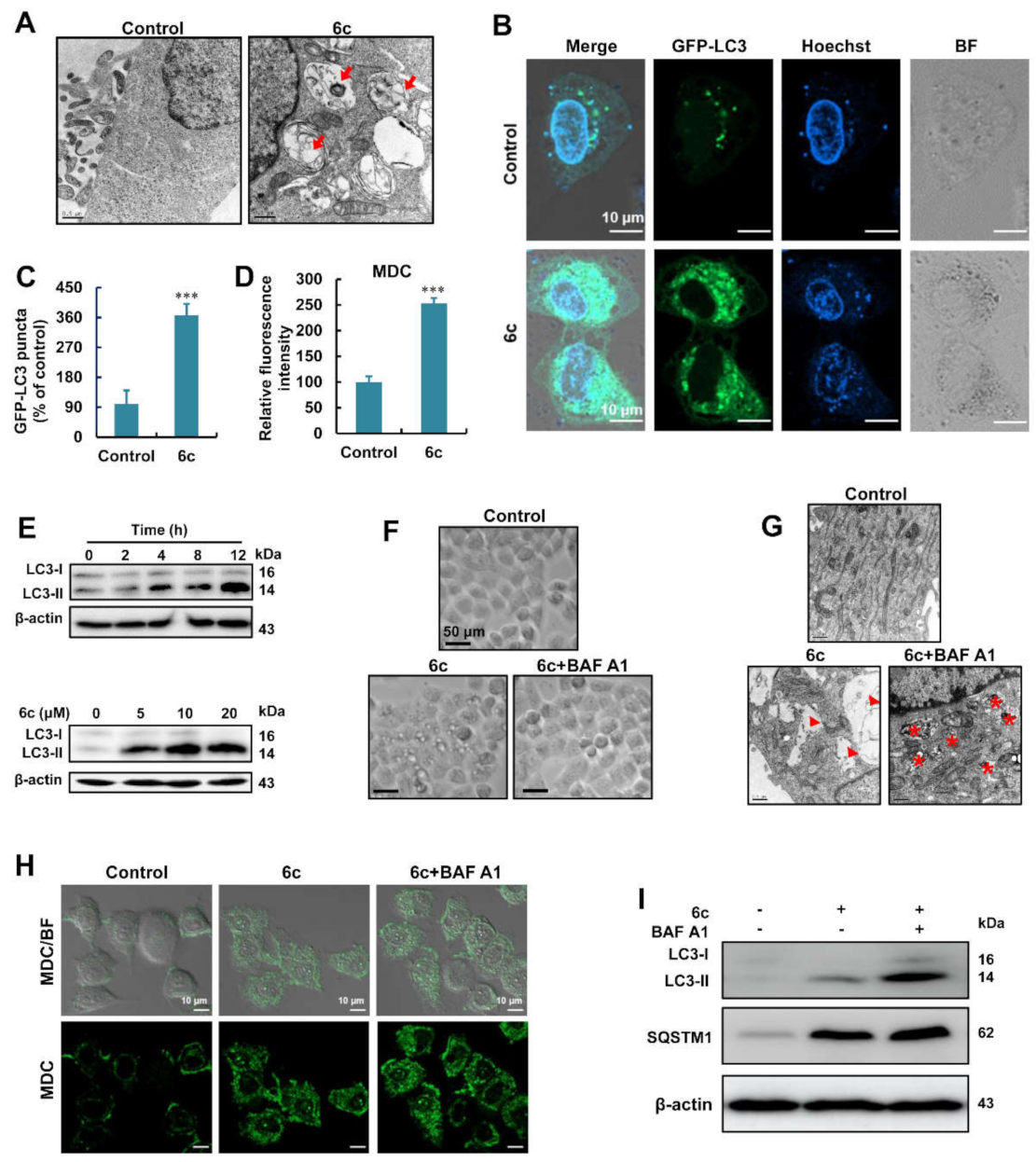
2.2. Compound 6c Induced the Formation of Autophagosome
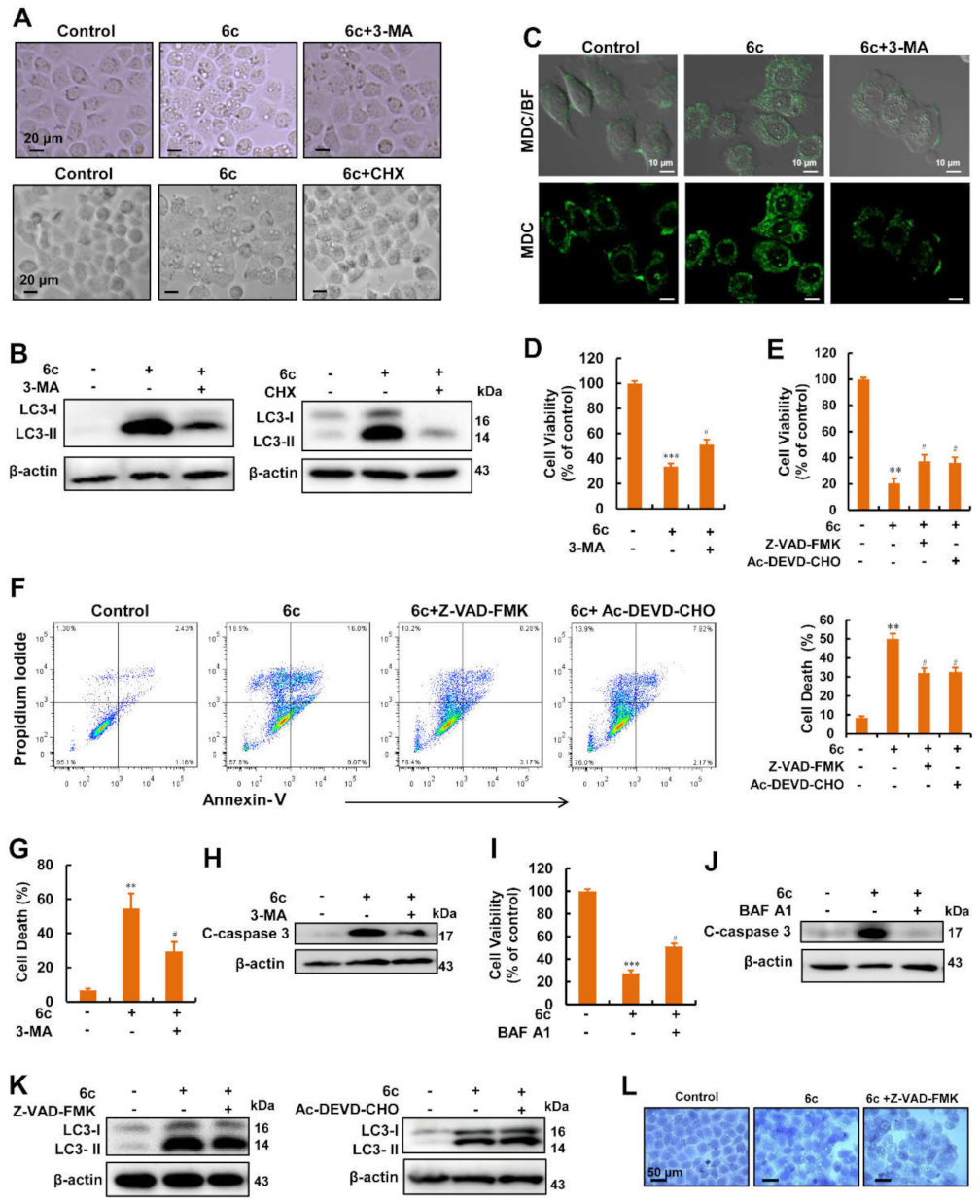
2.3. Autophagy Served as A Key Regulator in 6c-Induced Cell Death
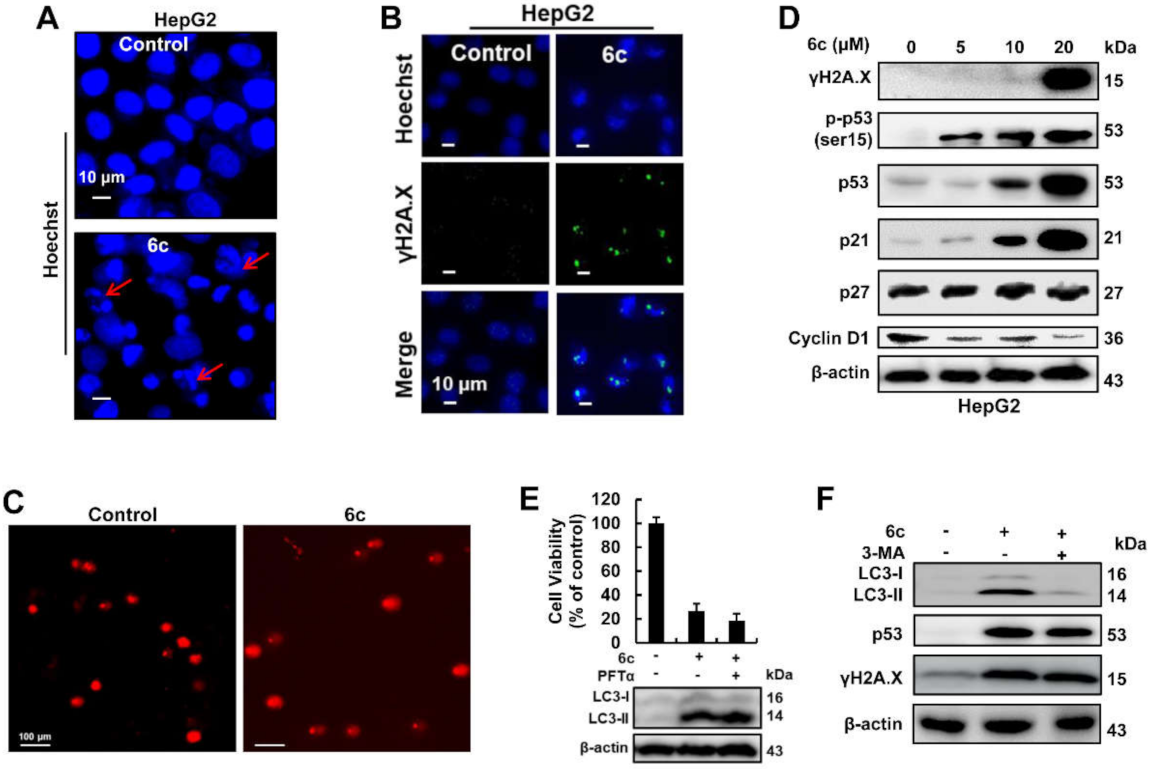
2.4. Compound 6c Induced DNA Damage in Autophagy-Independent Manner
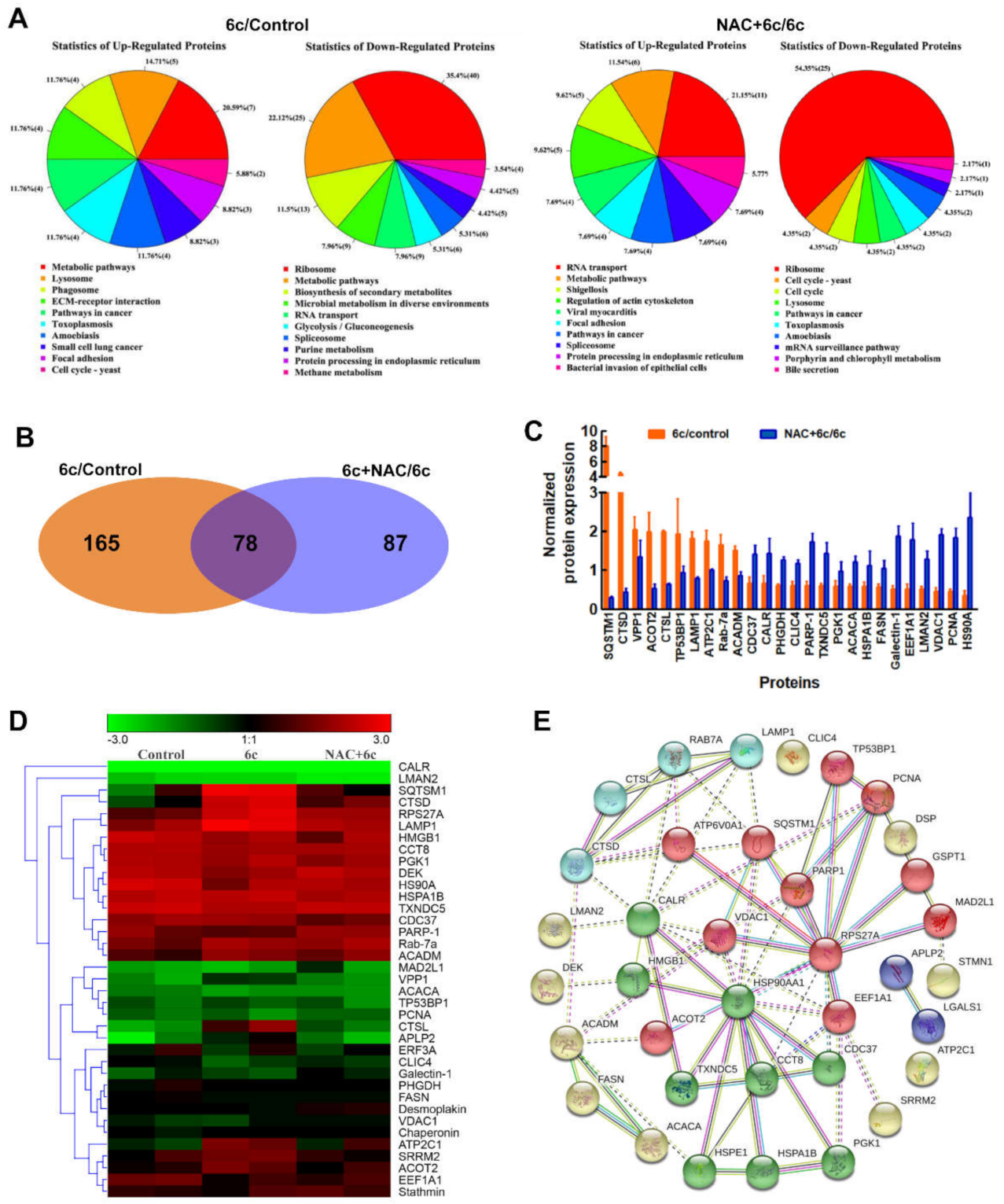
2.5. ROS Played an Important Role in 6c-Mediated Protein Expression
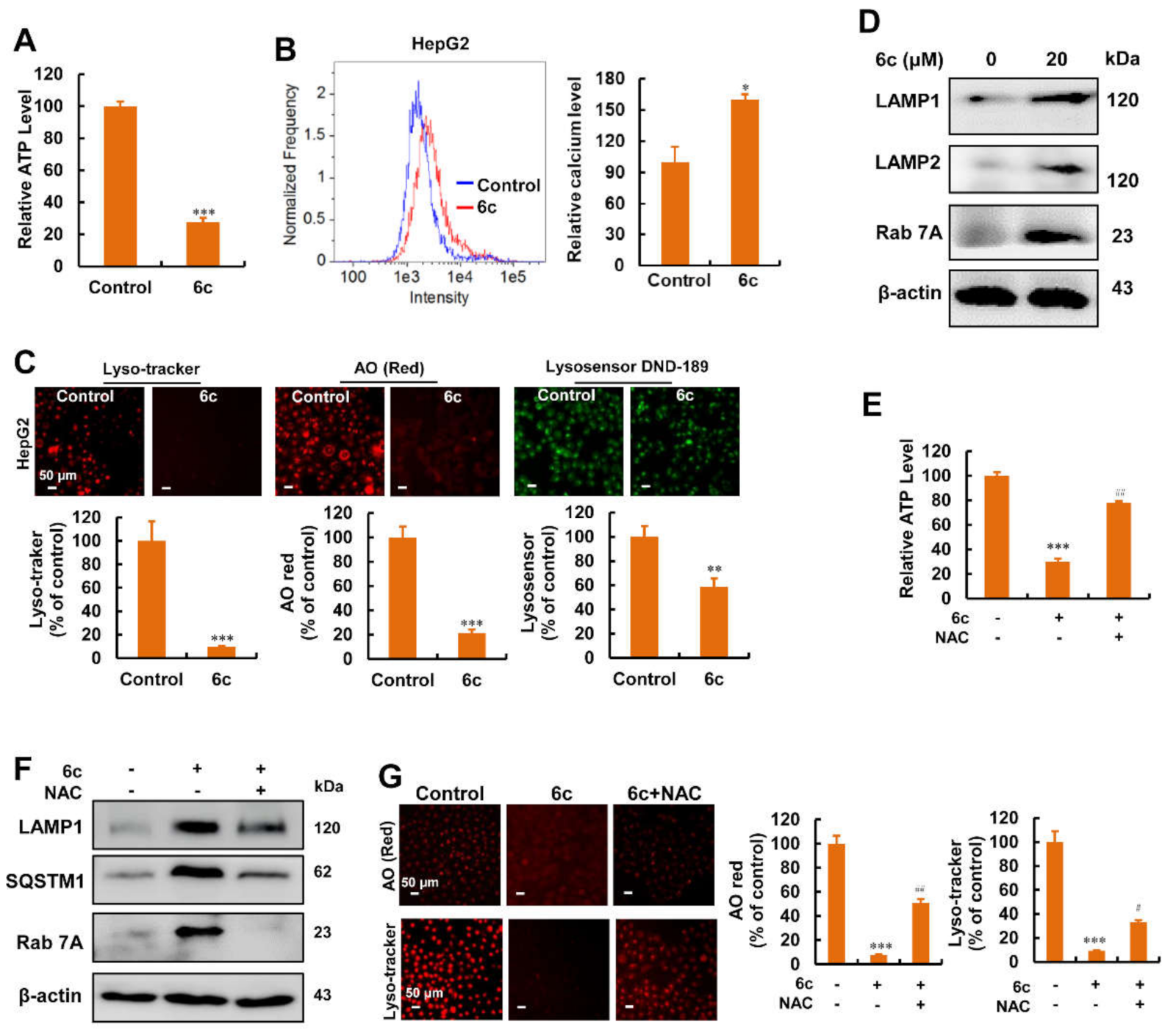
2.6. Compound 6c Induced Mitochondrial and Lysosomal Dysfunction Depending on ROS
2.7. Compound 6c-Induced Vacuolations, Autophagosome Formation, Cell Death, and DNA Damage Were Reversed by Antioxidants
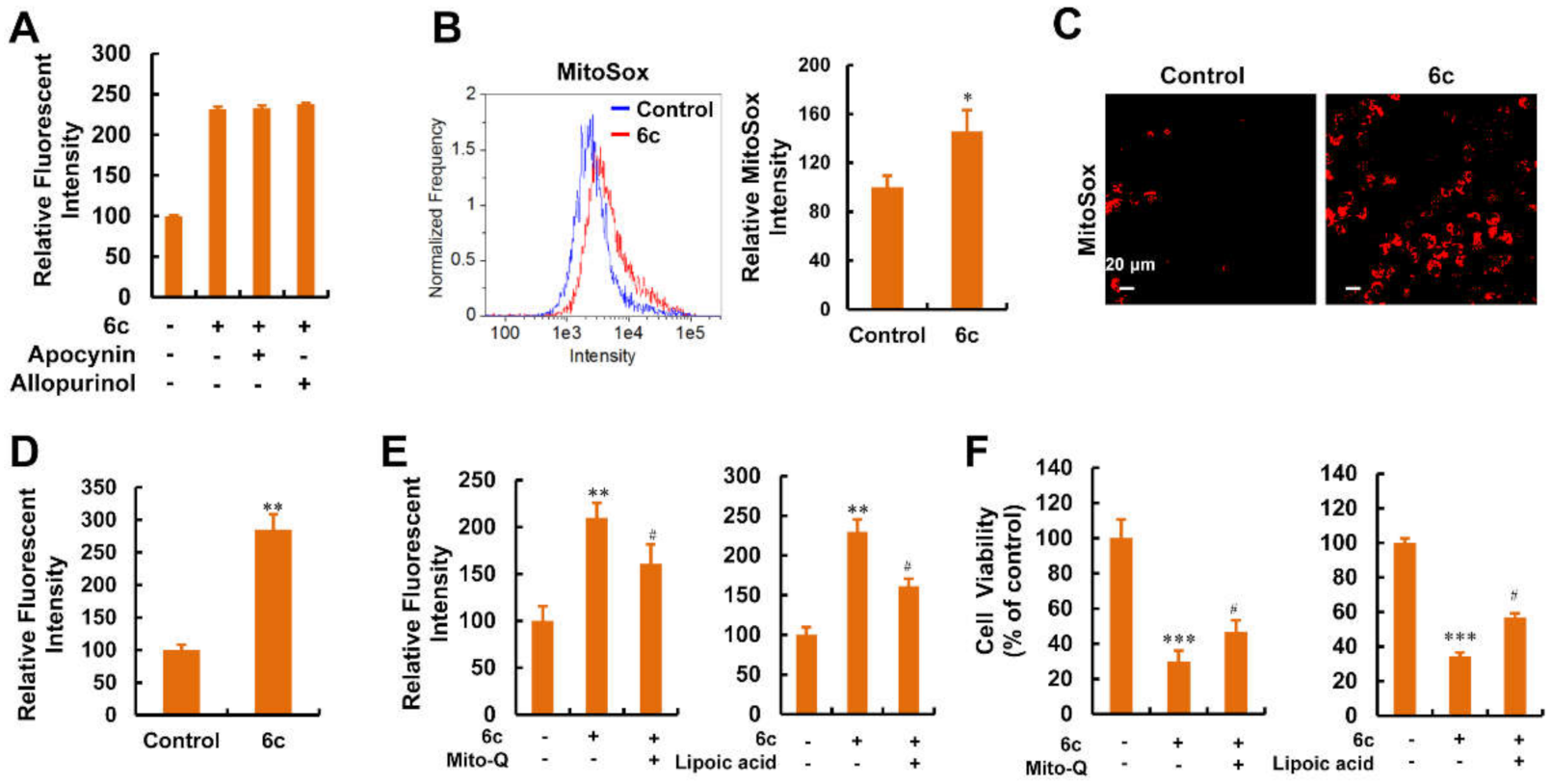
2.8. Mitochondria Were the Main Source of ROS Generation Induced by Compound 6c
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Cell Culture and Reagents
4.2. ATP Content Detection
4.3. GFP-LC3 Transfection
4.4. iTRAQ Quantification Analysis
4.5. MDC Staining
4.6. γH2A.X Staining
4.7. Annexin V-FITC/Propidium Iodide Staining
4.8. Reactive Oxygen Species Detection
4.9. Transmission Electron Microscope Assay
4.10. Lysosomal Acidification Detection
4.11. Western Blot
4.12. Mitochondrial Calcium Detection
4.13. Comet Assay
4.14. Data Analysis
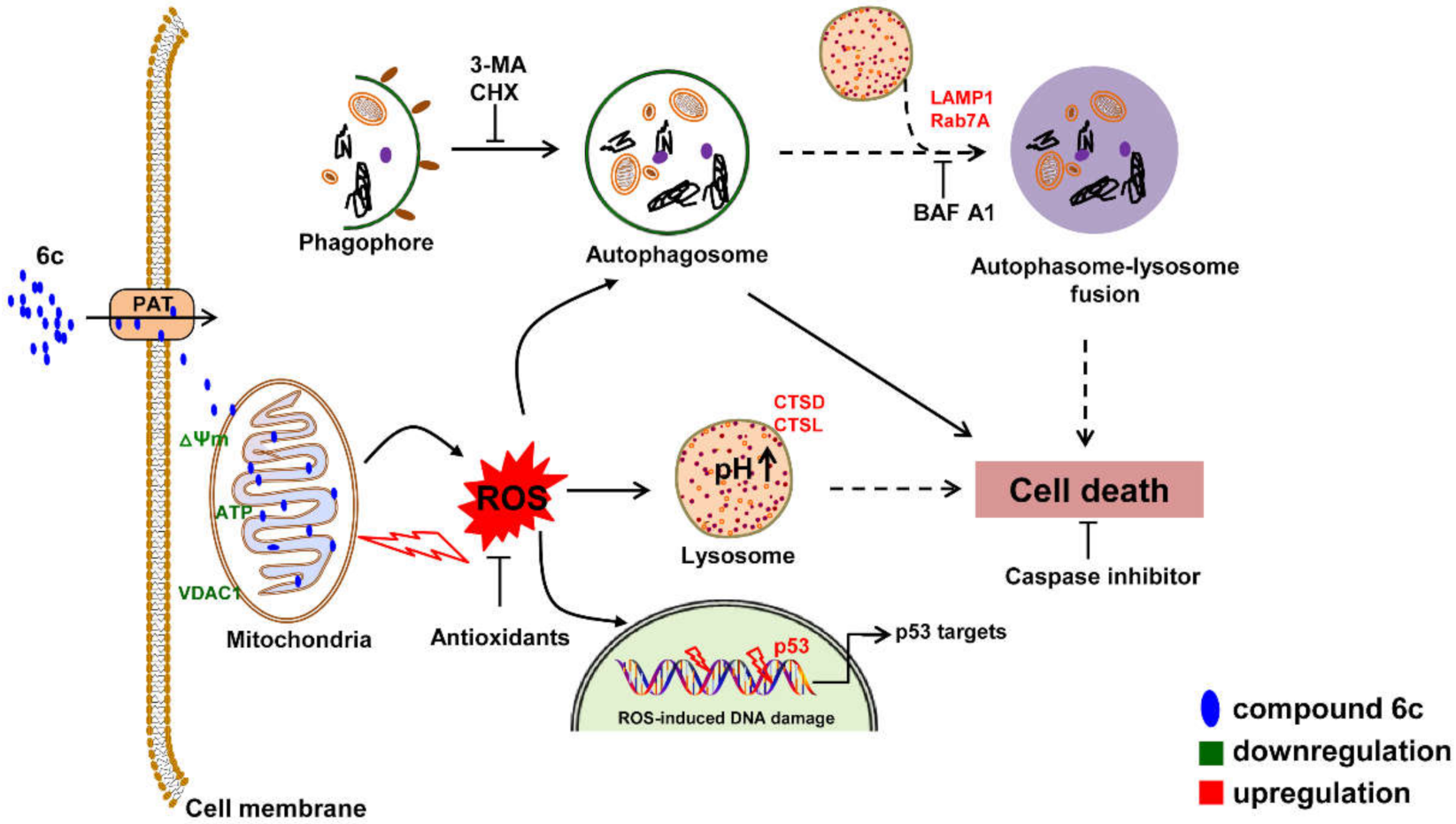
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Nonnenmacher, L.; Hasslacher, S.; Zimmermann, J.; Karpel-Massler, G.; La Ferla-Brühl, K.; Barry, S.E.; Burster, T.; Siegelin, M.D.; Brühl, O.; Halatsch, M.-E.; et al. Cell death induction in cancer therapy—Past, Present, and Future. Crit. Rev. Oncog. 2016, 21, 253–267. [Google Scholar] [CrossRef]
- Martins, I.; Raza, S.Q.; Voisin, L.; Dakhli, H.; Allouch, A.; Law, F.; Sabino, D.; De Jong, D.; Thoreau, M.; Mintet, E.; et al. Anticancer chemotherapy and radiotherapy trigger both non-cell-autonomous and cell-autonomous death. Cell Death Dis. 2018, 9, 1–18. [Google Scholar] [CrossRef]
- Fulda, S.; Kogel, D. Cell death by autophagy: Emerging molecular mechanisms and implications for cancer therapy. Oncogene 2015, 34, 5105–5113. [Google Scholar] [CrossRef]
- Kreuzaler, P.; Watson, C.J. Killing a cancer: What are the alternatives? Nat. Rev. Cancer 2012, 12, 411–424. [Google Scholar] [CrossRef]
- Yu, L.; Alva, A.; Su, H.; Dutt, P.; Freundt, E.; Welsh, S.; Baehrecke, E.H.; Lenardo, M.J. Regulation of an ATG7-beclin 1 Program of Autophagic Cell Death by Caspase-8. Science 2004, 304, 1500–1502. [Google Scholar] [CrossRef]
- Liu, Y.; Levine, B.L. Autosis and autophagic cell death: The dark side of autophagy. Cell Death Differ. 2014, 22, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 1–16. [Google Scholar] [CrossRef]
- Bork, U.; Lee, W.K.; Kuchler, A.; Dittmar, T.; Thevenod, F. Cadmium-induced DNA damage triggers G(2)/M arrest via chk1/2 and cdc2 in p53-deficient kidney proximal tubule cells. Am. J. Physiol. Renal. Physiol. 2010, 298, F255–F265. [Google Scholar] [CrossRef] [Green Version]
- Roos, W.; Thomas, A.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2015, 16, 20–33. [Google Scholar] [CrossRef]
- Noh, J.; Kwon, B.; Han, E.; Park, M.; Yang, W.; Cho, W.; Yoo, W.; Khang, G.; Lee, D. Amplification of oxidative stress by a dual stimuli-responsive hybrid drug enhances cancer cell death. Nat. Commun. 2015, 6, 6907. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Zhou, B.; Zhang, J.-Y.; Liu, X.-S.; Chen, H.-Z.; Ai, Y.-L.; Cheng, K.; Sun, R.-Y.; Zhou, D.; Han, J.; Wu, Q. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018, 28, 1171–1185. [Google Scholar] [CrossRef] [Green Version]
- Ivanova, D.; Bakalova, R.; Lazarova, D.; Gadjeva, V.; Zhelev, Z. The impact of reactive oxygen species on anticancer therapeutic strategies. Adv. Clin. Exp. Med. 2013, 22, 899–908. [Google Scholar]
- Raj, L.; Ide, T.; Gurkar, A.U.; Foley, M.; Schenone, M.; Li, X.; Tolliday, N.J.; Golub, T.R.; Carr, S.A.; Shamji, A.F.; et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 2011, 475, 231–234. [Google Scholar] [CrossRef]
- Pritchard, R.; Rodríguez-Enríquez, S.; Pacheco-Velázquez, S.C.; Bortnik, V.; Moreno-Sánchez, R.; Ralph, S. Celecoxib inhibits mitochondrial O2 consumption, promoting ROS dependent death of murine and human metastatic cancer cells via the apoptotic signalling pathway. Biochem. Pharmacol. 2018, 154, 318–334. [Google Scholar] [CrossRef] [Green Version]
- Dai, F.; Li, Q.; Wang, Y.; Ge, C.; Feng, C.; Xie, S.; He, H.; Xu, X.; Wang, C. Design, synthesis, and biological evaluation of mitochondria-targeted flavone-naphthalimide-polyamine conjugates with antimetastatic activity. J. Med. Chem. 2017, 60, 2071–2083. [Google Scholar] [CrossRef]
- Aki, T.; Nara, A.; Uemura, K. Cytoplasmic vacuolization during exposure to drugs and other substances. Cell Biol. Toxicol. 2012, 28, 125–131. [Google Scholar] [CrossRef]
- Jin, H.O.; Hong, S.E.; Park, J.A.; Chang, Y.H.; Hong, Y.J.; Park, I.C.; Lee, J.K. Inhibition of JNK-mediated autophagy enhances NSCLC cell sensitivity to mTORC1/2 inhibitors. Sci. Rep. 2016, 6, 28945. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, A.; Ordóñez, R.; Reiter, R.J.; González-Gallego, J.; Mauriz, J.L. Melatonin and endoplasmic reticulum stress: Relation to autophagy and apoptosis. J. Pineal Res. 2015, 59, 292–307. [Google Scholar] [CrossRef]
- Mizukami, H.; Takahashi, K.; Inaba, W.; Tsuboi, K.; Osonoi, S.; Yoshida, T.; Yagihashi, S. Involvement of oxidative stress-induced DNA damage, endoplasmic reticulum stress, and autophagy deficits in the decline of beta-cell mass in Japanese type 2 diabetic patients. Diabetes Care 2014, 37, 1966–1974. [Google Scholar] [CrossRef] [Green Version]
- Shieh, S.-Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA Damage-Induced Phosphorylation of p53 Alleviates Inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Murray-Zmijewski, F.; Slee, E.A.; Lu, X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat. Rev. Mol. Cell Biol. 2008, 9, 702–712. [Google Scholar] [CrossRef]
- Luo, Z.; Cui, R.; Tili, E.; Croce, C. Friend or Foe: MicroRNAs in the p53 network. Cancer Lett. 2018, 419, 96–102. [Google Scholar] [CrossRef]
- Jones, M.F.; Lal, A. MicroRNAs, wild-type and mutant p53: More questions than answers. RNA Biol. 2012, 9, 781–791. [Google Scholar] [CrossRef]
- Puissant, A.; Robert, G.; Fenouille, N.; Luciano, F.; Cassuto, J.-P.; Raynaud, S.; Auberger, P. Resveratrol Promotes Autophagic Cell Death in Chronic Myelogenous Leukemia Cells via JNK-Mediated p62/SQSTM1 Expression and AMPK Activation. Cancer Res. 2010, 70, 1042–1052. [Google Scholar] [CrossRef] [Green Version]
- Andrade, R.M.; Wessendarp, M.; Gubbels, M.-J.; Striepen, B.; Subauste, C.S. CD40 induces macrophage anti–Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J. Clin. Investig. 2006, 116, 2366–2377. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Stannard, K.; Gabutero, E.; Clark, A.; Neo, S.-Y.; Onturk, S.; Blanchard, H.; Ralph, S.J. Galectin-1 as a potent target for cancer therapy: Role in the tumor microenvironment. Cancer Metastasis Rev. 2012, 31, 763–778. [Google Scholar] [CrossRef] [Green Version]
- Pecorari, L.; Marin, O.; Silvestri, C.; Candini, O.; Rossi, E.; Guerzoni, C.; Cattelani, S.; Mariani, S.A.; Corradini, F.; Ferrari-Amorotti, G.; et al. Elongation Factor 1 alpha interacts with phospho-Akt in breast cancer cells and regulates their proliferation, survival and motility. Mol. Cancer 2009, 8, 58. [Google Scholar] [CrossRef]
- Jaäger, S.; Bucci, C.; Tanida, I.; Ueno, T.; Kominami, E.; Saftig, P.; Eskelinen, E.-L. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004, 117, 4837–4848. [Google Scholar] [CrossRef] [Green Version]
- Ben-Hail, D.; Begas-Shvartz, R.; Shalev, M.; Shteinfer-Kuzmine, A.; Gruzman, A.; Reina, S.; De Pinto, V.; Shoshan-Barmatz, V. Novel compounds targeting the mitochondrial protein VDAC1 inhibit apoptosis and protect against mitochondrial dysfunction. J. Biol. Chem. 2016, 291, 24986–25003. [Google Scholar] [CrossRef] [Green Version]
- Lao, Y.; Wan, G.; Liu, Z.; Wang, X.; Ruan, P.; Xu, W.; Xu, D.; Xie, W.; Zhang, Y.; Xu, H.; et al. The natural compound oblongifolin C inhibits autophagic flux and enhances antitumor efficacy of nutrient deprivation. Autophagy 2014, 10, 736–749. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Downs, D.; Ghosh, K.; Ghosh, A.K.; Staib, P.; Monod, M.; Tang, J. Candida albicans secreted aspartic proteases 4–6 induce apoptosis of epithelial cells by a novel Trojan horse mechanism. FASEB J. 2013, 27, 2132–2144. [Google Scholar] [CrossRef]
- Li, D.L.; Wang, Z.V.; Ding, G.; Tan, W.; Luo, X.; Criollo, A.; Xie, M.; Jiang, N.; May, H.; Kyrychenko, V.; et al. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation 2016, 133, 1668–1687. [Google Scholar] [CrossRef] [Green Version]
- Whelan, K.A.; Merves, J.F.; Giroux, V.; Tanaka, K.; Guo, A.; Chandramouleeswaran, P.M.; Benitez, A.J.; Dods, K.; Que, J.; Masterson, J.C.; et al. Autophagy mediates epithelial cytoprotection in eosinophilic oesophagitis. Gut 2016, 66, 1197–1207. [Google Scholar] [CrossRef]
- Leanza, L.; Romio, M.; Becker, K.A.; Azzolini, M.; Trentin, L.; Managò, A.; Venturini, E.; Zaccagnino, A.; Mattarei, A.; Carraretto, L.; et al. Direct pharmacological targeting of a mitochondrial ion channel selectively kills tumor cells in vivo. Cancer Cell 2017, 31, 516–531.e10. [Google Scholar] [CrossRef] [Green Version]
- Vaquero, E.C.; Edderkaoui, M.; Pandol, S.J.; Gukovsky, I.; Gukovskaya, A. Reactive oxygen species produced by NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. J. Biol. Chem. 2004, 279, 34643–34654. [Google Scholar] [CrossRef] [Green Version]
- Diebold, L.; Chandel, N.S. Mitochondrial ROS regulation of proliferating cells. Free. Radic. Biol. Med. 2016, 100, 86–93. [Google Scholar] [CrossRef]
- Barlow, J.L.; Jensen, V.H.; Affourtit, C. Uncoupling protein-2 attenuates palmitoleate protection against the cytotoxic production of mitochondrial reactive oxygen species in INS-1E insulinoma cells. Redox Biol. 2014, 4, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Liu, G.; Long, M.; Zou, H.; Cui, H. Alpha lipoic acid attenuates cadmium-induced nephrotoxicity via the mitochondrial apoptotic pathways in rat. J. Inorg. Biochem. 2018, 184, 19–26. [Google Scholar] [CrossRef]
- Kim, K.-H.; Lee, B.; Kim, Y.-R.; Kim, M.-A.; Ryu, N.; Jung, D.J.; Kim, U.-K.; Baek, J.-I.; Lee, K.-Y. Evaluating protective and therapeutic effects of alpha-lipoic acid on cisplatin-induced ototoxicity. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.-B.; Shi, Y.-H.; Zhou, J.; Qiu, S.-J.; Xu, Y.; Dai, Z.; Shi, G.-M.; Wang, X.-Y.; Ke, A.-W.; Wu, B.; et al. Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res. 2008, 68, 9167–9175. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Zhong, W.; Huang, H.; He, H.; Jiang, F.; Chen, Y.; Yue, F.; Zou, J.; Li, X.; He, Y.; et al. Autophagy defects suggested by low levels of autophagy activator MAP1S and high levels of autophagy inhibitor LRPPRC predict poor prognosis of prostate cancer patients. Mol. Carcinog. 2014, 54, 1194–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; Van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016, 30, 1704–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenfeldt, M.T.; Ryan, K.M. The role of autophagy in tumour development and cancer therapy. Expert Rev. Mol. Med. 2009, 11, e36. [Google Scholar] [CrossRef] [Green Version]
- Janku, F.; McConkey, D.J.; Hong, D.S.; Kurzrock, R. Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 2011, 8, 528–539. [Google Scholar] [CrossRef]
- Mindell, J.A. Lysosomal Acidification Mechanisms. Annu. Rev. Physiol. 2012, 74, 69–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, K.; Li, Y.; Wang, S.; Wang, X.; Liao, C.; Hu, X.; Fan, L.; Kang, Q.; Zeng, Y.; Wu, X.; et al. Inhibition of autophagosome-lysosome fusion by ginsenoside Ro via the ESR2-NCF1-ROS pathway sensitizes esophageal cancer cells to 5-fluorouracil-induced cell death via the CHEK1-mediated DNA damage checkpoint. Autophagy 2016, 12, 1593–1613. [Google Scholar] [CrossRef] [Green Version]
- Mauvezin, C.; Nagy, P.; Juhasz, G.; Neufeld, T.P. Autophagosome–lysosome fusion is independent of V-ATPase-mediated acidification. Nat. Commun. 2015, 6, 7007. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Zhang, N.; Dou, Y.; Mao, G.; Bi, C.; Pang, W.; Liu, X.; Song, D.; Deng, H. Lysosomal dysfunction and autophagy blockade contribute to IMB-6G-induced apoptosis in pancreatic cancer cells. Sci. Rep. 2017, 7, srep41862. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Li, G.; Zheng, Y.; Shen, H.-M.; Hu, X.; Ming, Q.-L.; Huang, C.; Li, P.; Gao, N. A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy 2015, 11, 1259–1279. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, Q.; Song, D.; Zen, R.; Zhang, L.; Wang, Y.; Yang, H.; Zhang, D.; Jia, J.; Zhang, J.; et al. Lysosomal dysfunction and autophagy blockade contribute to autophagy-related cancer suppressing peptide-induced cytotoxic death of cervical cancer cells through the AMPK/mTOR pathway. J. Exp. Clin. Cancer Res. 2020, 39, 1–18. [Google Scholar] [CrossRef]
- Cheng, X.-T.; Xie, Y.-X.; Zhou, B.; Huang, N.; Farfel-Becker, T.; Sheng, Z.-H. Revisiting LAMP1 as a marker for degradative autophagy-lysosomal organelles in the nervous system. Autophagy 2018, 14, 1472–1474. [Google Scholar] [CrossRef]
- Banerjee, S.; Veale, E.B.; Phelan, C.M.; Murphy, S.A.; Tocci, G.M.; Gillespie, L.J.; Frimannsson, D.O.; Kelly, J.M.; Gunnlaugsson, T. Recent advances in the development of 1,8-naphthalimide based DNA targeting binders, anticancer and fluorescent cellular imaging agents. Chem. Soc. Rev. 2013, 42, 1601–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barzilai, A.; Yamamoto, K.-I. DNA damage responses to oxidative stress. DNA Repair 2004, 3, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Czarny, P.; Pawlowska, E.; Bialkowska-Warzecha, J.; Kaarniranta, K.; Blasiak, J. Autophagy in DNA Damage Response. Int. J. Mol. Sci. 2015, 16, 2641–2662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, C.; Li, D.-J.; Jiang, Y.-J.; Tong, J.; Fu, H.; Wu, Y.-H.; Shen, F.-M. Vascular smooth muscle cell senescence and age-related diseases: State of the art. Biochim. Biophys. Acta aMol. Basis Dis. 2018, 1865, 1810–1821. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Xu, X.; Che, D.; Fan, R.; Gao, M.; Cao, Y.; Ge, C.; Feng, Y.; Li, J.; Xie, S.; et al. Reactive Oxygen Species Mediate 6c-Induced Mitochondrial and Lysosomal Dysfunction, Autophagic Cell Death, and DNA Damage in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 10987. https://doi.org/10.3390/ijms222010987
Wang S, Xu X, Che D, Fan R, Gao M, Cao Y, Ge C, Feng Y, Li J, Xie S, et al. Reactive Oxygen Species Mediate 6c-Induced Mitochondrial and Lysosomal Dysfunction, Autophagic Cell Death, and DNA Damage in Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2021; 22(20):10987. https://doi.org/10.3390/ijms222010987
Chicago/Turabian StyleWang, Senzhen, Xiaojuan Xu, Delu Che, Ronghui Fan, Mengke Gao, Yue Cao, Chaochao Ge, Yongli Feng, Jinghua Li, Songqiang Xie, and et al. 2021. "Reactive Oxygen Species Mediate 6c-Induced Mitochondrial and Lysosomal Dysfunction, Autophagic Cell Death, and DNA Damage in Hepatocellular Carcinoma" International Journal of Molecular Sciences 22, no. 20: 10987. https://doi.org/10.3390/ijms222010987