
The Regulatory Roles of PPARs in Skeletal Muscle Fuel Metabolism and Inflammation: Impact of PPAR Agonism on Muscle in Chronic Disease, Contraction and Sepsis



Abstract
:1. Introduction
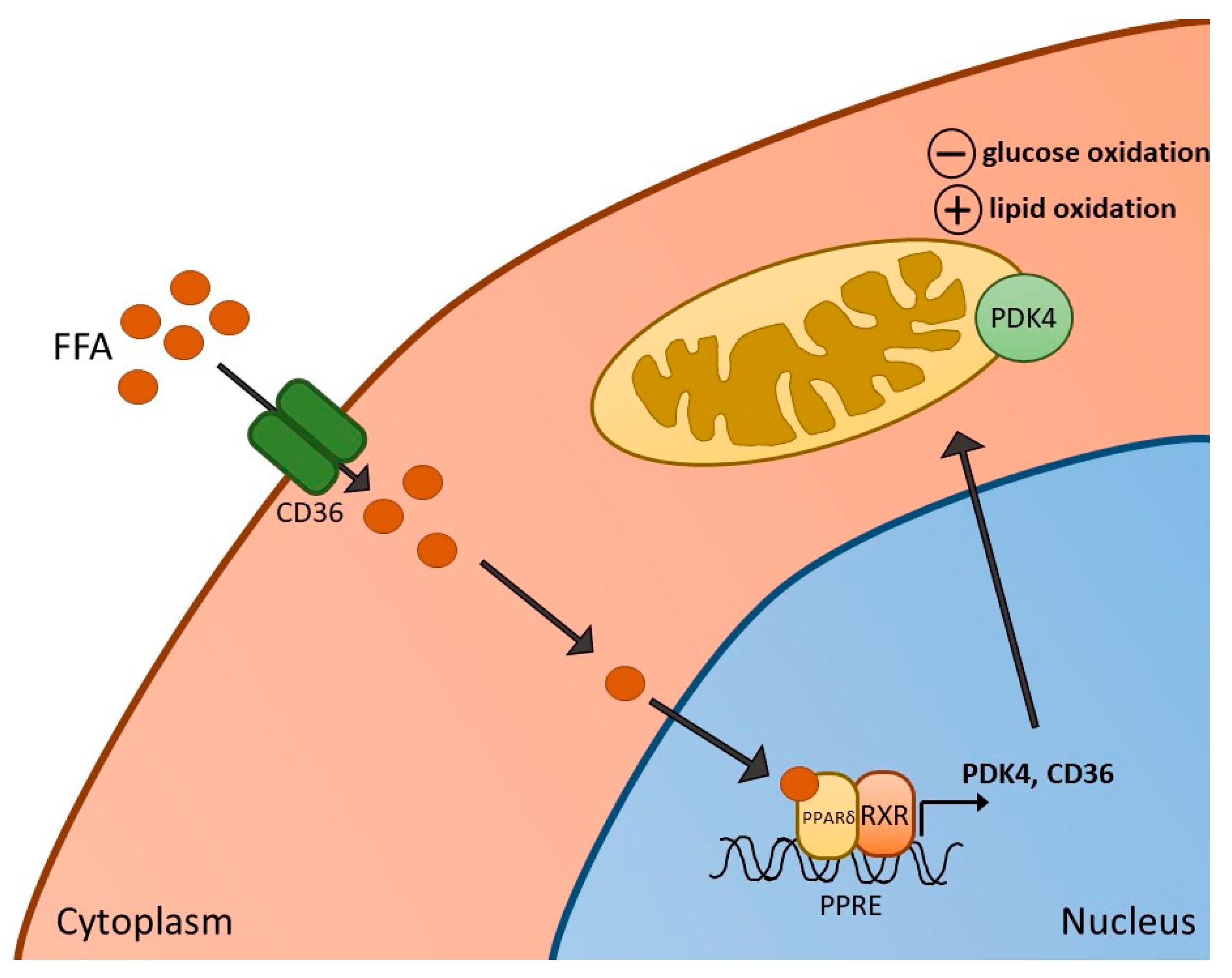
2. Metabolic Functions of PPARs and Their Actions in Skeletal Muscle
3. PPARδ Agonism and Skeletal Muscle Metabolism, Contractile Function and Inflammation
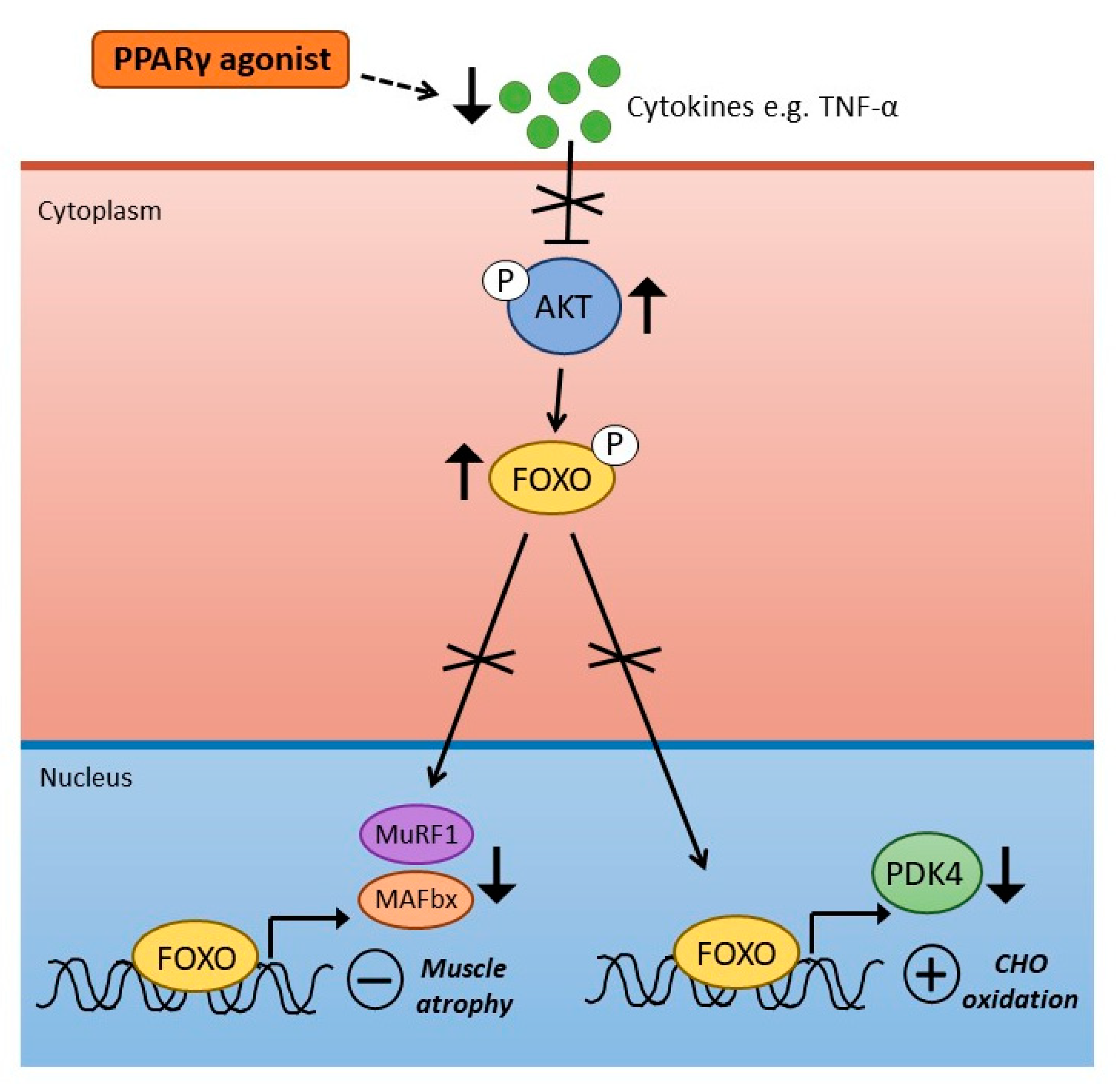
4. PPARγ Agonism and Skeletal Muscle Metabolism and Inflammation
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berger, J.; Wagner, J.A. Physiological and Therapeutic Roles of Peroxisome Proliferator-Activated Receptors. Diabetes Technol. Ther. 2002, 4, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Mukund, K.; Subramaniam, S. Skeletal muscle: A review of molecular structure and function, in health and disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puthucheary, Z.A.; Rawal, J.; McPhail, M.; Connolly, B.; Ratnayake, G.; Chan, P.; Hopkinson, N.S.; Padhke, R.; Dew, T.; Sidhu, P.S.; et al. Acute Skeletal Muscle Wasting in Critical Illness. JAMA 2013, 310, 1591–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varga, T.; Czimmerer, Z.; Nagy, L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2011, 1812, 1007–1022. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Olson, P.; Evans, R.M. Minireview: Lipid Metabolism, Metabolic Diseases, and Peroxisome Proliferator-Activated Receptors. Endocrinology 2003, 144, 2201–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Yamamoto, J.; Iwasaki, S.; Asaba, H.; Hamura, H.; Ikeda, Y.; Watanabe, M.; Magoori, K.; Ioka, R.X.; Tachibana, K.; et al. Activation of peroxisome proliferator-activated receptor δ induces fatty acid β-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, B.D. Review of the expression of peroxisome proliferator-activated receptors alpha (PPARα), beta (PPARβ), and gamma (PPARγ) in rodent and human development. Reprod. Toxicol. 2009, 27, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Baes, M.; Peeters, A. Role of PPARα in hepatic carbohydrate metabolism. PPAR Res. 2010, 2010, 572405. [Google Scholar]
- Muoio, D.M.; MacLean, P.S.; Lang, D.B.; Li, S.; Houmard, J.A.; Way, J.M.; Winegar, D.A.; Corton, J.C.; Dohm, G.L.; Kraus, W.E. Fatty Acid Homeostasis and Induction of Lipid Regulatory Genes in Skeletal Muscles of Peroxisome Proliferator-activated Receptor (PPAR) α Knock-out Mice. Evidence for compensatory regulation by PPARδ. J. Biol. Chem. 2002, 277, 26089–26097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueno, H.; Saitoh, Y.; Mizuta, M.; Shiiya, T.; Noma, K.; Mashiba, S.; Kojima, S.; Nakazato, M. Fenofibrate ameliorates insulin resistance, hypertension and novel oxidative stress markers in patients with metabolic syndrome. Obes. Res. Clin. Pr. 2011, 5, e335–e340. [Google Scholar] [CrossRef]
- Koh, K.K.; Han, S.H.; Quon, M.J.; Ahn, J.Y.; Shin, E.K. Beneficial Effects of Fenofibrate to Improve Endothelial Dysfunction and Raise Adiponectin Levels in Patients With Primary Hypertriglyceridemia. Diabetes Care 2005, 28, 1419–1424. [Google Scholar] [CrossRef] [Green Version]
- Fajas, L.; Auboeuf, D.; Raspé, E.; Schoonjans, K.; Lefebvre, A.-M.; Saladin, R.; Najib, J.; Laville, M.; Fruchart, J.-C.; Deeb, S.; et al. The Organization, Promoter Analysis, and Expression of the Human PPARγ Gene. J. Biol. Chem. 1997, 272, 18779–18789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Akoum, S. PPAR Gamma at the Crossroads of Health and Disease: A Masterchef in Metabolic Homeostasis. Endocrinol. Metab. Syndr. 2014, 3, 2161–1017. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, Y.; He, H.; Mandarino, L.J.; DeFronzo, R.A. Rosiglitazone improves downstream insulin receptor signaling in type 2 diabetic patients. Diabetes 2003, 52, 1943–1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gastaldelli, A.; Ferrannini, E.; Miyazaki, Y.; Matsuda, M.; Mari, A.; DeFronzo, R.A. Thiazolidinediones improve β-cell function in type 2 diabetic patients. Am. J. Physiol. Metab. 2007, 292, E871–E883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; et al. The fat-derived hormone adiponectin reverses insulin re-sistance associated with both lipoatrophy and obesity. Nat. Med. 2001, 7, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Ehrenborg, E.; Krook, A. Regulation of Skeletal Muscle Physiology and Metabolism by Peroxisome Proliferator-Activated Receptor δ. Pharmacol. Rev. 2009, 61, 373–393. [Google Scholar] [CrossRef] [Green Version]
- Tan, N.S.; Vázquez-Carrera, M.; Montagner, A.; Sng, M.K.; Guillou, H.; Wahli, W. Transcriptional control of physiological and pathological processes by the nuclear receptor PPARβ/δ. Prog. Lipid Res. 2016, 64, 98–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phua, W.W.T.; Wong, M.X.Y.; Liao, Z.; Tan, N.S. An apparent functional consequence in skeletal muscle physiology via pe-roxisome proliferator-activated receptors. Int. J. Mol. Sci. 2018, 19, 1425. [Google Scholar] [CrossRef] [Green Version]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dressel, U.; Allen, T.L.; Pippal, J.B.; Rohde, P.R.; Lau, P.; Muscat, G.E.O. The Peroxisome Proliferator-Activated Receptor β/δ Agonist, GW501516, Regulates the Expression of Genes Involved in Lipid Catabolism and Energy Uncoupling in Skeletal Muscle Cells. Mol. Endocrinol. 2003, 17, 2477–2493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, S.J.; Harris, R.A.; Heigenhauser, G.J.F.; Spriet, L.L. Muscle fiber type comparison of PDH kinase activity and isoform expression in fed and fasted rats. Am. J. Physiol. Integr. Comp. Physiol. 2001, 280, R661–R668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieland, O.H. The mammalian pyruvate dehydrogenase complex: Structure and regulation. Rev. Physiol. Biochem. Pharmacol. 1983, 96, 123–170. [Google Scholar] [CrossRef]
- Bowker-Kinley, M.M.; Davis, I.W.; Wu, P.; Harris, A.R.; Popov, M.K. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 1998, 329, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Gudi, R.; Wu, P.; Harris, R.A.; Hamilton, J.; Popov, K.M. Isoenzymes of Pyruvate Dehydrogenase Phosphatase. DNA-derived amino acid sequences, expression, and regulation. J. Biol. Chem. 1998, 273, 17680–17688. [Google Scholar] [CrossRef] [Green Version]
- Sugden, M.C.; Kraus, A.; Harris, R.A.; Holness, M.J. Fibre-type specific modification of the activity and regulation of skeletal muscle pyruvate dehydrogenase kinase (PDK) by prolonged starvation and refeeding is associated with targeted regulation of PDK isoenzyme 4 expression. Biochem. J. 2000, 346, 651–657. [Google Scholar] [CrossRef]
- Wu, P.; Inskeep, K.; Bowker-Kinley, M.M.; Popov, K.M.; Harris, R.A. Mechanism responsible for inactivation of skeletal muscle pyruvate dehydrogenase complex in starvation and diabetes. Diabetes 1999, 48, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Tsintzas, K.; Jewell, K.; Kamran, M.; Laithwaite, D.; Boonsong, T.; Littlewood, J.; Macdonald, I.; Bennett, A. Differential regulation of metabolic genes in skeletal muscle during starvation and refeeding in humans. J. Physiol. 2006, 575, 291–303. [Google Scholar] [CrossRef]
- Spriet, L.L.; Tunstall, R.J.; Watt, M.J.; Mehan, K.A.; Hargreaves, M.; Cameron-Smith, D. Pyruvate dehydrogenase activation and kinase expression in human skeletal muscle during fasting. J. Appl. Physiol. 2004, 96, 2082–2087. [Google Scholar] [CrossRef] [Green Version]
- Furuyama, T.; Kitayama, K.; Yamashita, H.; Mori, N. Forkhead transcription factor FOXO1 (FKHR)-dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem. J. 2003, 375, 365–371. [Google Scholar] [CrossRef] [Green Version]
- Wahli, W.; Michalik, L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Michalik, L.; Wahli, W. PPARs Mediate Lipid Signaling in Inflammation and Cancer. PPAR Res. 2008, 2008, 134059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop-Bailey, D.; Bystrom, J. Emerging roles of peroxisome proliferator-activated receptor-β/δ in inflammation. Pharmacol. Ther. 2009, 124, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Delerive, P.; De Bosscher, K.; Berghe, W.V.; Fruchart, J.-C.; Haegeman, G.; Staels, B. DNA Binding-Independent Induction of IκBα Gene Transcription by PPARα. Mol. Endocrinol. 2002, 16, 1029–1039. [Google Scholar] [CrossRef]
- Huang, W.; Glass, C.K. Nuclear receptors and inflammation control: Molecular mechanisms and pathophysiological rele-vance. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1542–1549. [Google Scholar] [CrossRef] [PubMed]
- Constantin, D.; Constantin-Teodosiu, D.; Layfield, R.; Tsintzas, K.; Bennett, A.J.; Greenhaff, P.L. PPARδ agonism induces a change in fuel metabolism and activation of an atrophy programme, but does not impair mitochondrial function in rat skeletal muscle. J. Physiol. 2007, 583, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Léger, B.; Cartoni, R.; Praz, M.; Lamon, S.; Dériaz, O.; Crettenand, A.; Gobelet, C.; Rohmer, P.; Konzelmann, M.; Luthi, F.; et al. Akt signalling through GSK-3β, mTOR and Foxo1 is involved in human skeletal muscle hypertrophy and atrophy. J. Physiol. 2006, 576, 923–933. [Google Scholar] [CrossRef]
- Chen, W.; Gao, R.; Xie, X.; Zheng, Z.; Li, H.; Li, S.; Dong, F.; Wang, L. A metabolomic study of the PPARδ agonist GW501516 for enhancing running endurance in Kunming mice. Sci. Rep. 2015, 5, 9884. [Google Scholar] [CrossRef] [Green Version]
- Constantin-Teodosiu, D.; Baker, D.J.; Constantin, D.; Greenhaff, P.L. PPARδ agonism inhibits skeletal muscle PDC activity, mitochondrial ATP production and force generation during prolonged contraction. J. Physiol. 2009, 587, 231–239. [Google Scholar] [CrossRef]
- Constantin-Teodosiu, D.; Cederblad, G.; Hultman, E. PDC activity and acetyl group accumulation in skeletal muscle during isometric contraction. J. Appl. Physiol. 1993, 74, 1712–1718. [Google Scholar] [CrossRef]
- Constantin-Teodosiu, D.; Constantin, D.; Stephens, F.; Laithwaite, D.; Greenhaff, P.L. The Role of FOXO and PPAR Transcription Factors in Diet-Mediated Inhibition of PDC Activation and Carbohydrate Oxidation During Exercise in Humans and the Role of Pharmacological Activation of PDC in Overriding These Changes. Diabetes 2012, 61, 1017–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantin-Teodosiu, D. Regulation of Muscle Pyruvate Dehydrogenase Complex in Insulin Resistance: Effects of Exercise and Dichloroacetate. Diabetes Metab. J. 2013, 37, 301–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, H.-C.; Greenhaff, P.L.; Constantin-Teodosiu, D. PPARδ and FOXO1 Mediate Palmitate-Induced Inhibition of Muscle Pyruvate Dehydrogenase Complex and CHO Oxidation, Events Reversed by Electrical Pulse Stimulation. Int. J. Mol. Sci. 2020, 21, 5942. [Google Scholar] [CrossRef]
- Grosset, J.-F.; Crowe, L.; de Vito, G.; O’Shea, D.; Caulfield, B. Comparative effect of a 1 h session of electrical muscle stimulation and walking activity on energy expenditure and substrate oxidation in obese subjects. Appl. Physiol. Nutr. Metab. 2013, 38, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Nieuwoudt, S.; Mulya, A.; Fealy, C.E.; Martelli, E.; Dasarathy, S.; Prasad, S.V.N.; Kirwan, J.P. In vitro contraction protects against palmitate-induced insulin resistance in C2C12 myotubes. Am. J. Physiol. Physiol. 2017, 313, C575–C583. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Moldawer, L.L.; Opal, S.M.; Reinhart, K.; Turnbull, I.R.; Vincent, J.L. Sepsis and septic shock. Nat. Rev. Dis. Prim. 2016, 2, 16045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, M. The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence 2014, 5, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Chioléro, R.; Revelly, J.P.; Tappy, L. Energy metabolism in sepsis and injury. Nutrition 1997, 13, 45–51. [Google Scholar] [CrossRef]
- Kamisoglu, K.; Haimovich, B.; Calvano, S.E.; Coyle, S.M.; Corbett, S.A.; Langley, R.J.; Kingsmore, S.F.; Androulakis, I.P. Human metabolic response to systemic inflammation: Assessment of the concordance between experimental endotoxemia and clinical cases of sepsis/SIRS. Crit. Care 2015, 19, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senol, S.P.; Temiz, M.; Guden, D.S.; Cecen, P.; Sari, A.N.; Sahan-Firat, S.; Falck, J.R.; Dakarapu, R.; Malik, K.U.; Tunctan, B. Contribution of PPARα/β/γ, AP-1, importin-α3, and RXRα to the protective effect of 5,14-HEDGE, a 20-HETE mimetic, against hypotension, tachycardia, and inflammation in a rat model of septic shock. Inflamm. Res. 2016, 65, 367–387. [Google Scholar] [CrossRef] [PubMed]
- Tunctan, B.; Kucukkavruk, S.P.; Temiz-Resitoglu, M.; Guden, D.S.; Sari, A.N.; Sahan-Firat, S. Bexarotene, a Selective RXRα Agonist, Reverses Hypotension Associated with Inflammation and Tissue Injury in a Rat Model of Septic Shock. Inflammation 2017, 41, 337–355. [Google Scholar] [CrossRef]
- Castillero, E.; Alamdari, N.; Aversa, Z.; Gurav, A.; Hasselgren, P.-O. PPARβ/δ Regulates Glucocorticoid- and Sepsis-Induced FOXO1 Activation and Muscle Wasting. PLoS ONE 2013, 8, e59726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drosatos, K.; Lymperopoulos, A.; Kennel, P.J.; Pollak, N.; Schulze, P.C.; Goldberg, I.J. Pathophysiology of Sepsis-Related Car-diac Dysfunction: Driven by Inflammation, Energy Mismanagement, or Both? Curr. Heart Fail. Rep. 2015, 12, 130–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feingold, K.; Kim, M.S.; Shigenaga, J.; Moser, A.; Grunfeld, C. Altered expression of nuclear hormone receptors and coactivators in mouse heart during the acute-phase response. Am. J. Physiol. Metab. 2004, 286, E201–E207. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Jiang, H.; Yang, X. PPARδ Activation protects H9c2 cardiomyoblasts from LPS-induced apoptosis through the heme oxygenase-1-mediated suppression of NF-κB activation. Mol. Med. Rep. 2017, 15, 3775–3780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benton, C.R.; Holloway, G.P.; Campbell, S.E.; Yoshida, Y.; Tandon, N.N.; Glatz, J.F.; Luiken, J.J.; Spriet, L.L.; Bonen, A. Rosiglitazone increases fatty acid oxidation and fatty acid translocase (FAT/CD36) but not carnitine palmitoyltransferase I in rat muscle mitochondria. J. Physiol. 2008, 586, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Mai, K.; Andres, J.; Bobbert, T.; Assmann, A.; Biedasek, K.; Diederich, S.; Graham, I.; Larson, T.R.; Pfeiffer, A.F.; Spranger, J. Rosiglitazone increases fatty acid Δ9-desaturation and decreases elongase activity index in human skeletal muscle in vivo. Metabolism 2012, 61, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Muurling, M.; Mensink, R.P.; Pijl, H.; Romijn, J.A.; Havekes, L.M.; Voshol, P.J. Rosiglitazone improves muscle insulin sensitivity, irrespective of increased triglyceride content, in ob/ob mice. Metabolism 2003, 52, 1078–1083. [Google Scholar] [CrossRef]
- Lessard, S.J.; Giudice, S.L.; Lau, W.; Reid, J.J.; Turner, N.; Febbraio, M.A.; Hawley, J.A.; Watt, M.J. Rosiglitazone Enhances Glucose Tolerance by Mechanisms Other than Reduction of Fatty Acid Accumulation within Skeletal Muscle. Endocrinology 2004, 145, 5665–5670. [Google Scholar] [CrossRef]
- Tan, L.; Song, A.; Ren, L.; Wang, C.; Song, G. Effect of pioglitazone on skeletal muscle lipid deposition in the insulin resistance rat model induced by high fructose diet under AMPK signaling pathway. Saudi J. Biol. Sci. 2020, 27, 1317–1323. [Google Scholar] [CrossRef]
- Mayerson, A.B.; Hundal, R.S.; Dufour, S.; Lebon, V.; Befroy, U.; Cline, G.W.; Enocksson, S.; Inzucchi, S.E.; Shulman, G.I.; Petersen, K.F. The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes 2002, 51, 797–802. [Google Scholar] [CrossRef] [Green Version]
- Coletta, D.K.; Sriwijitkamol, A.; Wajcberg, E.; Tantiwong, P.; Li, M.; Prentki, M.; Madiraju, M.; Jenkinson, C.P.; Cersosimo, E.; Musi, N.; et al. Pioglitazone stimulates AMP-activated protein kinase signalling and increases the expression of genes involved in adiponectin signalling, mitochondrial function and fat oxidation in human skeletal muscle in vivo: A randomised trial. Diabetologia 2009, 52, 723–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mensink, M.; Hesselink, M.K.C.; Russell, A.P.; Schaart, G.; Sels, J.-P.; Schrauwen, P. Improved skeletal muscle oxidative enzyme activity and restoration of PGC-1α and PPARβ/δ gene expression upon rosiglitazone treatment in obese patients with type 2 diabetes mellitus. Int. J. Obes. 2007, 31, 1302–1310. [Google Scholar] [CrossRef] [Green Version]
- Crossland, H.; Constantin-Teodosiu, D.; Gardiner, S.M.; Greenhaff, P.L. Peroxisome proliferator-activated receptor γ agonism attenuates endotoxaemia-induced muscle protein loss and lactate accumulation in rats. Clin. Sci. 2017, 131, 1437–1447. [Google Scholar] [CrossRef] [Green Version]
- Crossland, H.; Constantin-Teodosiu, D.; Gardiner, S.M.; Constantin, D.; Greenhaff, P.L. A potential role for Akt/FOXO sig-nalling in both protein loss and the impairment of muscle carbohydrate oxidation during sepsis in rodent skeletal muscle. J. Physiol. 2008, 586, 5589–5600. [Google Scholar] [CrossRef] [PubMed]
- Alamdari, N.; Constantin-Teodosiu, D.; Murton, A.J.; Gardiner, S.M.; Bennett, T.; Layfield, R.; Greenhaff, P.L. Temporal changes in the involvement of pyruvate dehydrogenase complex in muscle lactate accumulation during lipopolysaccharide infusion in rats. J. Physiol. 2008, 586, 1767–1775. [Google Scholar] [CrossRef]
- Murton, A.J.; Alamdari, N.; Gardiner, S.M.; Constantin-Teodosiu, D.; Layfield, R.; Bennett, T.; Greenhaff, P.L. Effects of Endotoxaemia on Protein Metabolism in Rat Fast-Twitch Skeletal Muscle and Myocardium. PLoS ONE 2009, 4, e6945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drosatos, K.; Khan, R.S.; Trent, C.M.; Jiang, H.; Son, N.-H.; Blaner, W.S.; Homma, S.; Schulze, P.C.; Goldberg, I.J. Peroxisome Proliferator–Activated Receptor-γ Activation Prevents Sepsis-Related Cardiac Dysfunction and Mortality In Mice. Circ. Hear. Fail. 2013, 6, 550–562. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.; Xu, J.; Ruan, W.; Li, S.; Xiao, F. PPAR-γ Activation Prevents Septic Cardiac Dysfunction via Inhibition of Apoptosis and Necroptosis. Oxidative Med. Cell. Longev. 2017, 2017, 8326749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.M.; Cai, X.F.; Ma, Y.L.; Lu, Q. Effect of rosiglitazone on myocardial injury in septic rats through NF-κB pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 452–460. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Skeletal Muscle | Liver | Adipose | |
|---|---|---|---|
| PPARδ | + FA oxidation −carbohydrate oxidation | + FA oxidation − lipogenesis | + FA oxidation |
| PPARγ | + FA oxidation + glucose uptake | + lipogenesis + lipid storage − glucose production | + adipogenesis + lipogenesis + lipid storage + adipokine production |
| PPARα | + FA oxidation | + FA oxidation − lipid storage |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crossland, H.; Constantin-Teodosiu, D.; Greenhaff, P.L. The Regulatory Roles of PPARs in Skeletal Muscle Fuel Metabolism and Inflammation: Impact of PPAR Agonism on Muscle in Chronic Disease, Contraction and Sepsis. Int. J. Mol. Sci. 2021, 22, 9775. https://doi.org/10.3390/ijms22189775
Crossland H, Constantin-Teodosiu D, Greenhaff PL. The Regulatory Roles of PPARs in Skeletal Muscle Fuel Metabolism and Inflammation: Impact of PPAR Agonism on Muscle in Chronic Disease, Contraction and Sepsis. International Journal of Molecular Sciences. 2021; 22(18):9775. https://doi.org/10.3390/ijms22189775
Chicago/Turabian StyleCrossland, Hannah, Dumitru Constantin-Teodosiu, and Paul L. Greenhaff. 2021. "The Regulatory Roles of PPARs in Skeletal Muscle Fuel Metabolism and Inflammation: Impact of PPAR Agonism on Muscle in Chronic Disease, Contraction and Sepsis" International Journal of Molecular Sciences 22, no. 18: 9775. https://doi.org/10.3390/ijms22189775