
The Role of Somatic Mutations on the Immune Response of the Tumor Microenvironment in Prostate Cancer

 ,
,  , and
, and
Abstract
:1. Introduction
2. Interactions between the Tumor Microenvironment and Cancer
3. Recent Lessons Learned about the Immune TME from Mouse Models of Human PCa
4. Role of Somatic Mutations in Shaping the Microenvironment of PCa
4.1. Phosphatase and Tensin Homologue (PTEN)
4.2. TP53
4.3. CDK12
4.4. AIRE
5. Single-Cell Descriptions of the Immune Ecosystem of the TME of Prostate Cancer
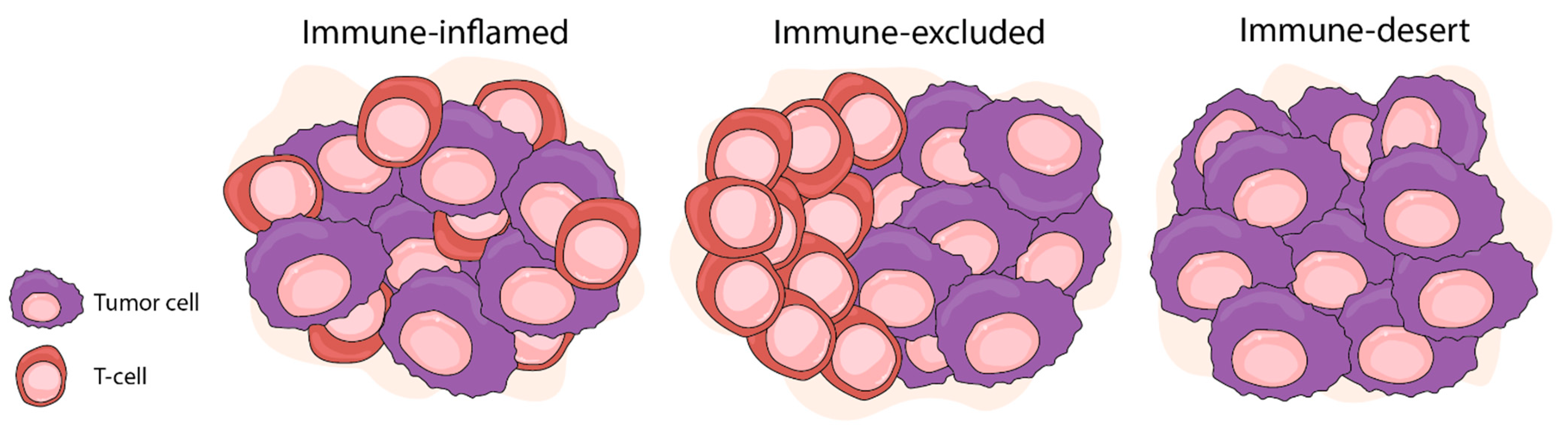
6. Spatial Analysis of the Immune Architecture of the TME of Prostate Cancer
7. Recent Immunotherapy Clinical Trials in Prostate Cancer
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Lu, S.; Stein, J.E.; Rimm, D.L.; Wang, D.W.; Bell, J.M.; Johnson, D.B.; Sosman, J.A.; Schalper, K.A.; Anders, R.A.; Wang, H.; et al. Comparison of Biomarker Modalities for Predicting Response to PD-1/PD-L1 Checkpoint Blockade: A Systematic Review and Meta-analysis. JAMA Oncol. 2019, 5, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Anandappa, A.J.; Wu, C.J.; Ott, P.A. Directing traffic: How to effectively drive t cells into tumors. Cancer Discov. 2020, 10, 185–197. [Google Scholar] [CrossRef]
- Abida, W.; Cheng, M.L.; Armenia, J.; Middha, S.; Autio, K.A.; Vargas, H.A.; Rathkopf, D.; Morris, M.J.; Danila, D.C.; Slovin, S.F.; et al. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncol. 2019, 5, 471–478. [Google Scholar] [CrossRef]
- Wu, Y.M.; Cieślik, M.; Lonigro, R.J.; Vats, P.; Reimers, M.A.; Cao, X.; Ning, Y.; Wang, L.; Kunju, L.P.; de Sarkar, N.; et al. Inactivation of CDK12 Delineates a Distinct Immunogenic Class of Advanced Prostate Cancer. Cell 2018, 173, 1770–1782. [Google Scholar] [CrossRef] [Green Version]
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate cancer. Nat. Rev. Dis. Prim. 2021, 7, 162–200. [Google Scholar]
- Wang, G.; Zhao, D.; Spring, D.J.; Depinho, R.A. Genetics and biology of prostate cancer. Genes Dev. 2018, 32, 1105–1140. [Google Scholar] [CrossRef] [Green Version]
- Cha, H.R.; Lee, J.H.; Ponnazhagan, S. Revisiting immunotherapy: A focus on prostate cancer. Cancer Res. 2020, 80, 1615–1623. [Google Scholar] [CrossRef] [Green Version]
- Petitprez, F.; Meylan, M.; de Reyniès, A.; Sautès-Fridman, C.; Fridman, W.H. The Tumor Microenvironment in the Response to Immune Checkpoint Blockade Therapies. Front. Immunol. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Frank, S.; Nelson, P.; Vasioukhin, V. Recent advances in prostate cancer research: Large-scale genomic analyses reveal novel driver mutations and DNA repair defects. F1000Research 2018, 7, 1173. [Google Scholar] [CrossRef] [PubMed]
- Jansen, C.S.; Prokhnevska, N.; Kissick, H.T. The requirement for immune infiltration and organization in the tumor microenvironment for successful immunotherapy in prostate cancer. Urol. Oncol. Semin. Orig. Investig. 2019, 37, 543–555. [Google Scholar] [CrossRef]
- Vitkin, N.; Nersesian, S.; Siemens, D.R.; Koti, M. The tumor immune contexture of prostate cancer. Front. Immunol. 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Albright, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, P.; et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 2017, 127. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Lanitis, E.; Dangaj, D.; Irving, M.; Coukos, G. Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann. Oncol. 2017, 28, xii18–xii32. [Google Scholar] [CrossRef] [PubMed]
- Maleki Vareki, S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J. Immunother. Cancer 2018, 6, 4–8. [Google Scholar] [CrossRef]
- Rådestad, E.; Egevad, L.; Jorns, C.; Mattsson, J.; Sundberg, B.; Nava, S.; Ericzon, B.G.; Henningsohn, L.; Levitsky, V.; Uhlin, M. Characterization of infiltrating lymphocytes in human benign and malignant prostate tissue. Oncotarget 2017, 8, 60257–60269. [Google Scholar] [CrossRef] [Green Version]
- Nardone, V.; Botta, C.; Caraglia, M.; Martino, E.C.; Ambrosio, M.R.; Carfagno, T.; Tini, P.; Semeraro, L.; Misso, G.; Grimaldi, A.; et al. Tumor infiltrating T lymphocytes expressing FoxP3, CCR7 or PD-1 predict the outcome of prostate cancer patients subjected to salvage radiotherapy after biochemical relapse. Cancer Biol. Ther. 2016, 17, 1213–1220. [Google Scholar] [CrossRef]
- Bronte, V.; Kasic, T.; Gri, G.; Gallana, K.; Borsellino, G.; Marigo, I.; Battistini, L.; Iafrate, M.; Prayer-Galetti, T.; Pagano, F.; et al. Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J. Exp. Med. 2005, 201, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.G.; Lehrer, J.; Chang, S.L.; Das, R.; Erho, N.; Liu, Y.; Sjöström, M.; Den, R.B.; Freedland, S.J.; Klein, E.A.; et al. The immune landscape of prostate cancer and nomination of PD-L2 as a potential therapeutic target. J. Natl. Cancer Inst. 2019, 111, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Ness, N.; Andersen, S.; Valkov, A.; Nordby, Y.; Donnem, T.; Al-Saad, S.; Busund, L.T.; Bremnes, R.M.; Richardsen, E. Infiltration of CD8+ lymphocytes is an independent prognostic factor of biochemical failure-free survival in prostate cancer. Prostate 2014, 74, 1452–1461. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Kiniwa, Y.; Miyahara, Y.; Wang, H.Y.; Peng, W.; Peng, G.; Wheeler, T.M.; Thompson, T.C.; Old, L.J.; Wang, R.F. CD8+ Foxp3+ regulatory T cells mediate immunosuppression in prostate cancer. Clin. Cancer Res. 2007, 13, 6947–6958. [Google Scholar] [CrossRef] [Green Version]
- Kaur, H.B.; Guedes, L.B.; Lu, J.; Maldonado, L.; Reitz, L.; Barber, J.R.; De Marzo, A.M.; Tosoian, J.J.; Tomlins, S.A.; Schaeffer, E.M.; et al. Association of tumor-infiltrating T-cell density with molecular subtype, racial ancestry and clinical outcomes in prostate cancer. Mod. Pathol. 2018, 31, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Apetoh, L.; Smyth, M.J.; Drake, C.G.; Abastado, J.P.; Apte, R.N.; Ayyoub, M.; Blay, J.Y.; Bonneville, M.; Butterfield, L.H.; Caignard, A.; et al. Consensus nomenclature for CD8+ T cell phenotypes in cancer. Oncoimmunology 2015, 4, 37–41. [Google Scholar] [CrossRef] [Green Version]
- Olson, B.; Li, Y.; Lin, Y.; Liu, E.T.; Patnaik, A. Mouse models for cancer immunotherapy research. Cancer Discov. 2018, 8, 1358–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidov, V.; Jensen, G.; Mai, S.; Chen, S.H.; Pan, P.Y. Analyzing One Cell at a TIME: Analysis of Myeloid Cell Contributions in the Tumor Immune Microenvironment. Front. Immunol. 2020, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Porta, C.; Morlacchi, S.; Banfi, S.; Strauss, L.; Rimoldi, M.; Totaro, M.G.; Riboldi, E. Origin and functions of Tumor-Associated Myeloid Cells (TAMCs). Cancer Microenviron. 2012, 5, 133–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezzi, M.; Seitzer, N.; Ishikawa, T.; Reschke, M.; Chen, M.; Wang, G.; Mitchell, C.; Ng, C.; Katon, J.; Lunardi, A.; et al. Diverse genetic-driven immune landscapes dictate tumor progression through distinct mechanisms. Nat. Med. 2018, 24, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 1–14. [Google Scholar] [CrossRef]
- Calcinotto, A.; Spataro, C.; Zagato, E.; Di Mitri, D.; Gil, V.; Crespo, M.; De Bernardis, G.; Losa, M.; Mirenda, M.; Pasquini, E.; et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature 2018, 559, 363–369. [Google Scholar] [CrossRef]
- Guillaume, H.; Janske, N.; Camille Corbier, C.; Nadege, T.; Stephanie, G. Tumor-associated macrophages: Shifting bad prognosis to improved efficacy in cancer therapies? Int. J. Immunother. Cancer Res. 2021, 7, 15–23. [Google Scholar] [CrossRef]
- Cassetta, L.; Baekkevold, E.S.; Brandau, S.; Bujko, A.; Cassatella, M.A.; Dorhoi, A.; Krieg, C.; Lin, A.; Loré, K.; Marini, O.; et al. Deciphering myeloid-derived suppressor cells: Isolation and markers in humans, mice and non-human primates. Cancer Immunol. Immunother. 2019, 68, 687–697. [Google Scholar] [CrossRef] [Green Version]
- Di Mitri, D.; Mirenda, M.; Vasilevska, J.; Calcinotto, A.; Delaleu, N.; Revandkar, A.; Gil, V.; Boysen, G.; Losa, M.; Mosole, S.; et al. Re-education of Tumor-Associated Macrophages by CXCR2 Blockade Drives Senescence and Tumor Inhibition in Advanced Prostate Cancer. Cell Rep. 2019, 28, 2156–2168. [Google Scholar] [CrossRef] [Green Version]
- Jiao, S.; Subudhi, S.K.; Aparicio, A.; Ge, Z.; Guan, B.; Miura, Y.; Sharma, P. Differences in Tumor Microenvironment Dictate T Helper Lineage Polarization and Response to Immune Checkpoint Therapy. Cell 2019, 179, 1177–1190. [Google Scholar] [CrossRef]
- Chen, S.; Zhu, G.; Yang, Y.; Wang, F.; Xiao, Y.-T.; Zhang, N.; Bian, X.; Zhu, Y.; Yu, Y.; Liu, F.; et al. Single-cell analysis reveals transcriptomic remodellings in distinct cell types that contribute to human prostate cancer progression. Nat. Cell Biol. 2021, 23, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kron, K.J.; Murison, A.; Zhou, S.; Huang, V.; Yamaguchi, T.N.; Shiah, Y.J.; Fraser, M.; Van Der Kwast, T.; Boutros, P.C.; Bristow, R.G.; et al. TMPRSS2-ERG fusion co-opts master transcription factors and activates NOTCH signaling in primary prostate cancer. Nat. Genet. 2017, 49, 1336–1345. [Google Scholar] [CrossRef]
- Suryawanshi, A.; Tadagavadi, R.K.; Swafford, D.; Manicassamy, S. Modulation of inflammatory responses by Wnt/β-catenin signaling in dendritic cells: A novel immunotherapy target for autoimmunity and cancer. Front. Immunol. 2016, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrandino, F.; Grazioli, P.; Bellavia, D.; Campese, A.F.; Screpanti, I.; Felli, M.P. Notch and NF-κB: Coach and players of regulatory T-Cell response in cancer. Front. Immunol. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, L.; Ji, T.; Su, X.; Shao, Q.; Du, T.; Zhang, S. TMPRSS2-ERG Fusion Promotes Recruitment of Regulatory T cells and Tumor Growth in Prostate Cancer. Am. J. Med. Sci. 2018, 356, 72–78. [Google Scholar] [CrossRef]
- Kalina, J.L.; Neilson, D.S.; Lin, Y.Y.; Hamilton, P.T.; Comber, A.P.; Loy, E.M.H.; Sahinalp, S.C.; Collins, C.C.; Hach, F.; Lum, J.J. Mutational analysis of gene fusions predicts novel MHC class I-restricted T-cell epitopes and immune signatures in a subset of prostate cancer. Clin. Cancer Res. 2017, 23, 7596–7607. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.R.; Alham, N.K.; Upton, E.; McIntyre, S.; Bryant, R.J.; Cerundolo, L.; Bowes, E.; Jones, S.; Browne, M.; Mills, I.; et al. Detailed Molecular and Immune Marker Profiling of Archival Prostate Cancer Samples Reveals an Inverse Association between TMPRSS2:ERG Fusion Status and Immune Cell Infiltration. J. Mol. Diagn. 2020, 22, 652–669. [Google Scholar] [CrossRef] [PubMed]
- Kumar-Sinha, C.; Tomlins, S.A.; Chinnaiyan, A.M. Recurrent gene fusions in prostate cancer. Nat. Rev. Cancer 2008, 8, 497–511. [Google Scholar] [CrossRef] [Green Version]
- Garcia, A.J.; Ruscetti, M.; Arenzana, T.L.; Tran, L.M.; Bianci-frias, D.; Sybert, E. Pten Null Prostate Epithelium Promotes Localized Myeloid-Derived Suppressor Cell Expansion and Immune Suppression during Tumor. Mol. Cell. Biol. 2017, 34, 2017–2028. [Google Scholar] [CrossRef] [Green Version]
- Vidotto, T.; Saggioro, F.P.; Jamaspishvili, T.; Chesca, D.L.; Reis, R.B.; de Albuquerque, C.G.P.; Graham, C.H.; Berman, D.M.; Siemens, D.R.; Squire, J.A.; et al. PTEN-deficient prostate cancer is associated with an immunosuppressive tumor microenvironment mediated by increased expression of IDO1 and infiltrating FoxP3+ T regulatory cells. Prostate 2019, 1, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vidotto, T.; Melo, C.M.; Castelli, E.; Koti, M.; dos Reis, R.B.; Squire, J.A. Emerging role of PTEN loss in evasion of the immune response to tumours. Br. J. Cancer 2020, 122, 1732–1743. [Google Scholar] [CrossRef] [PubMed]
- Toso, A.; Revandkar, A.; DiMitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; Pinton, S.; Zhang, J.; Kalathur, M.; Civenni, G.; et al. Enhancing chemotherapy efficacy in pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014, 9, 75–89. [Google Scholar] [CrossRef]
- Piro, G.; Carbone, C.; Carbognin, L.; Pilotto, S.; Ciccarese, C.; Iacovelli, R.; Milella, M.; Bria, E.; Tortora, G. Revising PTEN in the era of immunotherapy: New perspectives for an old story. Cancers 2019, 11, 1525. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Grasso, C.S.; Jordahl, K.M.; Kolb, S.; Nyame, Y.A.; Wright, J.L.; Ostrander, E.A.; Troyer, D.A.; Lance, R.; Feng, Z.; et al. Copy number alterations are associated with metastatic-lethal progression in prostate cancer. Prostate Cancer Prostatic Dis. 2020, 23, 494–506. [Google Scholar] [CrossRef]
- Muñoz-Fontela, C.; Mandinova, A.; Aaronson, S.A.; Lee, S.W. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat. Rev. Immunol. 2016, 16, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Kaur, H.B.; Lu, J.; Guedes, L.B.; Maldonado, L.; Reitz, L.; Barber, J.R.; De Marzo, A.M.; Tomlins, S.A.; Sfanos, K.S.; Eisenberger, M.; et al. TP53 missense mutation is associated with increased tumor-infiltrating T cells in primary prostate cancer. Hum. Pathol. 2019, 87, 95–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamid, A.A.; Gray, K.P.; Shaw, G.; MacConaill, L.E.; Evan, C.; Bernard, B.; Loda, M.; Corcoran, N.M.; Van Allen, E.M.; Choudhury, A.D.; et al. Compound Genomic Alterations of TP53, PTEN, and RB1 Tumor Suppressors in Localized and Metastatic Prostate Cancer. Eur. Urol. 2019, 76, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef]
- Wang, C.; Feng, Y.; Zhang, C.; Cheng, D.; Wu, R.; Yang, Y.; Sargsyan, D.; Kumar, D.; Kong, A.N. PTEN deletion drives aberrations of DNA methylome and transcriptome in different stages of prostate cancer. FASEB J. 2020, 34, 1304–1318. [Google Scholar] [CrossRef] [Green Version]
- Allott, E.H.; Ebot, E.M.; Stopsack, K.H.; Gonzalez-Feliciano, A.G.; Markt, S.C.; Wilson, K.M.; Ahearn, T.U.; Gerke, T.A.; Downer, M.K.; Rider, J.R.; et al. Statin use is associated with lower risk of PTEN-null and lethal prostate cancer. Clin Cances Res. 2020, 26, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.; Mota, J.M.; Nandakumar, S.; Stopsack, K.H.; Weg, E.; Rathkopf, D.; Morris, M.J.; Scher, H.I.; Kantoff, P.W.; Gopalan, A.; et al. Pan-cancer Analysis of CDK12 Alterations Identifies a Subset of Prostate Cancers with Distinct Genomic and Clinical Characteristics. Eur. Urol. 2020, 78, 671–679. [Google Scholar] [CrossRef]
- Rescigno, P.; Gurel, B.; Pereira, R.; Crespo, M.; Rekowski, J.; Rediti, M.; Barrero, M.; Mateo, J.; Bianchini, D.; Messina, C.; et al. Characterizing CDK12-mutated prostate cancers. Clin. Cancer Res. 2021, 27, 566–574. [Google Scholar] [CrossRef]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [Green Version]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [Green Version]
- Gamat, M.; McNeel, D.G. Androgen deprivation and immunotherapy for the treatment of prostate cancer. Endocr. Relat. Cancer 2017, 24, T297–T310. [Google Scholar] [CrossRef]
- Ben-Batalla, I.; Vargas-Delgado, M.E.; von Amsberg, G.; Janning, M.; Loges, S. Influence of Androgens on Immunity to Self and Foreign: Effects on Immunity and Cancer. Front. Immunol. 2020, 11, 1–20. [Google Scholar] [CrossRef]
- Wu, L.; Yi, B.; Wei, S.; Rao, D.; He, Y.; Naik, G.; Bae, S.; Liu, X.M.; Yang, W.; Sonpavde, G.; et al. Loss of FOXP3 and TSC1 Accelerates Prostate Cancer Progression through Synergistic Transcriptional and Posttranslational Regulation of c-MYC. Cancer Res. 2019, 79, 1413–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melis, M.H.M.; Nevedomskaya, E.; Van Burgsteden, J.; Van Zeeburg, H.J.T.; Song, J.; Zevenhoven, J.; Lukas, J.A.C. The adaptive immune system promotes initiation of prostate carcinogenesis in a human c-Myc transgenic mouse model. Oncotarget 2017, 8, 93867–93877. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, D.N.; Casiraghi, N.; Romanel, A.; Crespo, M.; Miranda, S.; Rescigno, P.; Figueiredo, I.; Riisnaes, R.; Carreira, S.; Sumanasuriya, S.; et al. Rb1 heterogeneity in advanced metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2019, 25, 687–697. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, E.S.; Pruitt, S.C.; Hershberger, P.A.; Witkiewicz, A.K.; Goodrich, D.W. Cell cycle and beyond: Exploiting new RB1 controlled mechanisms for cancer therapy. Trends Cancer 2019, 5, 308–324. [Google Scholar] [CrossRef] [PubMed]
- Ali, K.; Soond, D.R.; Pineiro, R.; Hagemann, T.; Pearce, W.; Friedman, L.; Turner, M.; Okkenhaug, K. Inactivation of the PI3K p110 δ breaks regulatory T cell-mediated immune tolerance to cancer. Nature 2015, 510, 407–411. [Google Scholar] [CrossRef] [Green Version]
- Jenzer, M.; Keß, P.; Nientiedt, C.; Endris, V.; Kippenberger, M.; Leichsenring, J.; Stögbauer, F.; Haimes, J.; Mishkin, S.; Kudlow, B.; et al. The BRCA2 mutation status shapes the immune phenotype of prostate cancer. Cancer Immunol. Immunother. 2019, 68, 1621–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linch, M.; Goh, G.; Hiley, C.; Shanmugabavan, Y.; Mcgranahan, N.; Rowan, A.; Wong, Y.N.S.; King, H.; Furness, A.; Freeman, A.; et al. Intratumoural evolutionary landscape of high-risk prostate cancer: The PROGENY study of genomic and immune parameters original article. Annals Oncol. 2017, 28, 2472–2480. [Google Scholar] [CrossRef]
- Squire, J.A. TMPRSS2-ERG and PTEN loss in prostate cancer. Nat. Genet. 2009, 41, 509–510. [Google Scholar] [CrossRef]
- Hägglöf, C.; Hammarsten, P.; Strömvall, K.; Egevad, L.; Josefsson, A.; Stattin, P.; Granfors, T.; Bergh, A. TMPRSS2-ERG expression predicts prostate cancer survival and associates with stromal biomarkers. PLoS ONE 2014, 9, e86824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, J.M.; Heinlein, C.; Kim, T.; Hernandez, S.A.; Malik, M.S.; True, L.D.; Morrissey, C.; Corey, E.; Montgomery, B.; Mostaghel, E.; et al. The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discov. 2014, 4, 1310–1325. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chi, P.; Rockowitz, S.; Iaquinta, P.J.; Shamu, T.; Shukla, S.; Gao, D.; Sirota, I.; Carver, B.S.; Wongvipat, J.; et al. ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss. Nat. Med. 2013, 19, 1023–1029. [Google Scholar] [CrossRef] [Green Version]
- Higgins, J.; Brogley, M.; Palanisamy, N.; Mehra, R.; Ittmann, M.M.; Li, J.Z.; Tomlins, S.A.; Robins, D.M. Interaction of the Androgen Receptor, ETV1, and PTEN Pathways in Mouse Prostate Varies with Pathological Stage and Predicts Cancer Progression. Horm. Cancer 2015, 6, 67–86. [Google Scholar] [CrossRef] [Green Version]
- Guo, G.; Cui, Y. New perspective on targeting the tumor suppressor p53 pathway in the tumor microenvironment to enhance the efficacy of immunotherapy. J. Immunother. Cancer 2015, 3, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, J.H.; Dewal, N.; Sokol, E.; Mathew, P.; Whitehead, R.; Millis, S.Z.; Frampton, G.M.; Bratslavsky, G.; Pal, S.K.; Lee, R.J.; et al. Prospective Comprehensive Genomic Profiling of Primary and Metastatic Prostate Tumors. JCO Precis. Oncol. 2019, 3, 1–23. [Google Scholar] [CrossRef]
- Lozano, R.; Castro, E.; Aragón, I.M.; Cendón, Y.; Cattrini, C.; López-Casas, P.P.; Olmos, D. Genetic aberrations in DNA repair pathways: A cornerstone of precision oncology in prostate cancer. Br. J. Cancer 2021, 124, 552–563. [Google Scholar] [CrossRef]
- Zhu, M.-L.; Bakhru, P.; Conley, B.; Nelson, J.S.; Free, M.; Martin, A.; Starmer, J.; Wilson, E.M.; Su, M.A. Sex bias in CNS autoimmune disease mediated by androgen control of autoimmune regulator. Nat. Commun. 2016, 7, 1–14. [Google Scholar] [CrossRef]
- Kalra, R.; Bhagyaraj, E.; Tiwari, D.; Nanduri, R.; Chacko, A.P.; Jain, M.; Mahajan, S.; Khatri, N.; Gupta, P. AIRE promotes androgen-independent prostate cancer by directly regulating IL-6 and modulating tumor microenvironment. Oncogenesis 2018, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Løvf, M.; Zhao, S.; Axcrona, U.; Johannessen, B.; Bakken, A.C.; Carm, K.T.; Hoff, A.M.; Myklebost, O.; Meza-Zepeda, L.A.; Lie, A.K.; et al. Multifocal Primary Prostate Cancer Exhibits High Degree of Genomic Heterogeneity. Eur. Urol. 2019, 75, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Melaiu, O.; Lucarini, V.; Cifaldi, L.; Fruci, D. Influence of the Tumor Microenvironment on NK Cell Function in Solid Tumors. Front. Immunol. 2020, 10, 3038. [Google Scholar] [CrossRef]
- Pasero, C.; Gravis, G.; Guerin, M.; Granjeaud, S.; Thomassin-Piana, J.; Rocchi, P.; Paciencia-Gros, M.; Poizat, F.; Bentobji, M.; Azario-Cheillan, F.; et al. Inherent and tumor-driven immune tolerance in the prostate microenvironment impairs natural killer cell antitumor activity. Cancer Res. 2016, 76, 2153–2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Guo, G. Immunomodulatory function of the tumor suppressor p53 in host immune response and the tumor microenvironment. Int. J. Mol. Sci. 2016, 17, 1942. [Google Scholar] [CrossRef] [Green Version]
- Chi, N.; Tan, Z.; Ma, K.; Bao, L.; Yun, Z. Increased circulating myeloid-derived suppressor cells correlate with cancer stages, interleukin-8 and -6 in prostate cancer. Int. J. Clin. Exp. Med. 2014, 7, 3181–3192. [Google Scholar] [PubMed]
- Bryant, G.; Wang, L.; Mulholland, D.J. Overcoming oncogenic mediated tumor immunity in prostate cancer. Int. J. Mol. Sci. 2017, 18, 1542. [Google Scholar] [CrossRef] [PubMed]
- Erlandsson, A.; Carlsson, J.; Lundholm, M.; Fält, A.; Andersson, S.O.; Andrén, O.; Davidsson, S. M2 macrophages and regulatory T cells in lethal prostate cancer. Prostate 2019, 79, 363–369. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Shaukat, F.; Isaacsson Velho, P.; Kaur, H.; Shenderov, E.; Pardoll, D.M.; Lotan, T.L. Clinical Features and Therapeutic Outcomes in Men with Advanced Prostate Cancer and DNA Mismatch Repair Gene Mutations. Eur. Urol. 2019, 75, 378–382. [Google Scholar] [CrossRef]
- Armstrong, C.W.D.; Maxwell, P.J.; Ong, C.W.; Redmond, K.M.; Mccann, C.; Neisen, J.; Ward, G.A.; Chessari, G.; Johnson, C.; Crawford, N.T.; et al. PTEN deficiency promotes macrophage infiltration and hypersensitivity of prostate cancer to IAP antagonist/radiation combination therapy. Oncotarget 2016, 7, 7885–7898. [Google Scholar] [CrossRef]
- Bonollo, F.; Thalmann, G.N.; De Julio, M.K.; Karkampouna, S. The Role of Cancer-Associated Fibroblasts in Prostate Cancer Tumorigenesis. Cancers 2020, 12, 1887. [Google Scholar] [CrossRef]
- Diaz-Montero, C.M.; Finke, J.; Montero, A.J. Myeloid-derived suppressor cells in cancer: Therapeutic, predictive, and prognostic implications. Semin. Oncol. 2014, 41, 174–184. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Qu, S.; Zhang, T.; Gao, Y.; Shi, H.; Song, K.; Chen, W.; Yin, W. Applications of Single-Cell Omics in Tumor Immunology. Front. Immunol. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Bandura, D.R.; Baranov, V.I.; Ornatsky, O.I.; Antonov, A.; Kinach, R.; Lou, X.; Pavlov, S.; Vorobiev, S.; Dick, J.E.; Tanner, S.D. Mass cytometry: Technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal. Chem. 2009, 81, 6813–6822. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, M.H.; Nolan, G.P. Mass Cytometry: Single Cells, Many Features. Cell 2016, 165, 780–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, B.; Lin, Y.; Navin, N. Advancing Cancer Research and Medicine with Single-Cell Genomics. Cancer Cell 2020, 37, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Guo, J.; Liu, K.; Chen, L.; Liu, D.; Dong, S.; Xia, J.; Long, Q.; Yue, Y.; Zhao, P.; et al. Identification of a distinct luminal subgroup diagnosing and stratifying early stage prostate cancer by tissue-based single-cell RNA sequencing. Mol. Cancer 2020, 19, 1–17. [Google Scholar] [CrossRef]
- Roditi, L.D.V.; Jacobs, A.; Rueschoff, J.H.; Bankhead, P.; Jackson, H.W.; Hermanns, T.; Fankhauser, C.D.; Poyet, C.; Chun, F.; Rupp, N.J.; et al. Single-Cell Proteomics Defines the Cellular Heterogeneity of Localized Prostate Cancer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Cuzick, J.; Swanson, G.P.; Fisher, G.; Brothman, A.R.; Berney, D.M.; Reid, J.E.; Mesher, D.; Speights, V.O.; Stankiewicz, E.; Foster, C.S.; et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: A retrospective study. Lancet Oncol. 2011, 12, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Xie, F.; Zhou, X.; Fang, M.; Li, H.; Su, P.; Tu, Y.; Zhang, L.; Zhou, F. Extracellular Vesicles in Cancer Immune Microenvironment and Cancer Immunotherapy. Adv. Sci. 2019, 6, 1901779. [Google Scholar] [CrossRef]
- Song, H.; Weinstein, H.N.W.; Allegakoen, P.; Wadsworth, M.H.; Xie, J.; Yang, H.; Feng, F.Y.; Carroll, P.R.; Wang, B.; Cooperberg, M.R.; et al. Single-cell analysis of human primary prostate cancer reveals the heterogeneity of tumor-associated epithelial cell states. bioRxiv 2020. [Google Scholar] [CrossRef]
- Vickman, R.E.; Broman, M.M.; Lanman, N.A.; Franco, O.E.; Sudyanti, P.A.G.; Ni, Y.; Ji, Y.; Helfand, B.T.; Petkewicz, J.; Paterakos, M.C.; et al. Heterogeneity of human prostate carcinoma-associated fibroblasts implicates a role for subpopulations in myeloid cell recruitment. Prostate 2019, 80, 1–13. [Google Scholar] [CrossRef]
- Zhao, D.; Cai, L.; Lu, X.; Liang, X.; Li, J.; Chen, P.; Ittmann, M.; Shang, X.; Jiang, S.; Li, H.; et al. Chromatin regulator chd1 remodels the immunosuppressive tumor microenvironment in pten-deficient prostate cancer. Cancer Discov. 2020, 10, 1374–1387. [Google Scholar] [CrossRef]
- Wagner, J.; Rapsomaniki, M.A.; Chevrier, S.; Anzeneder, T.; Langwieder, C.; Dykgers, A.; Rees, M.; Ramaswamy, A.; Muenst, S.; Soysal, S.D.; et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 2019, 177, 1330–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassez, A.; Vos, H.; Van Dyck, L.; Floris, G.; Arijs, I.; Desmedt, C.; Boeckx, B.; Vanden Bempt, M.; Nevelsteen, I.; Lambein, K.; et al. A single-cell map of intratumoral changes during anti-PD1 treatment of patients with breast cancer. Nat. Med. 2021, 27, 820–832. [Google Scholar] [CrossRef]
- Fu, T.; Dai, L.-J.; Wu, S.-Y.; Xiao, Y.; Ma, D.; Jiang, Y.-Z.; Shao, Z.-M. Spatial architecture of the immune microenvironment orchestrates tumor immunity and therapeutic response. J. Hematol. Oncol. 2021, 14, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Marx, V. Method of the Year: Spatially resolved transcriptomics. Nat. Methods 2021, 18, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X. Spatially resolved transcriptomics adds a new dimension to genomics. Nat. Methods 2021, 18, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Asp, M.; Bergenstråhle, J.; Lundeberg, J. Spatially Resolved Transcriptomes—Next Generation Tools for Tissue Exploration. BioEssays 2020, 42, 1–16. [Google Scholar] [CrossRef]
- Tan, W.C.C.; Nerurkar, S.N.; Cai, H.Y.; Ng, H.H.M.; Wu, D.; Wee, Y.T.F.; Lim, J.C.T.; Yeong, J.; Lim, T.K.H. Overview of multiplex immunohistochemistry/immunofluorescence techniques in the era of cancer immunotherapy. Cancer Commun. 2020, 40, 135–153. [Google Scholar] [CrossRef] [Green Version]
- Georgescu, I.; Gooding, R.J.; Doiron, R.C.; Day, A.; Selvarajah, S.; Davidson, C.; Berman, D.M.; Park, P.C. Molecular characterization of Gleason patterns 3 and 4 prostate cancer using reverse Warburg effect-associated genes. Cancer Metab. 2016, 4, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Berglund, E.; Maaskola, J.; Schultz, N.; Friedrich, S.; Marklund, M.; Bergenstråhle, J.; Tarish, F.; Tanoglidi, A.; Vickovic, S.; Larsson, L.; et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Brady, L.; Kriner, M.; Coleman, I.; Morrissey, C.; Roudier, M.; True, L.D.; Gulati, R.; Plymate, S.R.; Zhou, Z.; Birditt, B.; et al. Inter- and intra-tumor heterogeneity of metastatic prostate cancer determined by digital spatial gene expression profiling. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Wen, J.; Huang, G.; Liu, S.; Wan, J.; Wang, X.; Zhu, Y.; Kaliney, W.; Zhang, C.; Cheng, L.; Wen, X.; et al. Polymorphonuclear MDSCs are enriched in the stroma and expanded in metastases of prostate cancer. J. Pathol. Clin. Res. 2020, 6, 171–177. [Google Scholar] [CrossRef]
- Keam, S.P.; Halse, H.; Nguyen, T.; Wang, M.; Van Kooten Losio, N.; Mitchell, C.; Caramia, F.; Byrne, D.J.; Haupt, S.; Ryland, G.; et al. High dose-rate brachytherapy of localized prostate cancer converts tumors from cold to hot. J. Immunother. Cancer 2020, 8, e000792. [Google Scholar] [CrossRef] [PubMed]
- Claps, M.; Mennitto, A.; Guadalupi, V.; Sepe, P.; Stellato, M.; Zattarin, E.; Gillessen, S.S.; Sternberg, C.N.; Berruti, A.; De Braud, F.G.M.; et al. Immune-checkpoint inhibitors and metastatic prostate cancer therapy: Learning by making mistakes. Cancer Treat. Rev. 2020, 88, 102057. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.H.; Antonarakis, E.S. Emerging treatments for metastatic castration-resistant prostate cancer: Immunotherapy, PARP inhibitors, and PSMA-targeted approaches. Cancer Treat. Res. Commun. 2020, 23, 100164. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Mollica, V.; Cimadamore, A.; Santoni, M.; Scarpelli, M.; Giunchi, F.; Cheng, L.; Lopez-Beltran, A.; Fiorentino, M.; Montironi, R.; et al. Is There a Role for Immunotherapy in Prostate Cancer? Cells 2020, 9, 2051. [Google Scholar] [CrossRef]
- Graff, J.N.; Antonarakis, E.S.; Hoimes, C.J.; Tagawa, S.T.; Hwang, C.; Kilari, D.; Ten Tije, A.; Omlin, A.G.; McDermott, R.S.; Vaishampayan, U.N.; et al. Pembrolizumab (pembro) plus enzalutamide (enza) for enza-resistant metastatic castration-resistant prostate cancer (mCRPC): KEYNOTE-199 cohorts 4-5. J. Clin. Oncol. 2020, 38, 15. [Google Scholar] [CrossRef]
- Graff, J.N.; Beer, T.M.; Alumkal, J.J.; Slottke, R.E.; Redmond, W.L.; Thomas, G.V.; Thompson, R.F.; Wood, M.A.; Koguchi, Y.; Chen, Y.; et al. A phase II single-arm study of pembrolizumab with enzalutamide in men with metastatic castration-resistant prostate cancer progressing on enzalutamide alone. J. Immunother. Cancer 2020, 8, 1–11. [Google Scholar] [CrossRef]
- Yu, E.Y.; Piulats, J.M.; Gravis, G.; Laguerre, B.; Arija, J.A.A.; Oudard, S.; Fong, P.C.C.; Kolinsky, M.P.; Augustin, M.; Feyerabend, S.; et al. KEYNOTE-365 cohort A updated results: Pembrolizumab (pembro) plus olaparib in docetaxel-pretreated patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2020, 38, 5544. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.; Berger, E.; Small, E.J. Sipuleucel T immunotherapy for CRPC. N. Engl. J. 2010, 363, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Higano, C.S.; Schellhammer, P.F.; Small, E.J.; Burch, P.A.; Nemunaitis, J.; Yuh, L.; Provost, N.; Frohlich, M.W. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer 2009, 115, 3670–3679. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Schuetz, T.J.; Blumenstein, B.A.; Michael Glode, L.; Bilhartz, D.L.; Wyand, M.; Manson, K.; Panicali, D.L.; Laus, R.; Schlom, J.; et al. Overall survival analysis of a phase II randomized controlled trial of a poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2010, 28, 1099–1105. [Google Scholar] [CrossRef]
- Antonarakis, E.S. Cyclin-Dependent Kinase 12, Immunity, and Prostate Cancer. N. Engl. J. Med. 2018, 379, 1087–1089. [Google Scholar] [CrossRef]
- McGregor, B.A.; Xiao, X.W. CDK12 Loss in Prostate Cancer Imparts Poor Prognosis and Limited Susceptibility to Immune Checkpoint Inhibition. JCO Precis. Oncol. 2020, 4, 367–369. [Google Scholar] [CrossRef]
- Ratta, R.; Guida, A.; Scotté, F.; Neuzillet, Y.; Teillet, A.B.; Lebret, T.; Beuzeboc, P. PARP inhibitors as a new therapeutic option in metastatic prostate cancer: A systematic review. Prostate Cancer Prostatic Dis. 2020, 23, 549–560. [Google Scholar] [CrossRef]
- Risdon, E.N.; Chau, C.H.; Price, D.K.; Sartor, O.; Figg, W.D. PARP Inhibitors and Prostate Cancer: To Infinity and Beyond BRCA. Oncologist 2021, 26, e115. [Google Scholar] [CrossRef]
- Logtenberg, M.E.W.; Scheeren, F.A.; Schumacher, T.N. The CD47-SIRP α immune checkpoint. Immunity 2020, 52, 742–752. [Google Scholar] [CrossRef]
- Oronsky, B.; Carter, C.; Reid, T.; Brinkhaus, F.; Knox, S.J. Just eat it: A review of CD47 and SIRP-α antagonism. Semin. Oncol. 2020, 47, 117–124. [Google Scholar] [CrossRef]
- Chow, L.Q.M.; Gainor, J.F.; Lakhani, N.J.; Lee, K.W.; Chung, H.C.; Lee, J.; LoRusso, P.; Bang, Y.-J.; Hodi, F.S.; Santana-Davila, R.; et al. A phase I study of ALX148, a CD47 blocker, in combination with standard anticancer antibodies and chemotherapy regimens in patients with advanced malignancy. J. Clin. Oncol. 2020, 38, 3056. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Somatic Mutations | Frequency of PCa Mutated | Proposed Responses of Immune Effectors in PCa Microenvironment | References |
|---|---|---|---|
| Genes | |||
| ETS fusion genes | 20–30% | Activation NOTCH, WNT and NF-kB signaling; Induction of a pro-inflammatory gene signature (Tregs and dendritic cells recruitment). | [47,48,49,50,51,52,53] |
| PTEN loss | 20–40% | Increased PDL1 and IDO1, FoxP3+ Tregs, resting dendritic cells, M1 macrophages, MDSCs, and CAFs; PI3K pathway regulation; Negative regulation of cytokines. | [15,54,55,56,57,58,59] |
| TP53 loss | 20–30% | Induction of a pro-inflammatory and immunosuppressive TME (TAM, MDSCs, T cell); Increased PD1 and PDL-1 expression; Activation of NF-kB signaling; Increased genomic instability. | [35,55,60,61,62,63,64,65,66] |
| CDK12 inactivation | 1.2–6.9% | Secretion of pro-tumorigenic cytokines (CCL18, /IL-8); Immune checkpoints modulation; Increased neoantigen load and immune infiltration (T cell (higher CD4+ and lower CD8+), TAM and MDSCs). | [8,67,68] |
| SPOP/CHD1 | 6–15% | Upregulation of the AR, PI3K/mTOR, ERG fusion and PD-L1; Correlated to deletion of CHD1. | [13,69] |
| AR gene mutation | 1% | Increased immune infiltration (Tregs and IL-10) and lower CD3+ and CD68+ cells; Decreased in IFNγ-secretion by T cells; IL-23 secreted by MDSCs can cause downstream activation of AR target genes. | [70,71,72] |
| MYC amplification | 20% | Pro-inflammatory and immunosuppressive TME (recruitment of TAMs and MDSCs); Induce genomic instability. | [37,73,74] |
| RB1 loss | 28–30% | Activation of tumor cell cycle and antigen presentation; IFN response; Reduce the immune cells mobilization and NK cells. | [63,75,76] |
| Pathways | |||
| PI3K activation | 20–40% | Pro-inflammatory and immunosuppressive TME (recruitment of TAMs, MDSCs and PD-L1). | [37,77] |
| DNA repair | 19% | Accumulation of genetic aberrations, tumor evolution and progression; Inflamed TME phenotype; Increase IFN pathways and interactions between tumor-specific antigens and immune cells. | [78] |
| WNT/b-catenin activation | 20% | Immunosuppressive TME (recruitment of FOXP3+ regulatory T cell and lower CD8+:FoxP3+ ratio). | [79] |
| Agent | Function—Target | Description | NCT Number |
|---|---|---|---|
| Sipuleucel-T | Personalized, autologous, and cellular immunotherapy. Target the prostate specific antigen (PSA). | To determine if sipuleucel-T was effective for PCa treatment. Patients with localized PCa, metastatic castration sensitive PCa or mCRPC. | NCT00779402 NCT00065442 NCT03686683 NCT01133704 NCT00005947 |
| Ipilimumab | Humanized IgG1 monoclonal antibody that blocks cytotoxic T lymphocyte antigen-4 (CTLA-4) and removes an inhibitory signal from reducing the activity of T lymphocytes. | To evaluate whether the addition of radiotherapy or chemotherapy plus ADT improves the prognosis and survival of patients. Patients with localized PCa, metastatic castration sensitive PCa or mCRPC. | NCT00861614 NCT03879122 NCT01057810 |
| Aglatimagene Besadenovec + Valacyclovir | ProstAtak® kills tumor cells and stimulates a cancer vaccine effect. Killing tumor cells in an immune stimulatory environment induces the body’s immune system to detect and destroy cancer cells. | To evaluate the effectiveness of ProstAtak® immunotherapy in combination with radiation therapy for patients with intermediate-high risk localized prostate cancer. | NCT01436968 |
| Bevacizumab + Cisplatin + Gemcitabine + Hydrocloride | Humanized monoclonal IgG antibody and inhibits angiogenesis by binding and neutralizing VEGF-A. | To evaluate the effectiveness of bevacizumab to induce changes in body’s immune system and to interfere with the ability of tumor cells to grow and spread. Patients with metastatic castration sensitive PCa. | NCT00942331 |
| PROST-VAC-V + PROST-VAC-F + GM-CSF | Active immunotherapy vaccine that contains PSA as the tumor-associated antigen used to generate a T-cell response against prostate cancer. | To determine whether PROSTVAC alone or in combination with GM-CSF is effective in prolonging overall survival in men with few or no symptoms from metastatic, castrate-resistant prostate cancer. | NCT01322490 |
| DCVAC + Docetaxel + Taxotere | Cell therapy platform designed to improve efficacy compared to earlier generations of dendritic cell therapies by targeting multiple antigens and applying an immune-stimulatory technique. | To confirm the hypothesis that the combination of docetaxel with DCVAC/PCa followed by a maintenance therapy with DCVAC/PCa would improve overall survival in patients with metastatic castration-resistant prostate cancer. | NCT02111577 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melo, C.M.; Vidotto, T.; Chaves, L.P.; Lautert-Dutra, W.; Reis, R.B.d.; Squire, J.A. The Role of Somatic Mutations on the Immune Response of the Tumor Microenvironment in Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 9550. https://doi.org/10.3390/ijms22179550
Melo CM, Vidotto T, Chaves LP, Lautert-Dutra W, Reis RBd, Squire JA. The Role of Somatic Mutations on the Immune Response of the Tumor Microenvironment in Prostate Cancer. International Journal of Molecular Sciences. 2021; 22(17):9550. https://doi.org/10.3390/ijms22179550
Chicago/Turabian StyleMelo, Camila Morais, Thiago Vidotto, Luiz Paulo Chaves, William Lautert-Dutra, Rodolfo Borges dos Reis, and Jeremy Andrew Squire. 2021. "The Role of Somatic Mutations on the Immune Response of the Tumor Microenvironment in Prostate Cancer" International Journal of Molecular Sciences 22, no. 17: 9550. https://doi.org/10.3390/ijms22179550