
The Contribution of Autophagy and LncRNAs to MYC-Driven Gene Regulatory Networks in Cancers
 ,
,  , ,
, ,  , and
, and
Abstract
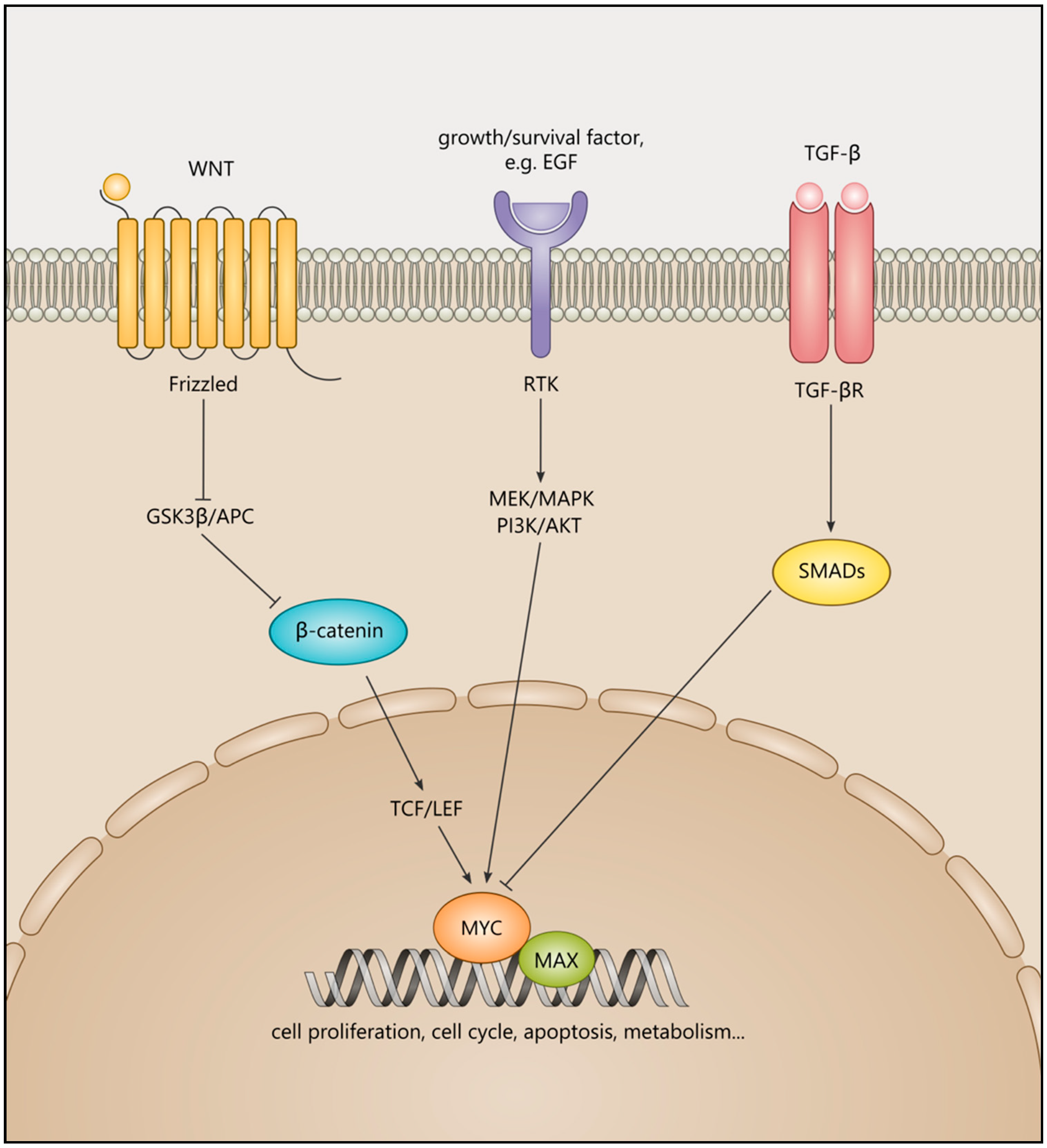
:1. Introduction to MYC Transcription Factors and Their Roles in Cancer
2. Cancers with MYC-Related Functions in Their Pathogenesis
2.1. Prostate Cancer
2.2. Pancreatic Ductal Adenocarcinoma
2.3. Medulloblastoma
2.4. Neuroblastoma
2.5. Colorectal Cancer
2.6. Rhabdomyosarcoma
2.7. Non-Small Cell Lung Cancer
3. MYC and Long Non-Coding RNAs (LncRNAs) in Cancer
3.1. LncRNAs in Cancer as Novel Biomarkers and Therapeutic Targets
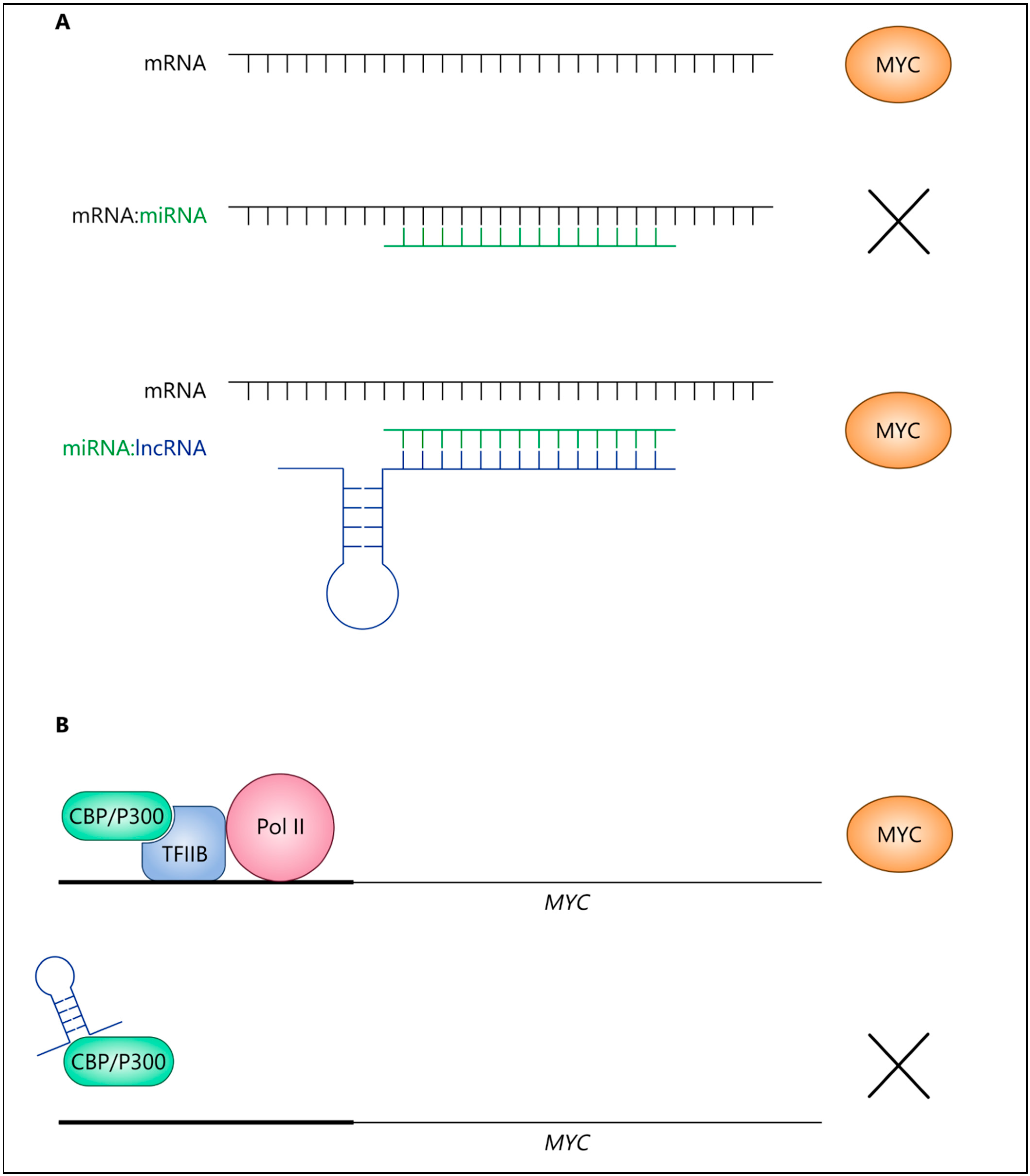
3.2. Oncogenic IncRNAs Promote MYC Expression and Activity in Cancer
3.3. LncRNAs Repress MYC under Certain Conditions
3.4. Mutual Regulation of IncRNAs and MYC via Feedback Loops
4. MYC and Autophagy
4.1. MYC, Autophagy, and Cellular and Molecular Processes in Cancers
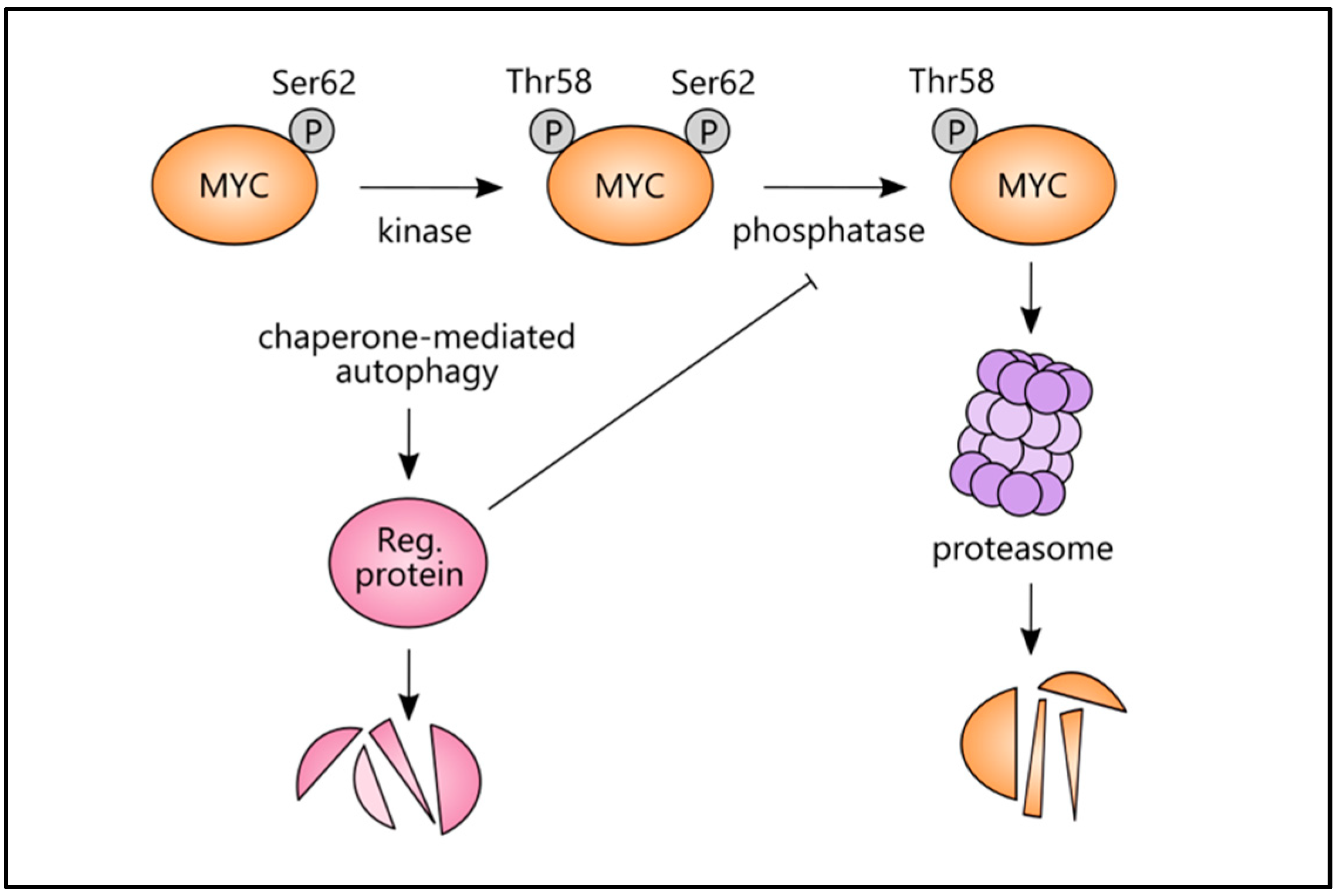
4.2. Chaperone-Mediated Autophagy (CMA) and Mitophagy Link with MYC
5. Discussion: Novel Treatment Options for MYC-Driven Malignancies in Light of the Topics Discussed
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | Adenomatous polyposis coli |
| AR | androgen receptor |
| ASOs | antisense oligonucleotides |
| Atg | autophagy-related protein |
| BBR | Berberine |
| bHLHZip | basic helix-loop-helix leucine zipper |
| Bif-1 | Bax-interacting factor 1 |
| BRD4 | BET bromodomain protein 4 |
| ceRNA | competing endogenous RNA |
| EZH2 | Enhancer of Zeste 2 Polycomb repressive complex 2 subunits |
| CMA | Chaperone-mediated autophagy |
| CRC | colorectal cancer cells |
| DLBCL | diffuse large B-cell lymphoma |
| EGF | epidermal growth factor |
| EMT | epithelial-to-mesenchymal transition |
| ER | endoplasmic reticulum |
| GSK3β | glycogen synthase kinase-3β |
| GRNs | Gene Regulatory Networks |
| H3K27 | Histone 3 lysine 27 |
| HEK293 cells | human embryonic kidney cells |
| IGF2BP1 | insulin-like growth factor 2 mRNA binding protein 1 |
| IGF2BP2 | insulin-like growth factor 2 mRNA binding protein 2 |
| LAPTM4B | lysosomal protein transmembrane 4 beta |
| LDHA | lactate dehydrogenase A |
| Lis/lin/GO | linamarase/linamarin/glucose oxidase system |
| LNAs | locked nucleic acids |
| LncRNAs | long non-coding RNAs |
| LPP-AS2 | LPP antisense RNA-2 |
| M6A | N6-methyl-adenosine |
| MEF | murine embryonic fibroblast |
| MM | multiple myeloma |
| MTSS1 | metastasis suppressor 1 |
| MZF1 | myeloid zinc finger 1 |
| NB | neuroblastoma |
| NCL | Nuclein |
| ncRNAs | non-coding RNAs |
| NCS | neural stem cell |
| NSCLC | non-small cell lung cancer |
| NSG | NOD SCID gamma mice |
| OXA | Oxaliplatin |
| PCYT1A | phosphate cytidylyltransferase 1 choline-α |
| P-TEFb | positive elongation factor complex b |
| PDAC | pancreatic ductal adenocarcinoma |
| PLK1 | polo-like kinase 1 |
| RMS | Rhabdomyosarcoma |
| ROS | reactive oxygen species |
| SHH | Sonic Hedgehog |
| STUB1 | STIP1 homology and U-box containing protein 1 |
| TCF/LEF | T-cell factor/ lymphoid enhancer factor |
| TEAD | TEA domain transcription factor |
| TF | transcription factors |
| TGF-β | transforming growth factor β |
| TH | tyrosine hydroxylase |
| TNM | tumour-node metastasis |
| UPR | unfolded protein response |
References
- Malynn, B.A.; de Alboran, I.M.; O’Hagan, R.C.; Bronson, R.; Davidson, L.; DePinho, R.A.; Alt, F.W. N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation. Genes Dev. 2000, 14, 1390–1399. [Google Scholar] [PubMed]
- Kohl, N.E.; Kanda, N.; Schreck, R.R.; Bruns, G.; Latt, S.A.; Gilbert, F.; Alt, F.W. Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell 1983, 35, 359–367. [Google Scholar] [CrossRef]
- Amati, B.; Littlewood, T.D.; Evan, G.I.; Land, H. The c-Myc protein induces cell cycle progression and apoptosis through dimerization with Max. EMBO J. 1993, 12, 5083–5087. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soucek, L.; Evan, G.I. The ups and downs of Myc biology. Curr. Opin. Genet. Dev. 2010, 20, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Kelly, K.; Cochran, B.H.; Stiles, C.D.; Leder, P. Cell-specific regulation of the c-myc gene by lymphocyte mitogens and platelet-derived growth factor. Cell 1983, 35, 603–610. [Google Scholar] [CrossRef]
- Satoh, K.; Yachida, S.; Sugimoto, M.; Oshima, M.; Nakagawa, T.; Akamoto, S.; Tabata, S.; Saitoh, K.; Kato, K.; Sato, S.; et al. Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc. Natl. Acad. Sci. USA 2017, 114, E7697–E7706. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.M.; Harris, A.W.; Pinkert, C.A.; Corcoran, L.M.; Alexander, W.S.; Cory, S.; Palmiter, R.D.; Brinster, R.L. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 1985, 318, 533–538. [Google Scholar] [CrossRef]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, metabolism, and cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef] [Green Version]
- He, T.-C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-MYC as a target of the APC pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swartling, F.J.; Savov, V.; Persson, A.I.; Chen, J.; Hackett, C.S.; Northcott, P.A.; Grimmer, M.R.; Lau, J.; Chesler, L.; Perry, A.; et al. Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell 2012, 21, 601–613. [Google Scholar] [CrossRef] [Green Version]
- Evan, G.I.; Littlewood, T.D. The role of c-myc in cell growth. Curr. Opin. Genet. Dev. 1993, 3, 44–49. [Google Scholar] [CrossRef]
- Ayer, D.E.; Eisenman, R.N. A switch from Myc:Max to Mad:Max heterocomplexes accompanies monocyte/macrophage differentiation. Genes Dev. 1993, 7, 2110–2119. [Google Scholar] [CrossRef] [Green Version]
- Laurenti, E.; Varnum-Finney, B.; Wilson, A.; Ferrero, I.; Blanco-Bose, W.E.; Ehninger, A.; Knoepfler, P.S.; Cheng, P.F.; MacDonald, H.R.; Eisenman, R.N.; et al. Hematopoietic stem cell function and survival depend on c-Myc and N-Myc activity. Cell Stem Cell 2008, 3, 611–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandarillas, A.; Watt, F. c-Myc promotes differentiation of human epidermal stem cells. Genes Dev. 1997, 11, 2869–2882. [Google Scholar] [CrossRef] [Green Version]
- Land, H.; Parada, L.F.; Weinberg, R.A. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 1983, 304, 596–602. [Google Scholar] [CrossRef]
- Mathew, P.; Valentine, M.B.; Bowman, L.C.; Rowe, S.T.; Nash, M.B.; Valentine, V.A.; Cohn, S.L.; Castleberry, R.P.; Brodeur, G.M.; Look, A.T. Detection of MYCN gene amplification in neuroblastoma by fluorescence in situ hybridization: A pediatric oncology group study. Neoplasia 2001, 3, 105–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, F.G.; McCarthy, B.J.; Freels, S.; Kupelian, V.; Bondy, M.L. The conditional probability of survival of patients with primary malignant brain tumors: Surveillance, epidemiology, and end results (SEER) data. Cancer 1999, 85, 485–491. [Google Scholar] [CrossRef]
- Dang, C.V. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef]
- Sodir, N.M.; Swigart, L.B.; Karnezis, A.N.; Hanahan, D.; Evan, G.I.; Soucek, L. Endogenous Myc maintains the tumor microenvironment. Genes Dev. 2011, 25, 907–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapp, U.R.; Korn, C.; Ceteci, F.; Karreman, C.; Luetkenhaus, K.; Serafin, V.; Zanucco, E.; Castro, I.; Potapenko, T. MYC is a metastasis gene for non-small-cell lung cancer. PLoS ONE 2009, 4, e6029. [Google Scholar] [CrossRef] [Green Version]
- Shchors, K.; Shchors, E.; Rostker, F.; Lawlor, E.R.; Brown-Swigart, L.; Evan, G.I. The Myc-dependent angiogenic switch in tumors is mediated by interleukin 1beta. Genes Dev. 2006, 20, 2527–2538. [Google Scholar] [CrossRef] [Green Version]
- Berger, A.; Brady, N.J.; Bareja, R.; Robinson, B.; Conteduca, V.; Augello, M.A.; Puca, L.; Ahmed, A.; Dardenne, E.; Lu, X.; et al. N-Myc-mediated epigenetic reprogramming drives lineage plasticity in advanced prostate cancer. J. Clin. Invest. 2019, 129, 3924–3940. [Google Scholar] [CrossRef]
- Murakami, S.; Nemazanyy, I.; White, S.M.; Chen, H.; Nguyen, C.D.K.; Graham, G.T.; Saur, D.; Pende, M.; Yi, C. A Yap-Myc-Sox2-p53 Regulatory Network Dictates Metabolic Homeostasis and Differentiation in Kras-Driven Pancreatic Ductal Adenocarcinomas. Dev. Cell 2019, 51, 113–128.e9. [Google Scholar] [CrossRef]
- Tao, R.; Murad, N.; Xu, Z.; Zhang, P.; Okonechnikov, K.; Kool, M.; Rivero-Hinojosa, S.; Lazarski, C.; Zheng, P.; Liu, Y.; et al. MYC Drives Group 3 Medulloblastoma through Transformation of Sox2+ Astrocyte Progenitor Cells. Cancer Res. 2019, 79, 1967–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Gao, S.; Wang, W.; Xia, Y.; Liang, J. Downregulation of N-Myc inhibits neuroblastoma cell growth via the Wnt/β-catenin signaling pathway. Mol. Med. Rep. 2018, 18, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Ooi, C.Y.; Carter, D.R.; Liu, B.; Mayoh, C.; Beckers, A.; Lalwani, A.; Nagy, Z.; De Brouwer, S.; Decaesteker, B.; Hung, T.T.; et al. Network Modeling of microRNA-mRNA Interactions in Neuroblastoma Tumorigenesis Identifies miR-204 as a Direct Inhibitor of MYCN. Cancer Res. 2018, 78, 3122–3134. [Google Scholar] [CrossRef] [Green Version]
- Pandey, P.R.; Chatterjee, B.; Olanich, M.E.; Khan, J.; Miettinen, M.M.; Hewitt, S.M.; Barr, F.G. PAX3-FOXO1 is essential for tumour initiation and maintenance but not recurrence in a human myoblast model of rhabdomyosarcoma. J. Pathol. 2017, 241, 626–637. [Google Scholar] [CrossRef] [Green Version]
- Kortlever, R.M.; Sodir, N.M.; Wilson, C.H.; Burkhart, D.L.; Pellegrinet, L.; Brown Swigart, L.; Littlewood, T.D.; Evan, G.I. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017, 171, 1301–1315.e14. [Google Scholar] [CrossRef] [PubMed]
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.J.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling Myc inhibition as a cancer therapy. Nature 2008, 455, 679–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, M.; Dong, S.; Hou, H.; Zhang, T.; Shen, M. Oncogenic Role of Long Noncoding RNAMALAT1 in Thyroid Cancer Progression through Regulation of the miR-204/IGF2BP2/m6A-MYC Signaling. Mol. Ther. Nucleic Acids 2020, 23, 1–12. [Google Scholar] [CrossRef]
- Crea, F.; Venalainen, E.; Ci, X.; Cheng, H.; Pikor, L.; Parolia, A.; Xue, H.; Nur Saidy, N.R.; Lin, D.; Lam, W.; et al. The role of epigenetics and long noncoding RNA MIAT in neuroendocrine prostate cancer. Epigenomics 2016, 8, 721–731. [Google Scholar] [CrossRef] [Green Version]
- Gargini, R.; García-Escudero, V.; Izquierdo, M.; Wandosell, F. Oncogene-mediated tumor transformation sensitizes cells to autophagy induction. Oncol. Rep. 2016, 35, 3689–3695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Escudero, V.; Gargini, R. Autophagy induction as an efficient strategy to eradicate tumors. Autophagy 2008, 4, 923–925. [Google Scholar] [CrossRef] [Green Version]
- Gargini, R.; García-Escudero, V.; Izquierdo, M. Therapy mediated by mitophagy abrogates tumor progression. Autophagy 2011, 7, 466–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Nandakumar, N.; Shi, Y.; Manzano, M.; Smith, A.; Graham, G.; Gupta, S.; Vietsch, E.E.; Laughlin, S.Z.; Wadhwa, M.; et al. Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci. Signal. 2014, 7, ra42. [Google Scholar] [CrossRef] [Green Version]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Driman, D.; Thorner, P.S.; Greenberg, M.L.; Chilton-MacNeill, S.; Squire, J. MYCN gene amplification in rhabdomyosarcoma. Cancer 1994, 73, 2231–2237. [Google Scholar] [CrossRef]
- Hachitanda, Y.; Toyoshima, S.; Akazawa, K.; Tsuneyoshi, M. N-myc gene amplification in rhabdomyosarcoma detected by fluorescence in situ hybridization: Its correlation with histologic features. Mod. Pathol. 1998, 11, 1222–1227. [Google Scholar]
- Williamson, D.; Lu, Y.J.; Gordon, T.; Sciot, R.; Kelsey, A.; Fisher, C.; Poremba, C.; Anderson, J.; Pritchard-Jones, K.; Shipley, J. Relationship between MYCN copy number and expression in rhabdomyosarcomas and correlation with adverse prognosis in the alveolar subtype. J. Clin. Oncol. 2005, 23, 880–888. [Google Scholar] [CrossRef]
- He, J.; Li, F.; Zhou, Y.; Hou, X.; Liu, S.; Li, X.; Zhang, Y.; Jing, X.; Yang, L. LncRNA XLOC_006390 promotes pancreatic carcinogenesis and glutamate metabolism by stabilizing c-Myc. Cancer Lett. 2020, 469, 419–428. [Google Scholar] [CrossRef]
- Xu, Z.; Liu, C.; Zhao, Q.; Lü, J.; Ding, X.; Luo, A.; He, J.; Wang, G.; Li, Y.; Cai, Z.; et al. Long non-coding RNA CCAT2 promotes oncogenesis in triple-negative breast cancer by regulating stemness of cancer cells. Pharmacol. Res. 2020, 152, 104628. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhu, P.; Lu, T.; Du, Y.; Wang, Y.; He, L.; Ye, B.; Liu, B.; Yang, L.; Wang, J.; et al. The long non-coding RNA LncHDAC2 drives the self-renewal of liver cancer stem cells via activation of Hedgehog signaling. J. Hepatol. 2019, 70, 918–929. [Google Scholar] [CrossRef]
- Crea, F.; Clermont, P.L.; Parolia, A.; Wang, Y.; Helgason, C.D. The non-coding transcriptome as a dynamic regulator of cancer metastasis. Cancer Metastasis Rev. 2014, 33, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Su, Y.; Liu, X.; Xu, M.; Chen, X.; Zhu, Y.; Guo, Z.; Bai, T.; Dong, L.; Wei, C.; et al. Serum and exosome long non coding RNAs as potential biomarkers for hepatocellular carcinoma. J. Cancer 2018, 9, 2631–2639. [Google Scholar] [CrossRef]
- Conigliaro, A.; Costa, V.; Lo Dico, A.; Saieva, L.; Buccheri, S.; Dieli, F.; Manno, M.; Raccosta, S.; Mancone, C.; Tripodi, M.; et al. CD90+ liver cancer cells modulate endothelial cell phenotype through the release of exosomes containing H19 lncRNA. Mol. Cancer 2015, 14, 155. [Google Scholar] [CrossRef] [PubMed]
- Roobol, M.J.; Schröder, F.H.; van Leeuwen, P.; Wolters, T.; van den Bergh, R.C.; van Leenders, G.J.; Hessels, D. Performance of the prostate cancer antigen 3 (PCA3) gene and prostate-specific antigen in prescreened men: Exploring the value of PCA3 for a first-line diagnostic test. Eur. Urol. 2010, 58, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Bellmunt, J.; Eigl, B.J.; Senkus, E.; Loriot, Y.; Twardowski, P.; Castellano, D.; Blais, N.; Sridhar, S.S.; Sternberg, C.N.; Retz, M.; et al. Borealis-1: A randomized, first-line, placebo-controlled, phase II study evaluating apatorsen and chemotherapy for patients with advanced urothelial cancer. Ann. Oncol. 2017, 28, 2481–2488. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.N.; Higano, C.S.; Blumenstein, B.; Ferrero, J.M.; Reeves, J.; Feyerabend, S.; Gravis, G.; Merseburger, A.S.; Stenzl, A.; Bergman, A.M.; et al. Custirsen in combination with docetaxel and prednisone for patients with metastatic castration-resistant prostate cancer (SYNERGY trial): A phase 3, multicentre, open-label, randomised trial. Lancet Oncol. 2017, 18, 473–485. [Google Scholar] [CrossRef]
- Chery, J. RNA therapeutics: RNAi and antisense mechanisms and clinical applications. Postdoc J. 2016, 4, 35–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.T.; Han, C.; Sun, Y.M.; Chen, T.Q.; Chen, Y.Q. Noncoding RNAs in cancer therapy resistance and targeted drug development. J. Hematol. Oncol. 2019, 12, 55. [Google Scholar] [CrossRef]
- Shen, X.; Corey, D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018, 46, 1584–1600. [Google Scholar] [CrossRef] [PubMed]
- Sekhon, H.S.; London, C.A.; Sekhon, M.; Iversen, P.L.; Devi, G.R. c-MYC antisense phosphosphorodiamidate morpholino oligomer inhibits lung metastasis in a murine tumor model. Lung Cancer 2008, 60, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Iversen, P.L.; Arora, V.; Acker, A.J.; Mason, D.H.; Devi, G.R. Efficacy of antisense morpholino oligomer targeted to c-myc in prostate cancer xenograft murine model and a Phase I safety study in humans. Clin. Cancer Res. 2003, 9, 2510–2519. [Google Scholar] [PubMed]
- Zhang, Z.; Lin, W.; Lin, Y.; Kang, M.; Zhu, J.; Tong, Z.; Wu, L.; Sun, J.; Lin, J. Long intergenic non-coding RNA Linc00485 promotes lung cancer progression by modulating miR-298/c-Myc axis. J. Cell Mol. Med. 2021, 25, 309–322. [Google Scholar] [CrossRef]
- Liu, S.; Zheng, Y.; Zhang, Y.; Zhang, J.; Xie, F.; Guo, S.; Gu, J.; Yang, J.; Zheng, P.; Lai, J.; et al. Methylation-mediated LINC00261 suppresses pancreatic cancer progression by epigenetically inhibiting c-Myc transcription. Theranostics 2020, 10, 10634–10651. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Niu, W.; Mu, M.; Hu, S.; Niu, C. Long non-coding RNA LPP-AS2 promotes glioma tumorigenesis via miR-7-5p/EGFR/PI3K/AKT/c-MYC feedback loop. J. Exp. Clin. Cancer Res. 2020, 39, 196. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, J.; Cao, W.; Xiao, X.; Liang, L.; Liu-Smith, F.; Wang, W.; Liu, H.; Zhou, P.; Ouyang, R.; et al. C-myc/miR-150/EPG5 axis mediated dysfunction of autophagy promotes development of non-small cell lung cancer. Theranostics 2019, 9, 5134–5148. [Google Scholar] [CrossRef] [PubMed]
- Cianfanelli, V.; Fuoco, C.; Lorente, M.; Salazar, M.; Quondamatteo, F.; Gherardini, P.F.; De Zio, D.; Nazio, F.; Antonioli, M.; D’Orazio, M.; et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol. 2015, 17, 20–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, L.S.; Cunningham, J.T.; Datta, T.; Dey, S.; Tameire, F.; Lehman, S.L.; Qiu, B.; Zhang, H.; Cerniglia, G.; Bi, M.; et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J. Clin. Invest. 2012, 122, 4621–4634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, L.R.; Menck, C.F.M.; Cuervo, A.M. Chaperone-mediated autophagy prevents cellular transformation by regulating MYC proteasomal degradation. Autophagy 2017, 13, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Zhai, S.; Xu, Z.; Xie, J.; Zhang, J.; Wang, X.; Peng, C.; Li, H.; Chen, H.; Shen, B.; Deng, X. Epigenetic silencing of LncRNA LINC00261 promotes c-myc-mediated aerobic glycolysis by regulating miR-222-3p/HIPK2/ERK axis and sequestering IGF2BP1. Oncogene 2021, 40, 277–291. [Google Scholar] [CrossRef]
- Shigeyasu, K.; Toden, S.; Ozawa, T.; Matsuyama, T.; Nagasaka, T.; Ishikawa, T.; Sahoo, D.; Ghosh, P.; Uetake, H.; Fujiwara, T. The PVT1 lncRNA is a novel epigenetic enhancer of MYC, and a promising risk-stratification biomarker in colorectal cancer. Mol. Cancer 2020, 19, 155. [Google Scholar] [CrossRef]
- Olivero, C.E.; Martínez-Terroba, E.; Zimmer, J.; Liao, C.; Tesfaye, E.; Hooshdaran, N.; Schofield, J.A.; Bendor, J.; Fang, D.; Simon, M.D.; et al. p53 Activates the Long Noncoding RNA Pvt1b to Inhibit Myc and Suppress Tumorigenesis. Mol. Cell 2020, 77, 761–774.e8. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, F.; Xu, F.; Fang, K.; Fang, Z.; Shuai, X.; Cai, K.; Chen, J.; Hu, P.; Chen, D.; et al. A reciprocal feedback of Myc and lncRNA MTSS1-AS contributes to extracellular acidity-promoted metastasis of pancreatic cancer. Theranostics 2020, 10, 10120–10140. [Google Scholar] [CrossRef]
- Usman, R.M.; Razzaq, F.; Akbar, A.; Farooqui, A.A.; Iftikhar, A.; Latif, A.; Hassan, H.; Zhao, J.; Carew, J.S.; Nawrocki, S.T.; et al. Role and mechanism of autophagy-regulating factors in tumorigenesis and drug resistance. Asia. Pac. J. Clin. Oncol. 2021, 17, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Toh, P.P.C.; Luo, S.; Menzies, F.M.; Raskó, T.; Wanker, E.E.; Rubinsztein, D.C. Myc inhibition impairs autophagosome formation. Hum. Mol. Genet. 2013, 22, 5237–5248. [Google Scholar] [CrossRef] [Green Version]
- Sears, R.C. The Life Cycle of C-Myc: From Synthesis to Degradation. Cell Cycle 2004, 3, 1131–1135. [Google Scholar] [CrossRef]
- Mo, H.; He, J.; Yuan, Z.; Wu, Z.; Liu, B.; Lin, X.; Guan, J. PLK1 contributes to autophagy by regulating MYC stabilization in osteosarcoma cells. Onco Targets Ther. 2019, 12, 7527–7536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kon, M.; Kiffin, R.; Koga, H.; Chapochnick, J.; Macian, F.; Varticovski, L.; Cuervo, A.M. Chaperone-mediated autophagy is required for tumor growth. Sci. Transl. Med. 2011, 3, 109ra117. [Google Scholar] [CrossRef] [Green Version]
- Vara-Perez, M.; Felipe-Abrio, B.; Agostinis, P. Mitophagy in Cancer: A Tale of Adaptation. Cells 2019, 8, 493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Nie, P.; Zhou, C.; Hu, Y.; Duan, S.; Gu, M.; Jiang, D.; Wang, Y.; Deng, Z.; Chen, J.; et al. Oxidative stress-induced mitophagy is suppressed by the miR-106b-93-25 cluster in a protective manner. Cell Death Dis. 2021, 12, 209. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Choi, K.S. A critical role of superoxide anion in selenite-induced mitophagic cell death. Autophagy 2008, 4, 76–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, Y.; Hori, T.; Cooper, T.K.; Liao, J.; Desai, N.; Serfass, J.M.; Young, M.M.; Park, S.; Izu, Y.; Wang, H.-G. Bif-1 haploinsufficiency promotes chromosomal instability and accelerates Myc-driven lymphomagenesis via suppression of mitophagy. Blood 2013, 121, 1622–1632. [Google Scholar] [CrossRef] [Green Version]
- Pelengaris, S.; Littlewood, T.; Khan, M.; Elia, G.; Evan, G. Reversible activation of c-Myc in skin: Induction of a complex neoplastic phenotype by a single oncogenic lesion. Mol. Cell 1999, 3, 565–577. [Google Scholar] [CrossRef]
- Bisgrove, D.A.; Mahmoudi, T.; Henklein, P.; Verdin, E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 13690–13695. [Google Scholar] [CrossRef] [Green Version]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.; Li, L.; Bai, Y.; Yu, B.; Xie, C.; Wu, H.; Zhang, Y.; Huang, L.; Yan, Y.; Li, X.; et al. The long noncoding RNA LUCAT1 promotes colorectal cancer cell proliferation by antagonizing Nucleolin to regulate MYC expression. Cell Death Dis. 2020, 11, 908. [Google Scholar] [CrossRef]
- Chen, Q.; Shen, H.; Zhu, X.; Liu, Y.; Yang, H.; Chen, H.; Xiong, S.; Chi, H.; Xu, W. A nuclear lncRNA Linc00839 as a Myc target to promote breast cancer chemoresistance via PI3K/AKT signaling pathway. Cancer Sci. 2020, 111, 3279–3291. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Li, J.; Zhou, L.; Han, J.; Liu, R.; Zhang, H.; Ning, T.; Gao, Z.; Liu, B.; Chen, X.; et al. The c-Myc/miR-27b-3p/ATG10 regulatory axis regulates chemoresistance in colorectal cancer. Theranostics 2020, 10, 1981–1996. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Wang, L.; Chen, D.; Chang, Y.; Zhang, M.; Xu, J.-J.; Zhou, R.; Zhang, Q.-Y. LAPTM4B: An oncogene in various solid tumors and its functions. Oncogene 2016, 35, 6359–6365. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wei, X.H.; Pan, Y.P.; Li, H.C.; Yang, H.; He, Q.H.; Pang, Y.; Shan, Y.; Xiong, F.X.; Shao, G.Z.; et al. LAPTM4B: A novel cancer-associated gene motivates multidrug resistance through efflux and activating PI3K/AKT signaling. Oncogene 2010, 29, 5785–5795. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.; Wang, L.; Fei, X.-C.; Jiang, X.-F.; Zheng, Z.; Zhao, Y.; Wang, C.-F.; Li, B.; Chen, S.-J.; Janin, A.; et al. MYC is a positive regulator of choline metabolism and impedes mitophagy-dependent necroptosis in diffuse large B-cell lymphoma. Blood Cancer J. 2017, 7, e582. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Examples of MYCN/MYC/MYCL-Associated Binding Partners, Upstream Regulators, Effectors, and Target Genes and Their Signalling Pathways |
|---|---|
| Prostate cancer | Co-occupation with AR at FOXA1 and HOXB13 loci [24] |
| Pancreatic ductal adenocarcinoma | KRAS and YAP [25] |
| Medulloblastoma | SOX2 [26] |
| Neuroblastoma | Wnt/ β-catenin, mir-204 [27,28] |
| Colorectal cancer | Wnt-APC, CAD, UMPS, and CTPS [7] |
| Rhabdomyosarcoma | PAX3-FOXO1 [29] |
| Non-small cell lung cancer | KRAS, IL-23, and CCL9 [30,31] |
| Target | Mechanism of Regulation | Outcome | Malignancy |
|---|---|---|---|
| MYC | Antisense oligonucleotide morpholinos | Reduction of pro-angiogenic proteins and metastasis | Lung cancer [55] |
| linc00485/miR-298/MYC network | linc00485 silencing | Reduction of cancer cell proliferation | Lung cancer [57] |
| Linc00261/p300/CBP/MYC network | Linc00261 overexpression | Inhibition of cancer cell proliferation, migration, and metastasis in vitro | Pancreatic cancer [58] |
| LPP-AS2/miR-7-5p/EGFR/MYC | LPP-AS2 knockdown | Tumour growth inhibition | Glioma [59] |
| MYC/miR-150/EPG5 (autophagy) | Inhibition of MYC/miR-150 expression | Reduced cell growth in vitro and in vivo | NSCLC [60] |
| AMBRA1-PP2A/MYC regulated by mTOR | mTOR inhibition | MYC protein degradation and reduction in cell proliferation | Breast and lung carcinoma cell lines [61] |
| MYC activation of PERK/eIF2α/ATF4 arm of the UPR | PERK inhibition Autophagy inhibition | Reduced MYC-induced autophagy and tumour formation, Increase in MYC-dependent apoptosis | Human P493-6B lymphoblastoid cell line and MEF cells [62] |
| CMA/ CIP2A/ MYC | CMA activation | Proteasomal degradation of MYC | MEF cells [63] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jahangiri, L.; Pucci, P.; Ishola, T.; Trigg, R.M.; Williams, J.A.; Pereira, J.; Cavanagh, M.L.; Turner, S.D.; Gkoutos, G.V.; Tsaprouni, L. The Contribution of Autophagy and LncRNAs to MYC-Driven Gene Regulatory Networks in Cancers. Int. J. Mol. Sci. 2021, 22, 8527. https://doi.org/10.3390/ijms22168527
Jahangiri L, Pucci P, Ishola T, Trigg RM, Williams JA, Pereira J, Cavanagh ML, Turner SD, Gkoutos GV, Tsaprouni L. The Contribution of Autophagy and LncRNAs to MYC-Driven Gene Regulatory Networks in Cancers. International Journal of Molecular Sciences. 2021; 22(16):8527. https://doi.org/10.3390/ijms22168527
Chicago/Turabian StyleJahangiri, Leila, Perla Pucci, Tala Ishola, Ricky M. Trigg, John A. Williams, Joao Pereira, Megan L. Cavanagh, Suzanne D. Turner, Georgios V. Gkoutos, and Loukia Tsaprouni. 2021. "The Contribution of Autophagy and LncRNAs to MYC-Driven Gene Regulatory Networks in Cancers" International Journal of Molecular Sciences 22, no. 16: 8527. https://doi.org/10.3390/ijms22168527